

Abstract
Acquired sensorineural hearing loss is one of the most prevalent chronic diseases, and aging and acoustic overexposure are common contributors. Decades of study in animals and humans have clarified the cellular targets and perceptual consequences of these forms of hearing loss, and preclinical studies have led to the development of therapeutics designed to slow, prevent or reverse them. Here, we review the histopathological changes underlying age-related and noise-induced hearing loss and the functional consequences of these pathologies. Based on these relations, we consider the ambiguities that arise in diagnosing underlying pathology from minimally invasive tests of auditory function, and how those ambiguities present challenges in the design and interpretation of clinical trials.
Keywords: Age-related hearing loss, Cochlear drug therapy, Cochlear synaptopathy, Noise-induced hearing loss Sensorineural hearing loss
1. Introduction
Two of the most common forms of acquired sensorineural hearing loss in humans are noise-induced hearing loss (NIHL) and age-related hearing loss (ARHL). Therapeutic approaches to either condition may, some day, include drugs to prevent the onset or retard the progression of these conditions, and possibly even treatments to reverse the impairments long after they are first observed. Both conditions are challenging to study, because the mechanisms underlying each are likely so diverse. For example, the functionally important structural changes underlying the NIHL caused by a single blast exposure producing peak sound pressures in excess of 180 dB SPL are likely fundamentally different from those underlying the hearing loss caused by 40 years of daily exposure to workplace noise at levels closer to 90 dB SPL. Similarly, the rate and nature of ARHL likely interact with lifetime doses of subtraumatic noise exposure, and both reflect contributions from known and unknown genetic factors (see Wang and Puel 2018 for review).
Nevertheless, the translation from animal models to human therapeutics is arguably more straightforward for NIHL than for conditions where the underlying insult is much less clear. For example, for a condition like Parkinson’s disease, the animal model is a model primarily in the sense that it recapitulates aspects of the phenotype. As such, there is no guarantee that addressing the phenotype in the model system will effectively address the disease process that it appears to mimic.
Our group studies both NIHL and ARHL in both humans and animal models, and here we discuss the progress and future directions for continued efforts to translate clinically relevant observations in animals to the human condition. We also discuss pitfalls that could cause results from animal experiments to fail to translate to the clinical issues they seek to address.
2. Diagnosing, preventing and reversing hair cell loss
The design of effective therapeutics to prevent or reverse NIHL or ARHL requires, at the most basic level, an understanding of the functionally important structural changes that should be targeted. Progress here also requires non-invasive assays of function that can be employed in differential diagnosis of pathology and assessment of treatment efficacy (see Rüttiger et al., 2017). Cochlear sensory function and sensory to neural communication can be assayed using complementary and non-invasive techniques that are applicable to both to laboratory animals and clinical patients. For example, on the input side, the function of outer hair cells (OHCs) can be assessed using distortion product otoacoustic emissions (DPOAEs), acoustic signals produced by the active and non-linear response of healthy OHCs in a healthy cochlea, as recorded with a microphone in the ear canal. On the output side, the auditory brainstem response (ABR) is a sound-evoked potential generated by neuronal circuits in the ascending auditory pathways that can be recorded via scalp or needle electrodes. The first wave of the ABR represents summed activity of cochlear nerve fibers synapsing with inner hair cells (IHCs). When combined and captured as both threshold and suprathreshold functions, these hair cell- and neural-based responses can help dissect sensory vs neural dysfunction in the auditory periphery.
2.1. Threshold shifts and hair-cell loss in NIHL and ARHL
The permanent deleterious effects of noise on human hearing include a classic pattern of threshold shifts often initially restricted to frequencies near 4 kHz, as seen in humans employed for many years in noisy professions (Passchier-Vermeer, 1974). Correspondingly, humans with 4-kHz audiometric notches, and a documented history of work-related acoustic overexposure, often show a mid-cochlear loss of outer hair cells (OHCs), where the epithelium is tuned to 4 kHz (e.g. McGill and Schuknecht, 1976). In the laboratory, countless animal studies have likewise documented that acoustic overstimulation causes loss of OHCs and elevates thresholds (Saunders et al., 1991). Since the OHCs function as biological amplifiers (Dallos, 2008), enhancing the sound-induced vibrations of the sensory epithelium, preventing or reversing OHC loss should prevent or reverse some of the threshold shifts in NIHL.
The apparent predilection of the 4-kHz region to human NIHL likely arises from a combination of factors including: 1) a general tendency for basal cochlear regions to be more vulnerable to injury (Fettiplace and Nam, 2018) such that damage potential increases with increasing stimulus frequency (Liberman and Kiang, 1978) and 2) an ear-canal resonance and middle-ear filter function that together emphasize transmission of mid-frequency energy to the cochlea (Geisler, 1998). Animal studies show that damage and threshold shift from narrow-band exposures tend to be tonotopically appropriate (Liberman and Kiang, 1978), with a small upward frequency shift in peak effects that is well explained by mechanical cochlear nonlinearities (Ruggero, 1992). In addition, there is a tonotopically inappropriate noise-induced loss of hair cells in the extreme basal (high-frequency) region of the cochlea (Liberman and Kiang, 1978). These “hook” lesions can appear before the damage in the tonotopically appropriate area (Wang et al., 2002). The mechanisms underlying such lesions are unclear, but they cannot be easily explained by cochlear mechanics (Overstreet et al., 2002). Clear histological evidence for noise-induced hook lesions is lacking in human cochleas, because the issue is clouded by the basal-turn hair cell loss that is ubiquitous in the aging ear, as discussed below. Audiometric evidence for these noise-induced hook lesions is suggested by the threshold elevations at extended high frequencies (> 8 kHz) in young human subjects with unprotected exposure to high-level recreational sound (Liberman et al., 2016). Similarly, the exaggerated threshold elevations above 8 kHz in older subjects with otherwise normal audiograms (Dubno et al., 2003) may also be due a lifetime accumulation of subtraumatic noise exposures.
Threshold-shift patterns in the aging human ear have been well characterized across the standard audiometric frequency range in many studies (e.g. Pearson et al., 1995), and a progressive high-tone hearing loss with smaller shifts at low frequencies are hallmarks of ARHL. Patterns of ARHL in animal models are more complex. In gerbils (Schmiedt et al., 2002), and in some mouse strains (Ohlemiller et al., 2010), ARHL is predominantly at the high frequency end of the animal’s hearing range. However, in aging rats, the threshold shifts are more evenly distributed across frequencies (Balogova et al., 2017; Cai et al., 2018). When compared at frequencies corresponding to the same cochlear locations (70% and 35% of the distance from the base) and at ages normalized to mean lifespan for each species, age-related threshold elevations are remarkably similar for CBA/CaJ mice and humans screened for NIHL and frank otopathology (Sergeyenko et al., 2013). This inbred strain is notable, among the many mouse strains commonly studied in auditory research, in that cochlear function is particularly well preserved with increasing age (Zheng et al., 1999). This is in contrast to the ARHL in another well-studied strain (C57BL/6) which shows early-onset threshold shifts that progress more rapidly (Zheng et al., 1999). The early onset ARHL in C57BL/6 arises from a mutation in the gene for a protein (cadherin 23) in the extracellular tip-links connecting adjacent stereocilia (Noben-Trauth et al., 2003). Such dramatic interstrain differences point to genetic makeup as a key variable in human ARHL; however, there is currently no compelling reason to assume that this cadherin 23 mutation, per se, has significant relevance to ARHL in the normal-aging human.
