

Abstract
Pyruvate dehydrogenase complex deficiency (PDCD) is a rare neurodegenerative disorder associated with abnormal mitochondrial metabolism. Structural brain abnormalities are common in PDCD. A case of a patient with PDCD with an unusual presentation is described. A 20-month-old boy with hypotonia and developmental delay, presented with hypoxia and respiratory distress due to bronchiolitis. During hospitalisation, he was prescribed PediaSure® feeds. Two days after starting these feeds, he developed respiratory arrest requiring intubation. His blood gas before arrest revealed lactate of 8.9 mmol/L despite normal haemodynamics. After stabilisation and a period of compulsory fasting, subsequent feeding with PediaSure® resulted in the recurrence of lactic acidosis. A metabolic workup revealed an elevated serum pyruvate level. Brain MRI was normal. Skeletal muscle biopsy confirmed PDCD. The most common cause of PDCD is a mutation in the X-linked PDHA1 gene. The severity of PDCD can range from neonatal death to more delayed onset of symptoms as in our index case. Normal brain MRI is reported in only 2% of patients with PDCD. There is no effective treatment for PDCD. In patients with proximal muscle weakness and feeding intolerance with glucose-containing feeds, the presence of lactic acidosis should raise the suspicion of PDCD irrespective of the patient's age and normal MRI.
Keywords: lactic acidosis, pyruvate dehydrogenase deficiency, ketogenic diet, magnetic resonance imaging, genetics, PediaSure®
Introduction
The pyruvate dehydrogenase complex (PDC) is a multienzyme complex located in the mitochondrial matrix, which serves as a catalyst for oxidative decarboxylation of pyruvate to acetyl CoA, the rate-limiting step in the Krebs cycle [1]. Pyruvate dehydrogenase complex deficiency (PDCD) is a rare genetic, potentially life-threatening mitochondrial disorder commonly associated with lactic acidosis, and progressive neurological and neuromuscular degeneration. Patients with PDCD usually present with hypotonia, lethargy and developmental delay. Definitive diagnosis is usually made by genetic testing. Structural abnormalities of the brain are quite common in PDCD. A case of a patient with a late-onset diagnosis of PDCD with proximal muscle weakness, lactic acidosis and a normal brain magnetic resonance imaging (MRI) with an unusual presentation is presented.
Case Report
A 20-month-old boy with a past medical history of developmental delay, hypotonia and feeding difficulty, presented to the emergency department (ED) with runny nose, cough, and increased work of breathing for five days. Birth history was insignificant. However, his developmental history was significant indicating he was unable to stand or crawl. He had just started to sit with support. Work up including audiology screen, fluorescent in-situ hybridisation, comparative genomic hybridisation microarray, and specific genetic testing for conditions like Prader-Willi syndrome as well as spinal muscular atrophy were normal. Neuroimaging included a brain MRI which was normal, except for an incidental finding of a small developmental venous anomaly in the right occipital lobe.
In the ED, vital signs revealed a temperature of 100.6° F (38.10C),a respiratory rate of 62 breaths per minute, heart rate 162 beats per min, blood pressure 111/60 mmHg, and 85% oxygen saturation at room air. On physical examination, he was in moderate respiratory distress with intercostal retractions, and coarse bilateral breath sounds with good perfusion. He received a fluid bolus and was placed on six litres high-flow nasal cannula which resulted in an improvement in his oxygen saturation. A chest radiograph was obtained which showed pulmonary hyper-expansion with bilateral streaky peripheral densities suggestive of viral lower airway disease (Figure 1). He was diagnosed with viral bronchiolitis and subsequently admitted for respiratory support. On day four after admission, his hypoxia worsened, raising the suspicion of a secondary pneumonia and intravenous (IV) Ampicillin (Athenex, Schaumburg, IL, USA) was started. Given his worsening respiratory status, he was made “nil per os” (NPO) and placed on IV fluids for hydration.
Fig. 1.
