

Abstract
Background:
Non-small cell lung carcinoma (NSCLC) is a major worldwide health threat due to its low cure rate and high lethality. Emerging evidence suggests that epidermal growth factor receptor (EGFR) plays vital roles in cancer initiation and progression, and is considered an important cancer-driving protein. However, how EGFR expression is regulated during NSCLC development remains to be fully elucidated.
Methods:
In NSCLC clinical samples, EGFR protein levels were measured by western blotting and qRT-PCR, respectively. Combining microRNA (miRNA) target prediction software and the pulldown assay, we predicted microRNAs (miRNAs) that targeted EGFR. Next, three NSCLC cell lines, A549 (p53 WT), H322 (p53 mutant), and H1299 (p53 null), were used to demonstrate the direct targeting of EGFR by miR-193a. In addition, we investigated the biological effects of EGFR inhibition by miR-193a in vitro using Cell Counting Kit-8, 5-Ethynyl-2′-deoxyuridine (EdU), transwell, and apoptosis assays. Then, using ChIP and luciferase assays, we demonstrated that miR-193a was directly activated by p53 at the transcriptional level and that p53-induced-miR-193a and EGFR form a double-negative feedback loop.
Results:
We found that EGFR mRNA and protein were upregulated in NSCLC. We predicted that EGFR was a target of miR-193a and validated that miR-193a bound directly to the 3′-UTR of the EGFR mRNA. Moreover, miR-193a inhibited NSCLC proliferation and invasion, and promotes NSCLC apoptosis by directly downregulating EGFR. Then, we demonstrated that p53 directly activated miR-193a transcription, whereas EGFR functioned as a transcriptional repressor to negatively control miR-193a expression, forming a feedback loop. The loop promoted NSCLC cell proliferation and migration and accelerated tumor growth in xenograft mice.
Conclusions:
This study highlights a double-negative feedback loop in NSCLC. The feedback loop is crucial because overexpressing EGFR strongly accelerated tumor growth, while miR-193a restoration blocked tumor growth in vivo. Our findings are in line with the emerging opinion that miRNAs and protein regulators form regulatory networks in critical biological processes and that their dysregulation can lead to cellular dysfunction. In conclusion, this study provides important insights into the molecular mechanisms of NSCLC progression and may help inform the development of new therapeutics for managing NSCLC.
Keywords: epidermal growth factor receptor, feedback, miRNA, non-small cell lung carcinoma
Introduction
Epidermal growth factor receptor (EGFR) is a transmembrane glycoprotein that possesses intrinsic tyrosine kinase activity. EGFR signaling dysregulation is vital to tumor progression.1 EGFR is considered a cancer-driving gene that affects numerous systems involved in oncogenesis, including cell proliferation, DNA synthesis, cell cycle progression, invasion, and metastasis.2 Hyperactivation of the EGFR pathway in NSCLC has been shown to result in the development of many cancer characteristics, such as increased cell proliferation, migration, angiogenesis, metastasis, and increased cell survival by blocking apoptosis.3–5 Many chemical inhibitors of EGFR and EGFR-neutralizing antibodies have been developed for cancer therapy.6,7 However, their effects in the treatment of NSCLC are not very satisfactory likely because the mechanisms driving NSCLC progression are not fully understood.8 Thus, the detailed molecular mechanisms of the role of EGFR-related pathways in NSCLC progression remain to be elucidated.
MicroRNAs (miRNAs) are small, noncoding RNAs and lead to mRNA cleavage or translational repression by binding to the 3′-untranslated region (3′-UTR) of target mRNAs.9 miRNAs can post-transcriptionally regulate diverse cellular functions, and play important roles in various physiological and pathological cellular processes, such as cell apoptosis, migration, differentiation, autophagy, development, and metabolism.10–12 During NSCLC development, numerous miRNAs expression deregulate significantly and affect NSCLC tumorigenesis, angiogenesis, proliferation, apoptosis, and metastasis.13–15 For example, recent studies reported that miR-483 inhibit proliferation and promote apoptosis in NSCLC.16 miR-128 and miR-193 suppress tumor angiogenesis, invasion, and metastasis, whereas miR-661, miR-137, on the contrary, promote NSCLC proliferation, invasion, and metastasis.17–20 In addition, miR-193a acts as a tumor suppressor in many human cancers, including NSCLC and breast cancer.21,22
Transcription factors regulate miRNA gene in a way like regulating protein-coding genes, that is, by combining with the binding sites in, or near, promoter regions upstream of miRNA genes.23,24 Thus, transcription factor, miRNA, and target gene form interconnected feedback and feedforward circuits.25 However, only a few miRNA transcription factors have been characterized. Thus, deciphering the contributions of miRNAs to phenotypic variations and diseases is challenging. The detailed mechanism underlying the transcriptional regulation of miRNAs remains unclear.