Hair-cell loss patterns in human ARHL are best characterized from studies of microdissected sensory epithelia (Bredberg, 1968; Wu et al., 2018), rather than from sectioned material (Schuknecht, 1974), where fractional hair cell loss is not well captured. Such studies of “normal-aging” humans, i.e. those without explicit otologic disease, show IHC loss restricted to the extreme basal end of the aging cochlea, whereas OHC loss progresses in both apical and basal ends of the cochlea, with selective sparing of mid-cochlear OHCs. Apical OHC losses are under-represented by the audiogram, because 1) OHCs contribute less to cochlear amplification at low frequencies than at high frequencies (Liberman et al., 2002; Schmiedt et al., 2002), 2) the apical extreme of the human cochlea is likely tuned to frequencies lower than the lowest frequency in the routine audiogram (250 Hz; Greenwood, 1990) and 3) the best thresholds of the most apical auditory nerve fibers are only slightly lower than the below-best-frequency thresholds of more basal fibers; the absence of the former can be masked by the latter, if they are still functional.
The hair cell survival patterns in animal models of ARHL are more varied. A basal-to-apical progression of IHC loss, coupled with both apical and basal foci of OHC loss, have been reported in mice i.e. CBA/CaJ and CBA/J (Ohlemiller et al., 2010; Sergeyenko et al., 2013). However, in two different rat strains (Balogova et al., 2017; Keithley and Feldman, 1982) as well as in the aging gerbil (Tarnowski et al., 1991) and rabbit (Bhattacharyya and Dayal, 1989), the OHC loss is much greater in the cochlear apex than in the base. This apparent discrepancy between normal-aging humans and other mammals raised in a controlled laboratory setting is consistent with the speculation that much of the high-frequency hearing loss in human ARHL arises from acoustic overstimulation. Such speculation is further reinforced by classic studies of human hearing in populations living in isolated non-industrialized societies, where the progressive high-tone hearing loss characterizing ARHL is not seen (Rosen et al., 1962). Comparing the rates of age-related OHC and IHC loss for CBA/CaJ mice vs. normal-aging humans is also interesting in this regard. The mouse data (Figure 1) show virtually no loss of OHCs or IHCs until well past the middle of the lifespan, whereas the human data, from two studies carried out 50 years apart (Bredberg, 1968; Wu et al., 2018) show steady loss, especially of OHCs, throughout life. Again, perhaps the accelerated hair cell loss in young and middle-aged humans reflects the steady stream of subtraumatic noise exposures that can be avoided in a controlled laboratory setting.
Figure 1: Age related loss of hair cells in human vs. mouse.
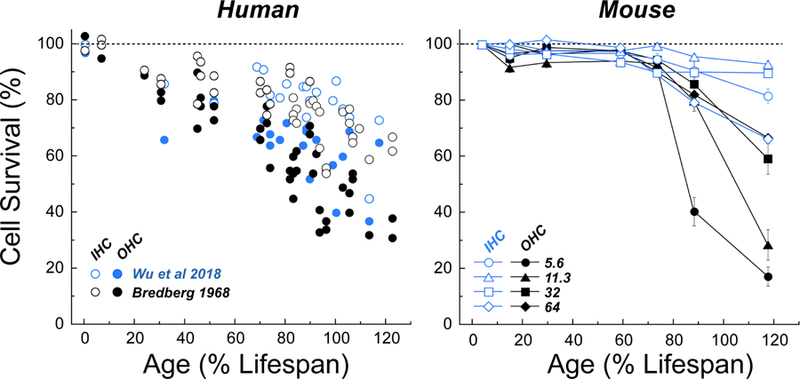
Hair cell data are normalized and expressed as % survival. Age is normalized and expressed as % lifespan, using a value of 76 yrs for humans and 2.1 yrs for mouse, as detailed in Sergeyenko et al. (2013). Mouse data (CBA/CaJ) are replotted from our prior work (Sergeyenko et al., 2013); human data are from two studies (Bredberg, 1968; Wu et al., 2018) and represent values averaged across the entire cochlear spiral.
2.2. Mechanisms of, and therapies for, hair cell loss
The longitudinal gradients of age-related OHC loss in human are intriguingly complementary to those of the cochlear projections from the olivocochlear efferent pathway, a sound-evoked negative feedback system known to protect the ear from NIHL (Guinan, 2006; Rajan, 1991). The density of these efferent terminals on OHCs peaks in mid-cochlear regions in all well-studied animal models, e.g. cat (Liberman et al., 2000), guinea pig (Liberman and Gao, 1995) and mouse (Maison et al., 2003). Interestingly, a study of the long-term effects of cochlear de-efferentation in mice showed acceleration of the age-related apical and basal loss of OHCs compared to normal-aging controls (Liberman et al., 2014). A similar mid-cochlear distribution of efferent terminals is seen in human cochleas (Liberman unpublished); it is therefore possible that therapeutics designed to enhance the magnitude or duration of efferent effects on cochlear function (e.g. Taranda et al., 2009) could be beneficial in protecting the ear from both NIHL and ARHL. To our knowledge, no clinical trials based on such approaches have been initiated.
Much of noise-induced OHC loss in animal models of NIHL appears to involve the apoptotic cell-death pathways (Furness, 2015). Much less is known about the mechanisms of cell death in ARHL. In guinea pigs, cochlear perfusion of a drug engineered to enter cell nuclei and block a key enzyme in the molecular cascade leading to apoptosis, prevented much of the noise-induced threshold shift and OHC loss seen in exposed animals perfused with vehicle only (Wang et al., 2003). The treatment was also successful in protecting hair cells and reducing threshold shifts after administration of an ototoxic antibiotic. The signaling pathways underlying apoptosis are highly conserved across animal species; thus, such treatments could also be successful in humans. Efficacy in the animal study was shown when drug delivery began 2 days prior to cochlear insult (Wang et al., 2003), which would be impractical for noise-exposure indications. However, hair cell loss continues for several days after acoustic overstimulation (Wang et al., 2002), and such a treatment might still be beneficial if delivered after the exposure. A clinical trial of this drug was recently completed in humans with idiopathic sudden sensorineural hearing loss of < 72 hrs duration (clinicaltrials.gov; drug name: AM111), although no results have been posted. Acute threshold elevations from noise or drug exposure were explicit exclusion criteria for entry into the study. Lack of success in such a trial could arise because the mechanisms underlying the target condition, sudden idiopathic sensorineural hearing loss, (including the role of apoptosis in the damage progression) are very different from those in noise damage or aminoglycoside ototoxicity. Furthermore, evidence suggests that cell death mechanisms shift from apoptosis to necrosis after the integrity of the epithelial barrier at the endolymphatic surface of the organ of Corti is breached, as supporting cells phagocytize the remains of apoptotic hair cells (Anttonen et al., 2014; Bohne and Rabbitt, 1983).
The last two decades have seen tremendous advances in our understanding of the signaling pathways underlying hair cell development in utero (Basch et al., 2016), and these insights have been exploited in animal models to design drug treatments to regenerate hair cells and restore some cochlear function after insult. Inspired by discoveries clarifying a role for the transcription factor atoh1 (math1) in the generation of hair cells from sensory cell progenitors (Bermingham et al., 1999), a guinea pig study showed that cochlear injection of a viral vector containing the atoh1 gene could partially transdifferentiate surviving supporting cells into hair-cell chimera, if delivered simultaneously with the ototoxic-drugs (Kawamoto et al., 2003). Similarly, developmental studies of the “notch” pathway, another key molecular signal in the control of hair-cell fate during development, inspired researchers to assay existing small-molecule inhibitors of this pathway from a drug library and showed that application of one such inhibitor to the round-window membrane within 24 hours after noise exposure could elicit transdifferentation of supporting cells into hair cells in the sensory epithelium, and small improvements of cochlear thresholds re untreated exposed controls (Mizutari et al., 2013). Since the myelinated auditory nerve fibers (ANFs) that carry the acoustic signals to the brain exclusively contact IHCs (Liberman, 1982), the OHCs presumably do not need to be reinnervated for such a regenerative therapy to be effective. However, efficacy of such treatments at trauma-treatment intervals of longer than 24 – 48 hrs is currently unknown. The long-term survivability of such chimeric cells, halfway between hair cells and supporting cells, is also untested.