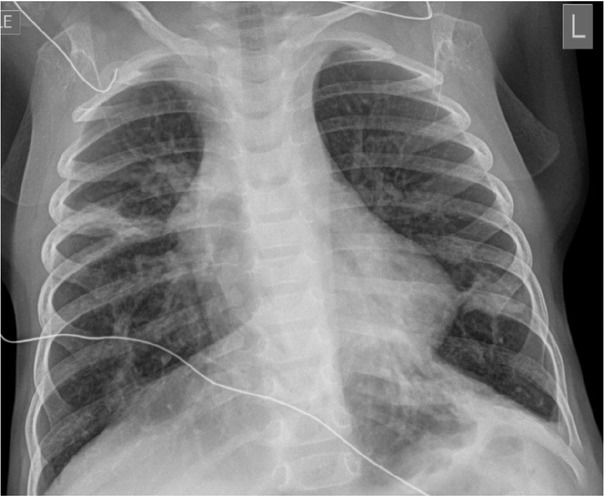
Chest radiograph on admission. The image shows pulmonary hyperexpansion with interstitial prominence and areas of patchy density which may be due to a viral airway disease with areas of atelectasis.
His respiratory status improved on day five after admission, and he was restarted on enteral nutrition with PediaSure®. IV fluids were discontinued. Within forty-eight hours of starting feeds with PediaSure®, his respiratory and mental status worsened. He was difficult to arouse and developed respiratory arrest requiring emergent intubation and subsequent transfer
to the paediatric intensive care unit (PICU). His capillary blood gas before arrest revealed a metabolic acidosis with respiratory compensation (Table 1). He had a significantly elevated lactic acid level of 8.9 mmol/L despite normal haemodynamics. A viral respiratory panel (VRP) and blood cultures were obtained, and antibiotics were switched to IV Piperacillin/Tazobactam (Fresenius Kabi, Lake Zurich, IL, USA) and Vancomycin (Hospira, Lake Forest, IL, USA) to cover a broader spectrum of organisms for an intended duration of ten days. His VRP came back positive for Metapneumovirus, which was thought to be the cause of his respiratory illness.
Table 1.
Pertinent laboratory results of the patient
| Labs | Values | |||||
|---|---|---|---|---|---|---|
| Time of collection | pH | pCO2 (mmHg) | pO2 (mmHg) | HCO3 (mmol/L) | Serum lactate (mmol/L) | |
| 2 hours prior to respiratory arrest (Hospital Day 6) (Capillary gas) | 7.42 | 22 | 81 | 14.2 | 8.9 | |
| Blood | 1 hour after respiratory arrest (Hospital Day 6) (Capillary gas) | 7.12 | 41.5 | 125 | 13.5 | 8.1 |
| gases | 36 hours after initiation of Pediasure® (Hospital Day 8) (Venous gas) | 7.31 | 51.1 | 45.7 | 25.4 | 9.3 |
| After development of ARDS (Hospital Day 13) (Arterial gas) | 7.42 | 52 | 55 | 33.5 | 1.3 | |
| Serum pyruvate level (mg/dL) | 2.42 (Reference range: 0.3-1.5) | |||||
| Blood lactate to pyruvate ratio | 14.87 | |||||
| Activated PDC level (nmol/min/mg protein) | 0.35 (Reference range: 1.2-6.52) | |||||
| Skeletal muscle biopsy | PDC/E3 ratio | 0.4 (Reference range: 0.82-4.54) | ||||
| Pyruvate dehydrogenase and mitochondrial complex panel | Positive for pyruvate dehydrogenase alpha 1 (PDHA1) gene mutation | |||||
In the PICU, he was again made NPO until he was stabilised. His repeat blood gases normalised over twelve hours and his lactate results improved. Feeding with PediaSure® was restarted two days after PICU admission. A rise in arterial lactate from 2.9 to 9.3 mmol/L after restarting enteral nutrition was noted (Figure 2). He was subsequently made NPO again.
Fig. 2.