The signaling molecules both upstream and downstream of the atoh1 and notch pathways are highly conserved; thus, a similar treatment could work in humans, under appropriate conditions. A clinical trial of intralabyrinthine perfusion of an adenovirus containing the cDNA for the human version of atoh1 is underway, with an expected completion date of 2021 (Clinicaltrials.gov; drug name: CGF166). The inclusion criteria are non-fluctuating severe-to-profound bilateral or unilateral hearing loss of indefinite duration. An important issue with longer trauma-treatment intervals is the question of survival of the supporting cells (Oesterle and Campbell, 2009). For transdifferentiation approaches to work, appropriately responsive supporting cell remnants must survive. In many long-deaf ears, the organ of Corti has been replaced by what appears to be a completely undifferentiated cuboidal epithelium, which may no longer be responsive to the relevant molecular signals (Izumikawa et al., 2008). Much more needs to be known about the condition of remaining epithelial cells in human ears with different deafness etiologies and durations (deTorres et al., 2018). Other challenges include a lack of knowledge about the relative permeability of the round window membrane to drugs in different species and the dynamics of drug distribution in the larger human cochlea (Kang et al., 2016).
3. Diagnosing, preventing and reversing stereocilia damage
3.1. Stereocilia damage and threshold shifts in NIHL and ARHL
Studies in cat, guinea pig, mouse and rabbit have all shown that permanent noise-induced damage to the stereocilia bundles on both IHCs and OHCs is an important contributor to noise-induced threshold shifts (Engstrom et al., 1983; Liberman and Dodds, 1984; Robertson, 1982; Wang et al., 2002). In all three species, reports have documented dramatic noise-induced permanent threshold shifts (PTSs), as great as 60 dB (Liberman and Dodds, 1984), in ears with virtually no loss of hair cells. In such cases, the threshold shift is well correlated with the extent and severity of stereocilia damage on either IHCs or OHCs, including disarray, fusion and loss. This type of stereocilia damage can be widespread on surviving hair cells at least as long as two years after noise exposure (Liberman and Mulroy, 1982). Although stereocilia dysfunction in the developing ear typically leads to hair cell death, as seen in mouse models with genetic abnormalities in specialized proteins involved in stereocilia function (e.g. Vreugde et al., 2002), it is clear that adult hair cells can survive with normal looking cytoplasmic organelles and at least some surviving synaptic connections (further discussion below) long after acoustic overexposure (Liberman and Mulroy, 1982).
3.2. Diagnosis and treatment of stereocilia damage in NIHL or ARHL
If the NIHL or ARHL is predominately due to stereocilia damage, preventing hair cell death, e.g. with anti-apoptotic drugs, will be ineffective in preventing the hearing loss. One study suggests that forced overexpression of atoh1, a key transcription factor in the generation of hair cells from progenitor cells during development, can restore hair bundles and cochlear function after noise damage in a guinea pig model (Yang et al., 2012). Atoh1 is highly conserved (Mulvaney and Dabdoub, 2012), thus a treatment based on this pathway in animal models could ultimately be applicable to humans.
In human ears, damaged stereocilia can be seen on surviving hair cells in cases with sensorineural hearing loss of various etiologies (Horner, 1992; Kimura et al., 1976) including ARHL (Scholtz et al., 2001). However, we have no quantitative data on the mix of hair cell loss vs. stereocilia damage in either NIHL or ARHL in humans, and there is currently no obvious way to unambiguously diagnose the mix from any physiological or psychophysical measure. Indeed, our understanding of the mix of these two fundamentally different mechanisms of NIHL is quite superficial, even in animal models. It is not unlikely that the relative importance of hair cell death vs. stereocilia damage in NIHL differs according to the spectrum, intensity and duration of exposure, much as these stimulus variables are key determinants of the magnitude of NIHL perse. Existing animal studies suggest, for example, that in aminoglycoside ototoxicity, few surviving hair cells show stereocilia damage and most of the threshold shift is due to hair cell death (Liberman and Dodds, 1984). Thus, the prevalence of stereocilia damage as an important contributor to hearing impairment differs substantially based on the etiology in ways that are incompletely understood at present.
4. Diagnosing, preventing and reversing noise-induced cochlear synaptopathy
4.1. Synaptopathy and hearing dysfunction in NIHL or ARHL
After decades of focus on the sensory (hair cell) component of sensorineural hearing loss, animal studies have recently turned attention to the status of the peripheral neural component, the ANFs. Recent studies have shown that the synaptic connections between ANFs and IHCs are the most vulnerable elements in the cochlea after acoustic overstimulation (Kujawa and Liberman, 2009) and that these synaptic connections also disappear before hair cells in ARHL (Sergeyenko et al., 2013). Dramatic cochlear synaptopathy has been demonstrated in mouse, guinea pig, chinchilla, rat and rhesus monkey (Hickox et al., 2017; Kujawa and Liberman, 2009; Lin et al., 2011; Valero et al., 2017), and even acoustic overexposures causing only a temporary threshold shift (TTS) can lead to a permanent loss of ANF peripheral synapses, despite no loss of IHCs or OHCs and no damage to stereocilia. The synapses disappear immediately after noise exposure (Liberman et al., 2015), and the peripheral axons of these bipolar ANFs degenerate within a few weeks, whereas the cell bodies and the central axons of ANFs can survive for years in animals (Fernandez et al., 2015; Kujawa and Liberman, 2009), and perhaps for decades in humans (Liu et al., 2015). Similarly, when normalized to total lifespan for each species, age-related loss of ANF cell bodies (i.e. spiral ganglion cells) progresses similarly and reaches a similar degree of loss at the oldest ages (Makary et al., 2011; Sergeyenko et al., 2013). The long-term survival of these spiral ganglion cells and their central axons is amply demonstrated by the long term effectiveness of cochlear implants in humans (Shannon, 2012). Since each ANF contacts only a single IHC via a single synaptic connection, each lost synapse translates directly into a silenced ANF that no longer carries any information about acoustic stimuli.
In animal models, the synapse loss can be as high as 50% following exposures causing a large initial (40 – 50 dB), but ultimately reversible, threshold shift. Dramatic synaptopathy is also present in ears with PTS and hair cell loss (Valero et al., 2017), and further experiments characterizing the influence of hair cell damage on synaptic and neural degeneration are underway. Although it is not yet clear the extent to which synaptopathy is present in human ears with an acoustic trauma history, a classic study of 14 cases with definitive acoustic trauma history showed several cases with spiral ganglion cell losses in cochlear regions with minimal IHC loss (McGill and Schuknecht, 1976). In one more recent study of a 55 yr-old with a classic 4 kHz notch and a corresponding mid-cochlear patch of missing OHCs, almost 70% of the spiral ganglion cells in the 4 kHz region had no peripheral axons, despite survival of many of the IHCs they formerly contacted (Liu et al., 2015).
The prevalence of primary neural degeneration in human ears was recently documented in 23 normal-aging specimens, i.e. cases without explicit otologic disease (Wu et al., 2018). As shown in Figure 2, the age-related loss of ANF peripheral axons was almost three times faster than the loss of the IHCs that they originally contacted, and the degree of synaptopathy was even more dramatic than that originally demonstrated in mouse. Thus, in the normal-aging human, many surviving IHCs are partly de-afferented, and the disconnected ANFs are silenced, though their cell bodies and peripheral axons may survive for decades.
Figure 2: Age-related primary neural degeneration in human vs. mouse.
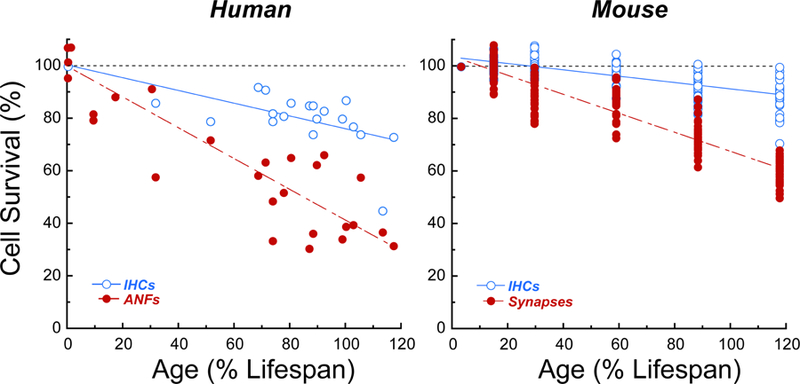
Data are normalized as described for Figure 1. Human data are for IHCs and ANF peripheral axon counts, averaged across cochlear regions at half-octave intervals spanning the audiometric frequencies (0.25 to 8.0 kHz; Wu et al., 2018); mouse data are for IHCs and ANF synaptic counts averaged across 5 cochlear regions (5.6, 11.3, 22, 32 and 64 kHz), pooled from Parthasarathy and Kujawa (2018a) and Sergeyenko et al., (2013).
Years ago, it was shown that primary neural degeneration has little effect on behavioral thresholds in the absence of hair cell damage (Schuknecht and Woellner, 1955). More recently, it was shown that thresholds do not elevate significantly (i.e. by more than 20 dB) until more than 80% of ANFs are disconnected from their IHC inputs (Lobarinas et al., 2013). The phenomenon of cochlear synaptopathy has been given the nickname hidden hearing loss, because it hides behind the audiogram (Schaette and McAlpine, 2011). Thus, the reason that each IHC is innervated by 10 – 30 ANFs, depending on species and cochlear location (Liberman, 2017), is not to provide exquisite threshold sensitivity. The working hypothesis, which was first articulated decades before the discovery of noise-induced cochlear synaptopathy, is that this redundant innervation is required to extract a weak signal out of a noisy background by essentially averaging the outputs of these multiple ANF channels. It is, therefore, tempting to speculate that cochlear synaptopathy is a major contributor to problems hearing in noise that characterize the impairment in NIHL and ARHL (see below). Recent animal behavioral studies have added support to this notion (Lobarinas et al., 2016).
4.2. Diagnosis and treatment for synaptopathy in NIHL and ARHL
In the mouse studies, the degree of noise-induced or age-related cochlear synaptopathy could be diagnosed with reasonable accuracy by measuring the suprathreshold amplitude of the first wave of the auditory brainstem response (ABR wave 1), which represents the summed sound-evoked activity of ANFs (Kujawa and Liberman, 2009; Sergeyenko et al., 2013). As predicted, given the similarity of the single-ANF contributions to the summed action potential currents recorded at the round window regardless of their point of origin along the cochlear spiral (Kiang et al., 1976), a loss of 50% of the ANF synapses translated into a 50% reduction of the measured suprathreshold amplitudes of ABR wave 1 (Sergeyenko et al., 2013). Of course, this diagnostic power was present only if cochlear amplifier (i.e. OHC) function was completely normal, as threshold elevation will also decrease the suprathreshold amplitudes of ABR wave 1. Mouse studies have also suggested that other electrophysiological measures of sound-evoked activity, such as envelope-following responses (EFRs) to amplitude-modulated tones or tests of middle-ear muscle reflexes, may provide important diagnostic clues to underlying cochlear synaptopathy and its functional consequences (Parthasarathy and Kujawa, 2018a; Parthasarathy et al., 2018b; Shaheen et al., 2015; Valero et al., 2018).
These animal-based studies of hidden hearing loss have inspired numerous laboratories to look for evidence of the phenomenon in human subjects. The general approach has been to select participants with normal audiometric thresholds (0.25 to 8.0 kHz), then measure the suprathreshold amplitudes of ABR wave 1 or a type of EFR, and look for correlations with noise-exposure history and/or performance on a speech-in-noise task (Bramhall et al., 2017; Guest et al., 2017; 2018; Liberman et al., 2016). Results have been mixed: some report a correlation between cochlear electrophysiology and estimated noise exposure (Bramhall et al., 2017; Liberman et al., 2016) while others don’t (Guest et al., 2017; 2018). Similarly, some found electrophysiological evidence for a cochlear contribution to reduced speech-in-noise performance among “normal-hearing” listeners with higher self-reported noise exposure (Liberman et al., 2016), while others did not (Guest et al., 2018). Even when there is a significant correlation in the population data consistent with a contribution of primary ANF degeneration to differences in speech-in-noise performance in normal-hearing listeners, a diagnosis of cochlear synaptopathy on an individual basis is not possible, because there is too much intersubject variability (Liberman et al., 2016). It remains possible that better assays can be devised and/or that the existing assays could be used in individual subjects to track synaptopathy longitudinally, and thereby to assess the effectiveness of a drug treatment in an individual subject in a clinical trial.
It is also possible that functionally important primary neural degeneration in human NIHL (or ARHL) will be most obvious once the cochlear pathology has progressed to include hair cell damage/loss, and therefore significant threshold elevations at audiometric frequencies. Indeed, as shown in Figure 2, we observed loss of >60% of ANF peripheral axons throughout the cochlear regions tuned to audiometric frequencies in half (8/16) of the subjects between 55 and 85 yrs of age (Wu et al., 2018), ages at which significant ARHL can often be observed audiometrically (Pearson et al., 1995). It seems likely that this degree of de-afferentation would significantly compromise hearing function on complex listening tasks at low signal-to-noise ratios, adding to the compromised fidelity of stimulus coding in the surviving ANFs due to OHC damage.
Because the pathology in hidden hearing loss involves only the synaptic connections and peripheral axons of ANFs, repair would not require cell division or transdifferentiation. Furthermore, in guinea pigs with drug-induced deafening, adult spiral ganglion cells will send out new peripheral axons towards the organ of Corti in response to cochlear perfusion of neurotrophins, the endogenous signaling molecules that are key to neuronal development and maintenance in the ear (Wise et al., 2005). Inspired by those findings, we recently showed in adult mice that round-window delivery of neurotrophins (i.e. NT-3) in a slow-release gel, could regenerate synaptic connections and ABR wave 1 amplitudes when delivered 24 hrs after exposure to noise (Suzuki et al., 2016). At this trauma-treatment interval, the neural degeneration likely includes only the synapse itself and the unmyelinated dendritic portion of the ANF within the organ of Corti (Liberman et al., 2015). Although some have reported that virally driven NT-3 overexpression can disrupt the normal innervation patterns in an undamaged cochlea (Lee et al., 2016), we have seen no signs of this in our experiments (Hashimoto et al., 2019). At longer traumatreatment intervals (i.e. 1 wk) no regenerative response is seen (Liberman unpublished), suggesting that higher drug concentrations or more sustained delivery periods may be required. Nonetheless, the fundamentally conserved nature of this signaling system (the amino acid sequence of NT-3 is identical in mouse and man) suggests that a treatment developed in mouse is likely to be translatable to human, subject to the proper selection of subjects, and the relative efficacy of the delivery route through the round window membrane in mouse vs. human.