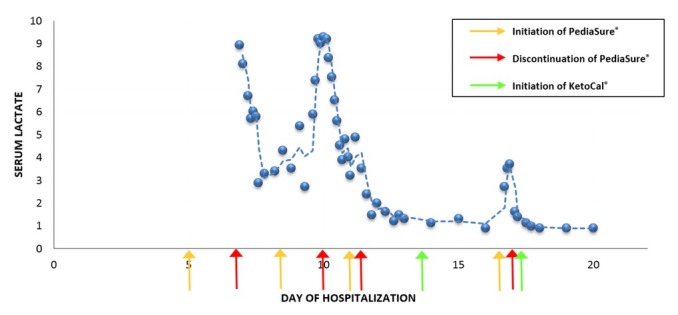
Change in lactate associated with introduction of feeds. The figure describes an increase in lactate after each time the patient is started on PediaSure® feeds. The level decreased to normal after discontinuation of PediaSure®. The figure also describes no change in lactate level after starting the patient on KetoCal® feeds.
Further trials of enteral administration of PediaSure® were unsuccessful, secondary to recurrent episodes of lactic acidosis (Figure 2). A geneticist was consulted due to concerns regarding the patient’s hypotonia and persistent lactic acidosis despite good perfusion. A metabolic workup that included a funduscopic examination, plasma amino acids, acylcarnitine, carnitine, urine organic acids and pyruvate level was recommended. Also recommended was obtaining a whole exome sequencing and a myotonia congenita panel. All results were normal except for an elevated pyruvate level with a blood lactate to pyruvate ratio of 14.87. This was suggestive of pyruvate dehydrogenase complex deficiency (PDCD) (Table 1). A skeletal muscle biopsy showed low levels of activated PDC and PDC/E3 ratio (Table 1) as well as a pyruvate dehydrogenase and mitochondrial complex panel testing positive for pyruvate dehydrogenase alpha 1 (PDHA1) gene mutation confirming the diagnosis of PDCD.
The patient was given a trial of a ketogenic diet (KD) which did not result in an elevation of lactate levels (Figure 2). He remained on prolonged mechanical ventilation secondary to the development of acute respiratory distress syndrome (ARDS). On the thirteenth day of hospitalisation, he developed severe ARDS with an oxygenation index of 32.7. His chest radiograph, after the development of ARDS, is shown in Figure 3. He also required inhaled nitric oxide, oscillator and prone positioning during his ICU stay. On the thirty-eighth day of hospitalisation he was extubated and subsequently transferred out of the PICU to the general inpatient paediatrics service for further management.
Fig. 3.
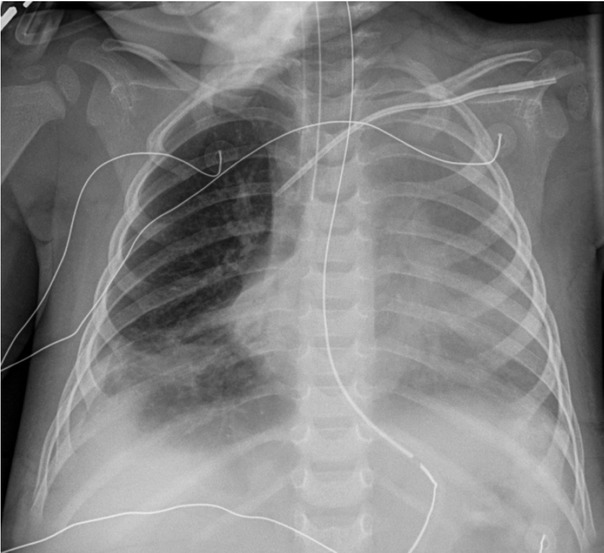
Chest radiograph on hospital day 13. The image shows generalized opacification of the left hemithorax with no mediastinal shift. This could represent a combination of pleural fluid and atelectasis. Persistent right lower lobe atelectasis is seen along with right pleural fluid.
He was discharged home from hospital on day fifty-two, and a KetoCal® 4:1 formula diet was recommended.
Discussion
PDCD is a rare genetic disorder resulting from an abnormal mitochondrial metabolism and is characterised by lactic acidosis as well as features of neurological damage including hypotonia, poor feeding, developmental delay and lethargy. PDC is located in the mitochondrial matrix and is a multi-enzyme complex whose primary function is to act as a catalyst for the conversion of pyruvate to acetyl CoA via the process of oxidative decarboxylation [1]. PDC is made up of three catalytic and one structural domain. The catalytic domains consist of E1, is a heterotetramer of two alpha and two beta subunits, E2 and E3 domains [2]. The structural domain is identified as the E3-binding protein [3, 4]. Thus, in PDCD, decrease in the availability of acetyl CoA reduces the production of cofactors nicotinamide adenine dinucleotide and flavin adenine dinucleotide, whose primary function is to provide the respiratory chain with electrons for oxidative phosphorylation. This promotes the cytoplasmic reduction of acetyl CoA to lactate or transamination to alanine [5]. Therefore, a defect in any component of PDC, congenital or acquired, can lead to elevated lactate levels in the body and inability to produce energy at the cellular level, with resultant neurological and neuromuscular dysfunction.