5. Other issues in translating NIHL or ARHL findings from animals to humans
5.1. Strial degeneration in NIHL and AHL
According to our studies of acoustic overstimulation in cat (Liberman and Mulroy, 1982), guinea pig (Liberman and Gao, 1995) and mouse (Wang et al., 2002), strial damage does not make a major contribution to the PTS in NIHL. The stria is much less vulnerable to noise than the hair cells, and, by the time there is functionally important strial damage, the relevant cochlear regions are typically non-responsive due to hair cell loss (Liberman and Mulroy, 1982).
On the other hand, both human studies and animal work suggest that strial degeneration might be an important contributor to ARHL. Classic work on otopathology in human temporal bones proposed four major types of ARHL, a.k.a. presbycusis (Schuknecht, 1974): 1) sensory, where the threshold shift is well explained by hair cell loss, 2) neural, where there is minimal hair cell loss and the threshold shift is caused by ANF loss, 3) strial, where there is minimal hair cell loss and the flat audiometric loss is caused by strial damage and associated reductions in the endolymphatic potential, and 4) cochlear conductive, where there is minimal hair cell loss and the gently down-sloping audiogram is caused by thickening of the basilar membrane.
The cochlear conductive subtype was abandoned (and renamed “indeterminate”) after quantitative analysis failed to find basilar membrane alterations that correlated with the threshold elevations (Bhatt et al., 2001). The hypothesis of a neural subtype is complicated by the emergent view that even massive IHC loss (and the associated ANF inactivation) does not cause threshold elevations (Lobarinas et al., 2013). An alternative view is that “neural presbycusis” is actually a combination of ARHL and NIHL, where the exaggerated ANF degeneration is due to noise-induced cochlear synaptopathy and the threshold shift is “explained” by stereocilia damage that is not assessed in standard otopathological analysis.
The potential importance of a strial subtype of presbycusis is underscored by observations in animals, although there is little if any preclinical research focused on the prevention or repair of strial damage. Several species, i.e. Mongolian gerbil (Suryadevara et al., 2001), CBA/CaJ mice (Ohlemiller et al., 2010), and Fischer 344 rats (Balogova et al., 2017) show age-related strial degeneration, coupled with an age-related reduction in the endolymphatic potential, which would compromise cochlear thresholds in any frequency regions where the reductions are observed. A strial role in presbycusis must be strongly influenced by genetics, given that it is present in only one of two very closely related inbred strains of mouse (Ohlemiller et al., 2010). It is also affected by gender, at least in rats where the strial degeneration is strikingly worse in male than females (Balogova et al., 2017), although in CBA/CaJ mice, the gender sensitivity was reversed (Ohlemiller et al 2010). In our own studies in CBA/CaJ mice, the onset and progression of OHC-based sensitivity losses with age was strikingly parallel to the onset and progression of OHC loss in the same ears (Sergeyenko et al., 2013), arguing against a major influence of strial degeneration on ARHL in this inbred strain.
If strial degeneration is indeed a major driver of ARHL in humans, as some have suggested (Dubno et al., 2013), it would cause therapeutic failure in any clinical trial designed to address hair cell damage or loss. The challenge would be to identify those with strial damage and exclude them from the trial. It is not clear that unambiguous diagnosis is possible at present, although animal work suggests that a reduction in endolymphatic potential elevates ABR thresholds more than DPOAEs (Mills, 2003), because it compromises cochlear amplifier function (attenuating both DPOAEs and ABRs), as well as IHC function (affecting only the ABRs).
Although the classic otopathological literature (Pauler et al., 1988), as well as animal research on ears with chronic reduction of the endolymphatic potential (Schmiedt et al., 2002), suggest that strial degeneration leads to a flat or gently sloping audiometric loss, it is not clear that flat audiograms are strong indicators of strial pathology. Indeed, in a recent study of aged human cases with flat audiometric patterns, only one of six such cases showed significant strial pathology (Nelson and Hinojosa, 2003). It is also far from clear how common strial pathology is in a random sample of humans with presbycusis.
The classic human otopathology literature, as discussed above, suggests that “sensory” presbycusis, where threshold shifts are well explained by hair cell loss, is relatively rare. If true, therapies designed to regenerate hair cells would be inappropriate for a large proportion of the presbycusic population. This may not be the case. Our ongoing reanalysis of the histopathology in archival presbycusis cases at the Massachusetts Eye and Ear suggests that the degree of hair cell loss has been dramatically underestimated in past publications (Wu et al., 2019). Techniques for the generation of cytocochleograms from serial sections, promulgated in otopathology laboratories for close to 100 years (Guild, 1921), teach that hair cells in each section can only be scored as “present” or “absent”, thereby insuring that only regions of complete hair cell loss are noted in published cytocochleograms. Thus, it may be that, in all four patterns of ARHL, threshold shifts are well explained by hair cell loss.
5.2. Vulnerability and critical intensity in NIHL
For most well-studied types of stimuli in NIHL, there is a critical range of intensities over which the resultant threshold shift (either temporary or permanent) grows rapidly when all other stimulus variables (i.e. spectrum, duration and duty cycle) are held constant. For example, in mice exposed to an 8–16 kHz octave-band of noise for two hours, the PTS rises from 0 to 40 dB as the exposure SPL rises by just 5 dB, from 95 to 100 dB SPL (Yoshida et al., 2000). This yields a slope of > 7 dB threshold shift per dB exposure level for this particular stimulus spectrum. As level increases well beyond this critical range, the threshold shift tends to saturate for a number of reasons, the most important being the spread of excitation from the test frequency in a test such as the ABR to cochlear regions distant from the damage focus.
Many studies of protection from acoustic trauma choose their exposure to be in the middle of this critical intensity range, to allow the outcome measure (threshold shift) to move, either up or down, depending on the intervention. The steep slope of the relation between exposure and PTS in this region tends to exaggerate the size of the effect, since a protection of 20 dB is, in some sense, only equivalent to reducing the exposure SPL by 3 dB. If the exposure levels experienced in a human clinical trial are not in the critical range, an intervention that showed a dramatic effect in animals might not be significant in humans. Such considerations may contribute to the observation that anti-oxidant therapies that have been shown to be protective in animal studies (Le Prell et al., 2007) failed to show effectiveness in human trials (Le Prell et al., 2011).
In considering the likelihood that hidden hearing loss is a widespread problem in humans, the issue of relative NIHL vulnerability of humans vs. common animal models is important. Humans (and possibly primates in general) appear less vulnerable to NIHL than other well-studied, smaller experimental animals such as cat, mouse, guinea pig and chinchilla, at least for exposures producing only temporary changes in threshold sensitivity. Such comparisons are difficult given 1) the dearth of controlled overstimulation experiments in humans, 2) the fact that human work is constrained to exposures designed to produce only TTS, and 3) the complexity of the exposure parameter space (spectrum, intensity, duration, duty cycle etc.). Nevertheless, a recent report discussing the adequacy of federal exposure guidelines in light of new discoveries re hidden hearing loss (Dobie and Humes, 2017) points out that 2-hr octave band exposures in the middle of the hearing frequency range in mouse (Fernandez et al., 2015) vs. human (Ward, 1960) produced a similar TTS (30 dB) at 91 vs. 105 dB exposure levels, respectively: a vulnerability difference of 15 dB. A similar conclusion is reached comparing human to a species with a similar frequency range: in experiments tracking effects of long exposures to octave-band noise centered near 0.7 kHz, chinchillas showed 21 dB TTS after 16 hrs of exposure at 80 dB SPL (Clark, 1991), whereas human TTS reached 21 dB at 16 hrs with exposure at 95 dB SPL (Melnick, 1974) - again a 15 dB difference in vulnerability.
Such noise risk comparisons are based on threshold measures, however, and recent animal work clearly demonstrates the insensitivity of thresholds to even dramatic synaptopathy (Fernandez et al., 2015). Details of what is necessary and sufficient to produce synaptopathy remains poorly understood, even in animals. Importantly, there is no evidence that risk of synaptopathy scales simply with threshold shift magnitude. At present, exposure parameters required to produce synaptopathy in humans are completely uncharacterized.
Despite apparent differences in threshold shift vulnerability, the existing data suggest that the same progression of histopathological changes is seen with increasing exposure severity, i.e. IHC synapses more vulnerable than stereocilia, which are damaged before OHC death that occurs before IHC death. The species differences are mainly in the sound pressure levels required to progress from one type of damage to the next. If true, it remains likely that treatments developed in pre-clinical NIHL studies in animals will be relevant to human populations in clinical trials.
Highlights.
Noise exposure and aging are major causes of acquired sensorineural hearing loss
Threshold shifts can be due to hair cell loss, stereocilia or strial damage
IHC-ANF synapse loss contributes to hearing impairment without elevating thresholds
Preclinical studies showing cochlear regeneration are spawning clinical trials
Trial design is complicated by crude non-invasive diagnosis of underlying pathology
Acknowledgments
This work was supported by grants from the DoD (W81XWH-15-1-0103), the NIH (R01 DC 000188, P30 DC 005209 and P50 DC 015857), and the ONR (N00014-16-1-2867).
Abbreviations:
- ABR
Auditory Brainstem Response
- ANF
Auditory Nerve Fiber
- ARHL
Age-Related Hearing Loss
- IHC
Inner Hair Cell
- NIHL
Noise-Induced Hearing Loss
- OHC
Outer Hair Cell
- PTS
Permanent Threshold Shift
- SGC
Spiral Ganglion Cell
- SNHL
Sensorineural Hearing Loss
- SPL
Sound Pressure Level
- TTS
Temporary Threshold Shift
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- Anttonen T, Belevich I, Kirjavainen A, Laos M, Brakebusch C, Jokitalo E, Pirvola U 2014. How to bury the dead: elimination of apoptotic hair cells from the hearing organ of the mouse. J Assoc Res Otolaryngol 15, 975–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balogova Z, Popelar J, Chiumenti F, Chumak T, Burianova JS, Rybalko N, Syka J 2017. Age-related differences in hearing function and cochlear morphology between male and female Fischer 344 rats. Front Aging Neurosci 9, 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basch ML, Brown RM 2nd, Jen HI, Groves AK 2016. Where hearing starts: the development of the mammalian cochlea. J Anat 228, 233–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermingham NA, Hassan BA, Price SD, Vollrath MA, Ben-Arie N, Eatock RA, Bellen HJ,Lysakowski A, Zoghbi HY 1999. Math1: an essential gene for the generation of inner ear hair cells. Science 284, 1837–41. [DOI] [PubMed] [Google Scholar]
- Bhatt KA, Liberman MC, Nadol JB Jr. 2001. Morphometric analysis of age-related changes in the human basilar membrane. Ann Otol Rhinol Laryngol 110, 1147–53. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya TK, Dayal VS 1989. Influence of age on hair cell loss in the rabbit cochlea. Hear Res 40, 179–83. [DOI] [PubMed] [Google Scholar]
- Bohne BA, Rabbitt KD 1983. Holes in the reticular lamina after noise exposure: Implication for continuing damage in the organ of corti. Hearing Res 11, 41–53. [DOI] [PubMed] [Google Scholar]
- Bramhall NF, Konrad-Martin D, McMillan GP, Griest SE 2017. Auditory brainstem response altered in humans with noise exposure despite normal outer hair cell function. Ear Hear 38, e1–e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredberg G 1968. Cellular pattern and nerve supply of the human organ of Corti. Acta Otolaryngol, Suppl 236:1+. [PubMed] [Google Scholar]
- Cai R, Montgomery SC, Graves KA, Caspary DM, Cox BC 2018. The FBN rat model of aging: investigation of ABR waveforms and ribbon synapse changes. Neurobiol Aging 62, 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark WW 1991. Recent studies of temporary threshold shift (TTS) and permanent threshold shift (PTS) in animals. J Acoust Soc Am 90, 155–63. [DOI] [PubMed] [Google Scholar]
- Dallos P 2008. Cochlear amplification, outer hair cells and prestin. Curr Opin Neurobiol 18, 370–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deTorres A, Olszewski RT, Lopez IA, Ishiyama A, Linthicum FH Jr., Hoa M 2018. Supporting cell survival after cochlear implant surgery. Laryngoscope. Jan;129(1):E36–E40. 10.1002/lary.27539. Epub 2018 Oct 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobie RA, Humes LE 2017. Commentary on the regulatory implications of noise-induced cochlear neuropathy. Int J Audiol 56, 74–78. [DOI] [PubMed] [Google Scholar]
- Dubno JR, Horwitz AR, Ahlstrom JB 2003. Recovery from prior stimulation: masking of speech by interrupted noise for younger and older adults with normal hearing. J Acoust Soc Am 113, 208494. [DOI] [PubMed] [Google Scholar]
- Dubno JR, Eckert MA, Lee FS, Matthews LJ, Schmiedt RA 2013. Classifying human audiometric phenotypes of age-related hearing loss from animal models. J Assoc Res Otolaryngol 14, 687–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstrom B, Flock A, Borg E 1983. Ultrastructural studies of stereocilia in noise-exposed rabbits. Hear Res 12, 251–64. [DOI] [PubMed] [Google Scholar]
- Fernandez KA, Jeffers PW, Lall K, Liberman MC, Kujawa SG 2015. Aging after noise exposure: acceleration of cochlear synaptopathy in “recovered” ears. J Neurosci 35, 7509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fettiplace R, Nam JH 2018. Tonotopy in calcium homeostasis and vulnerability of cochlear hair cells. Hear Res November 16 pii: S0378-5955(18)30394-0. [DOI] [PMC free article] [PubMed]
- Furness DN 2015. Molecular basis of hair cell loss. Cell Tissue Res 361, 387–99. [DOI] [PubMed] [Google Scholar]
- Geisler CD 1998. From Sound to Synapse: Physiology of the Mammalian Ear Oxford University Press, Oxford. [Google Scholar]
- Greenwood DD 1990. A cochlear frequency-position function for several species−-29 years later. J Acoust Soc Am 87, 2592–605. [DOI] [PubMed] [Google Scholar]
- Guest H, Munro KJ, Prendergast G, Howe S, Plack CJ 2017. Tinnitus with a normal audiogram: Relation to noise exposure but no evidence for cochlear synaptopathy. Hear Res 344, 265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guest H, Munro KJ, Prendergast G, Millman RE, Plack CJ 2018. Impaired speech perception in noise with a normal audiogram: No evidence for cochlear synaptopathy and no relation to lifetime noise exposure. Hear Res 364, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guild SR 1921. A graphic reconstruction method for the study of the organ of Corti. Anatomical Record 22, 141–157. [Google Scholar]
- Guinan JJ Jr. 2006. Olivocochlear efferents: anatomy, physiology, function, and the measurement of efferent effects in humans. Ear and hearing 27, 589–607. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Hickman T, Ji L, Corfas G, Liberman MC 2019. Virally-mediated NT3 overexpression minimizes noise-induced cochlear synaptopathy., Midwinter Meeting of the Association for Research in Otolaryngology, Vol. 42, Baltimore MD: pp. 331. [Google Scholar]