Despite being reported in the literature for four decades, the exact incidence of PDCD remains unknown [6]. More than fifty mutations have been reported in patients with PDCD [7]. The most commonly identified mutations are found in the PDHA1 gene, which is an X-linked gene encoding the E1α subunit, and the majority of these mutations are de novo mutations [8, 9].
Given the X-linked nature of PDCD, affected males are generally symptomatic. In contrast, heterozygous females have variable X-inactivation pattern in different tissues and thus may present with varying symptoms [10]. Disease severity in PDCD ranges from the most severe form leading to neonatal death due to persistent lactic acidosis to a more subtle and delayed onset of symptoms as seen in the current reported patient. Most patients with PDCD manifest with impairment of neurological and motor function. Additional symptoms may include developmental delay, hypotonia, seizures and encephalopathy. There are a few reports of ataxia of childhood-onset or isolated neuropathy with normal cognition. Structural abnormalities of the brain are common in PDCD and can include cortical atrophy, ventriculomegaly, dysgenesis of the corpus callosum and subacute necrotising encephalomyopathy [4]. Normal brain MRI in patients with PDCD, is rarely reported in the literature. A study on 371 patients with PDCD reported normal MRI in only 7 (2%) patients, all of whom had E1α deficiency [4]. Ventriculomegaly was the most common abnormality reported in this study.
The patient in this report had very nonspecific symptoms of developmental delay, hypotonia and remote history of feeding intolerance but no other findings or symptoms which would have suggested a metabolic cause. Symptoms of PDCD presented late, at twenty months of age, as these usual present within the first year of life. Any suspicion of PDCD or any other metabolic disorder was very low as he presented with a normal brain MRI. What makes the case even more unique is that the diagnosis was made based on lactic acidosis associated with PediaSure® feeds in a haemodynamically stable patient. PediaSure® contains glucose which leads to the production of pyruvate upon glycolysis. As previously mentioned, in PDCD, there is decreased conversion of pyruvate to acetyl CoA which leads to an increase in pyruvate level which in turn, leads to an increase in lactate.
Unfortunately, there is no effective treatment for PDCD. Several strategies have been employed, with variable success [11]. Thiamine pyrophosphate supplementation is often tried since it is an obligate cofactor for PDC's E1 component. Individuals who respond to thiamine usually have a better outcome. As in the present patient, lactic acidosis may be decreased by giving a high fat/low carbohydrate, i.e. KD, typically with fats to carbohydrate plus protein ratio of 3–4 to 1 [4]. It is hypothesised that this diet induces ketosis, which provides an alternative source of acetyl CoA [4]. KD does not alleviate the neurological symptoms, though some case reports have shown improvement in the clinical course [12]. A less restrictive KD such as the modified Atkins and low glycaemic index diets may be tried in those with compliance issues to the standard KD [13]. Oral citrate can also be used for acidosis. Dichloroacetate (DCA) inhibits pyruvate dehydrogenase kinases and activates PDC. Randomised control trials have shown that the use of DCA only blunts postprandial increase in lactate but does not improve clinical outcome [14].
Although the natural history of PDCD does not appear to be affected by gender, there are suggestions that mortality might be higher in males [4]. The majority of the individuals with PDCD die in childhood, though there are reports of some surviving into adolescence and young adulthood [4].
Conclusion
In patients with proximal muscle weakness and feeding intolerance especially with glucose-containing feeds, one should consider metabolic workup including blood lactate level. If recurrent lactic acidosis is present in a patient with normal haemodynamics, genetics should be consulted irrespective of the patient's age. The presence of a normal MRI does not exclude the possibility of PDCD and work up should be considered if clinical suspicion is strong.