- Hickox AE, Larsen E, Heinz MG, Shinobu L, Whitton JP 2017. Translational issues in cochlear synaptopathy. Hear Res 349, 164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner KC 1992. Cochlear and vestibular epithelia from a patient with Meniere’s disease: a case study. Scanning Microsc 6, 1115–27; discussion 1127–8. [PubMed] [Google Scholar]
- Izumikawa M, Batts SA, Miyazawa T, Swiderski DL, Raphael Y 2008. Response of the flat cochlear epithelium to forced expression of Atoh1. Hear Res 240, 52–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang WS, Nguyen K, McKenna CE, Sewell WF, McKenna MJ, Jung DH 2016. Intracochlear drug delivery through the oval window in fresh cadaveric human temporal bones. Otol Neurotol 37, 218–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto K, Ishimoto S, Minoda R, Brough DE, Raphael Y 2003. Math1 gene transfer generates new cochlear hair cells in mature guinea pigs in vivo. J Neurosci 23, 4395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keithley EM, Feldman ML 1982. Hair cell counts in an age-graded series of rat cochleas. Hear Res 8, 249–62. [DOI] [PubMed] [Google Scholar]
- Kiang NYS, Moxon EC, Kahn AR 1976. The relationship of gross potentials recorded from the cochlea to single unit activity in the auditory nerve. In: Ruben RJ, Eberling C, Solomon G, (Eds.), Electrocochleography University Park, Baltimore. [Google Scholar]
- Kimura RS, Ota CY, Schuknecht HF, Takahashi T 1976. Electron microscopic cochlear observations in bilateral Meniere’s disease. Ann Otol Rhinol Laryngol 85, 791–801. [DOI] [PubMed] [Google Scholar]
- Kujawa SG, Liberman MC 2009. Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J Neurosci 29, 14077–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Prell CG, Hughes LF, Miller JM 2007. Free radical scavengers vitamins A, C, and E plus magnesium reduce noise trauma. Free Radic Biol Med 42, 1454–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Prell CG, Johnson AC, Lindblad AC, Skjonsberg A, Ulfendahl M, Guire K, Green GE, Campbell KC, Miller JM 2011. Increased vitamin plasma levels in Swedish military personnel treated with nutrients prior to automatic weapon training. Noise Health 13, 432–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MY, Kurioka T, Nelson MM, Prieskorn DM, Swiderski DL, Takada Y, Beyer LA,Raphael Y 2016. Viral-mediated Ntf3 overexpression disrupts innervation and hearing in nondeafened guinea pig cochleae. Mol Ther Methods Clin Dev August 3;3:16052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman LD, Suzuki J, Liberman MC 2015. Dynamics of cochlear synaptopathy after acoustic overexposure. J Assoc Res Otolaryngol 16, 205–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman MC 1982. Single-neuron labeling in the cat auditory nerve. Science 216, 1239–1241. [DOI] [PubMed] [Google Scholar]
- Liberman MC 2017. Noise-induced and age-related hearing loss: new perspectives and potential therapies. F1000Res 6, 927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman MC, Kiang NY 1978. Acoustic trauma in cats. Cochlear pathology and auditory-nerve activity. Acta oto-laryngologica 358, 1–63. [PubMed] [Google Scholar]
- Liberman MC, Mulroy MJ 1982. Acute and chronic effects of acoustic trauma: Cochlear pathology and auditory nerve pathophysiology. In: Hamernik RP, Henderson D, Salvi R, (Eds.), New Perspectives on Noise-Induced Hearing Loss pp. 105–136.
- Liberman MC, Dodds LW 1984. Single-neuron labeling and chronic cochlear pathology. III. Stereocilia damage and alterations of threshold tuning curves. Hear Res 16, 55–74. [DOI] [PubMed] [Google Scholar]
- Liberman MC, Gao WY 1995. Chronic cochlear de-efferentation and susceptibility to permanent acoustic injury. Hear Res 90, 158–68. [DOI] [PubMed] [Google Scholar]
- Liberman MC, Liberman LD, Maison SF 2014. Efferent feedback slows cochlear aging. J Neurosci 34, 4599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman MC, O’Grady DF, Dodds LW, McGee J, Walsh EJ 2000. Afferent innervation of outer and inner hair cells is normal in neonatally de-efferented cats. J Comp Neurol 423, 132–9. [PubMed] [Google Scholar]
- Liberman MC, Epstein MJ, Cleveland SS, Wang H, Maison SF 2016. Toward a differential diagnosis of hidden hearing loss in humans. PLoS One 11, e0162726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman MC, Gao J, He DZ, Wu X, Jia S, Zuo J 2002. Prestin is required for electromotility of the outer hair cell and for the cochlear amplifier. Nature 419, 300–4. [DOI] [PubMed] [Google Scholar]
- Lin HW, Furman AC, Kujawa SG, Liberman MC 2011. Primary neural degeneration in the Guinea pig cochlea after reversible noise-induced threshold shift. J Assoc Res Otolaryngol 12, 605–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Edin F, Atturo F, Rieger G, Lowenheim H, Senn P, Blumer M, Schrott-Fischer A,Rask-Andersen H, Glueckert R 2015. The pre- and post-somatic segments of the human type I spiral ganglion neurons--structural and functional considerations related to cochlear implantation. Neuroscience 284, 470–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobarinas E, Salvi R, Ding D 2013. Insensitivity of the audiogram to carboplatin induced inner hair cell loss in chinchillas. Hear Res 302, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobarinas E, Salvi R, Ding D 2016. Selective inner hair cell dysfunction in chinchillas impairs hearing-in-noise in the absence of outer hair cell loss. J Assoc Res Otolaryngol 17, 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maison SF, Adams JC, Liberman MC 2003. Olivocochlear innervation in the mouse: immunocytochemical maps, crossed versus uncrossed contributions, and transmitter colocalization. J Comp Neurol 455, 406–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makary CA, Shin J, Kujawa SG, Liberman MC, Merchant SN 2011. Age-related primary cochlear neuronal degeneration in human temporal bones. J Assoc Res Otolaryngol 12, 711–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill TJ, Schuknecht HF 1976. Human cochlear changes in noise induced hearing loss. Laryngoscope 86, 1293–1302. [DOI] [PubMed] [Google Scholar]
- Melnick W 1974. Human temporary threshold shift from 16-hour noise exposures. Arch Otolaryngol 100, 180–9. [DOI] [PubMed] [Google Scholar]
- Mills DM 2003. Differential responses to acoustic damage and furosemide in auditory brainstem and otoacoustic emission measures. J Acoust Soc Am 113, 914–24. [DOI] [PubMed] [Google Scholar]
- Mizutari K, Fujioka M, Hosoya M, Bramhall N, Okano HJ, Okano H, Edge AS 2013. Notch inhibition induces cochlear hair cell regeneration and recovery of hearing after acoustic trauma. Neuron 77, 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvaney J, Dabdoub A 2012. Atoh1, an essential transcription factor in neurogenesis and intestinal and inner ear development: function, regulation, and context dependency. J Assoc Res Otolaryngol 13, 281–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson EG, Hinojosa R 2003. Presbycusis: a human temporal bone study of individuals with flat audiometric patterns of hearing loss using a new method to quantify stria vascularis volume. Laryngoscope 113, 1672–86. [DOI] [PubMed] [Google Scholar]