Footnotes
Conflict of interest None to declare.
Ethical Review This case report was exempt from our hospital's Ethics Board review, and verbal consent of the parents was obtained to publish this case.
References
- 1.Patel MS, Nemeria NS, Furey W, Jordan F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem. 2014;289:16615. doi: 10.1074/jbc.R114.563148. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maj MC, Cameron JM, Robinson BH. Pyruvate dehydrogenase phosphatase deficiency: orphan disease or an under-diagnosed condition? Mol Cell Endocrinol. 2006;249:1. doi: 10.1016/j.mce.2006.02.003. –. [DOI] [PubMed] [Google Scholar]
- 3.Kara B, Genc HM, Uyur-Yalcin E. Pyruvate dehydrogenase-E1alpha deficiency presenting as recurrent acute proximal muscle weakness of upper and lower extremities in an 8-year-old boy. Neuromuscul Disord. 2017;27:94. doi: 10.1016/j.nmd.2016.11.001. et al. –. [DOI] [PubMed] [Google Scholar]
- 4.Patel KP, O'Brien TW, Subramony SH, Shuster J, Stacpoole PW. The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab. 2012;106:385. doi: 10.1016/j.ymgme.2012.03.017. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stenlid MH, Ahlsson F, Forslund A, von Dobeln U, Gustafsson J. Energy substrate metabolism in pyruvate dehydrogenase complex deficiency. J Pediatr Endocrinol Metab. 2014;27:1059. doi: 10.1515/jpem-2013-0423. –. [DOI] [PubMed] [Google Scholar]
- 6.DeBrosse SD, Okajima K, Zhang S. Spectrum of neurological and survival outcomes in pyruvate dehydrogenase complex [PDC] deficiency: lack of correlation with genotype. Mol Genet Metab. 2012;107:394. doi: 10.1016/j.ymgme.2012.09.001. et al. –. [DOI] [PubMed] [Google Scholar]
- 7.Imbard A, Boutron A, Vequaud C. Molecular characterization of 82 patients with pyruvate dehydrogenase complex deficiency. Structural implications of novel amino acid substitutions in E1 protein. Mol Genet Metab. 2011;104:507. doi: 10.1016/j.ymgme.2011.08.008. et al. –. [DOI] [PubMed] [Google Scholar]
- 8.Brown RM, Head RA, Boubriak II, Leonard JV, Thomas NH, Brown GK. Mutations in the gene for the E1beta subunit: a novel cause of pyruvate dehydrogenase deficiency. Hum Genet. 2004;115:123. doi: 10.1007/s00439-004-1124-8. –. [DOI] [PubMed] [Google Scholar]
- 9.Ciara E, Rokicki D, Halat P. Difficulties in recognition of pyruvate dehydrogenase complex deficiency on the basis of clinical and biochemical features. The role of next-generation sequencing. Mol Genet Metab Rep. 2016;7:70. doi: 10.1016/j.ymgmr.2016.03.004. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Meirleir L. Defects of pyruvate metabolism and the Krebs cycle. J Child Neurol. 2002;17:3S26. Suppl 3. –. [PubMed] [Google Scholar]
- 11.Kerr DS. Treatment of congenital lactic acidosis: a review. Int Pediatr. 1995;10:75. –. [Google Scholar]
- 12.Kossoff EH, Zupec-Kania BA, Amark PE. Optimal clinical management of children receiving the ketogenic diet: recommendations of the International Ketogenic Diet Study Group. Epilepsia. 2009;50:304. doi: 10.1111/j.1528-1167.2008.01765.x. et al. –. [DOI] [PubMed] [Google Scholar]
- 13.El-Gharbawy AH, Boney A, Young SP, Kishnani PS. Follow-up of a child with pyruvate dehydrogenase deficiency on a less restrictive ketogenic diet. Mol Genet Metab. 2011;102:214. doi: 10.1016/j.ymgme.2010.11.001. –. [DOI] [PubMed] [Google Scholar]
- 14.Stacpoole PW, Kerr DS, Barnes C. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006;117:1519. doi: 10.1542/peds.2005-1226. et al. –. [DOI] [PubMed] [Google Scholar]