- Noben-Trauth K, Zheng QY, Johnson KR 2003. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat Genet 35, 21–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesterle EC, Campbell S 2009. Supporting cell characteristics in long-deafened aged mouse ears. J Assoc Res Otolaryngol 10, 525–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlemiller KK, Dahl AR, Gagnon PM 2010. Divergent aging characteristics in CBA/J and CBA/CaJ mouse cochleae. J Assoc Res Otolaryngol 11, 605–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet EH 3rd, Temchin AN, Ruggero MA 2002. Basilar membrane vibrations near the round window of the gerbil cochlea. J Assoc Res Otolaryngol 3, 351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthasarathy A, Kujawa SG 2018a. Synaptopathy in the aging cochlea: Characterizing early-neural deficits in auditory temporal envelope processing. J Neurosci 38, 7108–7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthasarathy A, Bartlett EL, Kujawa SG 2018b. Age-related changes in neural coding of envelope cues: Peripheral declines and central compensation. Neuroscience [DOI] [PMC free article] [PubMed]
- Passchier-Vermeer W 1974. Hearing loss due to continuous exposure to steady-state broad-band noise. J Acoust Soc Am 56, 1585–93. [DOI] [PubMed] [Google Scholar]
- Pauler M, Schuknecht HF, White JA 1988. Atrophy of the stria vascularis as a cause of sensorineural hearing loss. Laryngoscope 98, 754–9. [DOI] [PubMed] [Google Scholar]
- Pearson JD, Morrell CH, Gordon-Salant S, Brant LJ, Metter EJ, Klein LL, Fozard JL 1995. Gender differences in a longitudinal study of age-associated hearing loss. J Acoust Soc Am 97, 1196–205. [DOI] [PubMed] [Google Scholar]
- Rajan R 1991. Protective functions of the efferent pathways to the mammalian cochlea: A review. Mosby Year Book, St. Louis. [Google Scholar]
- Robertson D 1982. Effects of acoustic trauma on stereocilia structure and spiral ganglion cell tuning properties in the guinea pig cochlea. Hear Res 7, 55–74. [DOI] [PubMed] [Google Scholar]
- Rosen S, Bergman M, Plester D, El-Mofty A, Satti MH 1962. Presbycusis study of a relatively noise-free population in the Sudan. Ann Otol Rhinol Laryngol 71, 727–43. [DOI] [PubMed] [Google Scholar]
- Ruggero MA 1992. Responses to sound of the basilar membrane of the mammalian cochlea. Curr Opin Neurobiol 2, 449–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüttiger L, Zimmermann U, Knipper M 2017. Biomarkers for hearing dysfunction: Facts and outlook. ORL J Otorhinolaryngol Relat Spec 79, 93–111. [DOI] [PubMed] [Google Scholar]
- Saunders JC, Cohen YE, Szymko YM 1991. The structural and functional consequences of acoustic injury in the cochlea and peripheral auditory system: a five year update. J Acoust Soc Am 90, 136–46. [DOI] [PubMed] [Google Scholar]
- Schaette R, McAlpine D 2011. Tinnitus with a normal audiogram: physiological evidence for hidden hearing loss and computational model. J Neurosci 31, 13452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiedt RA, Lang H, Okamura HO, Schulte BA 2002. Effects of furosemide applied chronically to the round window: a model of metabolic presbyacusis. J Neurosci 22, 9643–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholtz AW, Kammen-Jolly K, Felder E, Hussl B, Rask-Andersen H, Schrott-Fischer A 2001. Selective aspects of human pathology in high-tone hearing loss of the aging inner ear. Hear Res 157, 77–86. [DOI] [PubMed] [Google Scholar]
- Schuknecht HF 1974. Pathology of the Ear Harvard Universtiy Press, Cambrideg, MA. [Google Scholar]
- Schuknecht HF, Woellner RC 1955. An experimental and clinical study of deafness from lesions of the cochlear nerve. J Laryngol Otol 69, 75–97. [DOI] [PubMed] [Google Scholar]
- Sergeyenko Y, Lall K, Liberman MC, Kujawa SG 2013. Age-related cochlear synaptopathy: an early-onset contributor to auditory functional decline. J Neurosci 33, 13686–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen LA, Valero MD, Liberman MC 2015. Towards a diagnosis of cochlear neuropathy with envelope following responses. J Assoc Res Otolaryngol 16, 727–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon RV 2012. Advances in auditory prostheses. Curr Opin Neurol 25, 61–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryadevara AC, Schulte BA, Schmiedt RA, Slepecky NB 2001. Auditory nerve fibers in young and quiet-aged gerbils: morphometric correlations with endocochlear potential. Hear Res 161, 45–53. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Corfas G, Liberman MC 2016. Round-window delivery of neurotrophin 3 regenerates cochlear synapses after acoustic overexposure. Sci Rep 6, 24907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taranda J, Maison SF, Ballestero JA, Katz E, Savino J, Vetter DE, Boulter J, Liberman MC, Fuchs PA, Elgoyhen AB 2009. A point mutation in the hair cell nicotinic cholinergic receptor prolongs cochlear inhibition and enhances noise protection. PLoS Biol 7, e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnowski BI, Schmiedt RA, Hellstrom LI, Lee FS, Adams JC 1991. Age-related changes in cochleas of mongolian gerbils. Hear Res 54, 123–34. [DOI] [PubMed] [Google Scholar]
- Valero MD, Hancock KE, Maison SF, Liberman MC 2018. Effects of cochlear synaptopathy on middle-ear muscle reflexes in unanesthetized mice. Hear Res 363, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valero MD, Burton JA, Hauser SN, Hackett TA, Ramachandran R, Liberman MC 2017. Noise-induced cochlear synaptopathy in rhesus monkeys (Macaca mulatta). Hear Res 353, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreugde S, Erven A, Kros CJ, Marcotti W, Fuchs H, Kurima K, Wilcox ER, Friedman TB, Griffith AJ, Balling R, Hrabe De Angelis M, Avraham KB, Steel KP 2002. Beethoven, a mouse model for dominant, progressive hearing loss DFNA36. Nat Genet 30, 257–8. [DOI] [PubMed] [Google Scholar]
- Wang J, Puel JL 2018. Toward cochlear therapies. Physiol Rev 98, 2477–2522. [DOI] [PubMed] [Google Scholar]
- Wang J, Van De Water TR, Bonny C, de Ribaupierre F, Puel JL, Zine A 2003. A peptide inhibitor of c-Jun N-terminal kinase protects against both aminoglycoside and acoustic trauma-induced auditory hair cell death and hearing loss. J Neurosci 23, 8596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Hirose K, Liberman MC 2002. Dynamics of noise-induced cellular injury and repair in the mouse cochlea. J Assoc Res Otolaryngol 3, 248–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward GA 1960. Recovery from highh values of temporary threshold shift. J. Acoust. Soc Am 32, 497500. [Google Scholar]
- Wise AK, Richardson R, Hardman J, Clark G, O’Leary S 2005. Resprouting and survival of guinea pig cochlear neurons in response to the administration of the neurotrophins brain-derived neurotrophic factor and neurotrophin-3. J Comp Neurol 487, 147–65. [DOI] [PubMed] [Google Scholar]
- Wu PZ, Quesnel AM, O’Malley JT, Liberman MC 2019. Techniques for assessing fractional hair cell survival in archhival human temporal bones: New insights from old specimens., Midwinter Meeting of the Association for Research in Otolaryngology, Vol. 42. [Google Scholar]
- Wu PZ, Liberman LD, Bennett K, de Gruttola V, O’Malley JT, Liberman MC 2018. Primary neural degeneration in the human cochlea: Evidence for hidden hearing loss in the aging ear. Neuroscience August 10 pii: S0306-4522(18)30537-2. [DOI] [PMC free article] [PubMed]
- Yang SM, Chen W, Guo WW, Jia S, Sun JH, Liu HZ, Young WY, He DZ 2012. Regeneration of stereocilia of hair cells by forced Atoh1 expression in the adult mammalian cochlea. PLoS One 7, e46355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida N, Hequembourg SJ, Atencio CA, Rosowski JJ, Liberman MC 2000. Acoustic injury in mice: 129/SvEv is exceptionally resistant to noise-induced hearing loss. Hear Res 141, 97–106. [DOI] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR, Erway LC 1999. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res 130, 94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]