In our study, we identified that miR-193a directly targets EGFR. In addition, we characterized the crosstalk between miR-193a and EGFR and determined that EGFR negatively regulates miR-193a expression via specific EGFR-binding motifs. Moreover, the tumor suppressor p53 directly activates miR-193a at the transcription level. Thus, the p53-regulated miR-193a and EGFR form a double-negative feedback loop that contributes to NSCLC tumorigenesis.
Methods
Cells and patients
Human NSCLC cell lines A549, H322 and H1299, and 293T were purchased from Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM and RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) at 37°C and 5% CO2. In total, 27 paired NSCLC cancer and normal adjacent tissues and 4 paired NSCLC and nontumorous tissues samples were acquired from patients undergoing a surgical procedure at the First Affiliated Hospital of USTC. All patients had signed the written informed consent on the study, in which the key information including voluntary participation, collection and use of patients’ samples only for scientific research, publication of their individual-level data, detailed design of the study and no risk or influence on their following clinical diagnosis and treatment, was clearly described. And all experiments were performed in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki). The study (including the collection and use of patients’ samples and the in vivo study) was approved by the Ethics Committee of the First Affiliated Hospital of USTC (ID: 2012220). The clinical features of these NSCLC patients are listed in Table 1.
Table 1.
Clinical features of the studied patients with non-small cell lung carcinoma (NSCLC).
| Number | Age | Gender | Pathological stage |
|---|---|---|---|
| Case #1 | 60 | Male | I |
| Case #5 | 48 | Male | I |
| Case #12 | 69 | Male | I |
| Case #19 | 55 | Male | I |
| Case #26 | 59 | Female | I |
| Case #2 | 68 | Male | II |
| Case #4 | 60 | Male | II |
| Case #6 | 54 | Male | II |
| Case #11 | 66 | Male | II |
| Case #18 | 69 | Male | II |
| Case #20 | 69 | Female | II |
| Case #21 | 69 | Male | II |
| Case #22 | 64 | Male | II |
| Case #27 | 66 | Female | II |
| Case #3 | 50 | Male | III |
| Case #7 | 59 | Male | III |
| Case #8 | 49 | Female | III |
| Case #9 | 58 | Male | III |
| Case #10 | 76 | Male | III |
| Case #13 | 73 | Male | III |
| Case #14 | 46 | Male | III |
| Case #15 | 78 | Female | III |
| Case #16 | 79 | Female | III |
| Case #17 | 83 | Male | III |
| Case #23 | 66 | Female | III |
| Case #24 | 40 | Male | III |
| Case #25 | 45 | Male | III |
| Summary | Average: 62.14 ± 11.23 | Male (74.07%) | I (18.52%) II (29.63%) III (51.85%) |
Transfection
The miRNA mimics, inhibitors, and negative control RNAs were designed by Ruibobio Company (Guangzhou, China). In EGFR and p53 overexpression assays, mammalian expression plasmids encoding EGFR and p53 open reading frame were designed by GeneCopoeia (Germantown, MD, USA). A549, H322, or H1299 cells were seeded in 6-well plates, and then 100 pmol synthetic RNAs or 2.5 μg plasmids were transfected into the cells by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocols.
RNA extraction and reverse transcriptase quantitative real-time PCR
The RNA extraction, reverse transcription and quantitative real-time polymerase chain reaction (qRT-PCR) were performed as described previously.26 The primers sequences were as follows: EGFR (forward), TTGCCGCAAAGTGTGTAACG and EGFR (reverse), GTCACCCCTAAATGCCACCG; and GAPDH (forward), CGAGCCACATCGCT CAGACA and GAPDH (reverse), GTGGTGAAGACGCCAGTGGA.
Pulldown assay
Pulldown of mRNA was performed as previously described.26 In brief, DNA probes complementary to EGFR mRNA were labeled with biotin at the 3′ terminus (Genescript, Nanjing, China). Scrambled biotinylated probe served as negative control. The probes (8 pmol/μl) were incubated with streptavidin-coated magnetic beads (New England BioLabs, S1420S) at 25°C for 1 h to generate probe-coated magnetic beads. The A549 cells were harvested in lysis buffer. Then, the lysate was incubated with probe-coated beads at 37°C for 3 h, with constant rotation. After incubation, two washes with lysis buffer were performed, and RNA was extracted with Trizol (Invitrogen). The extracted RNAs were analyzed by qRT-PCR.
Luciferase assay
The EGFR 3′-UTR sequences containing the presumed miR-193a-3p binding sites or mutated binding sites were inserted into the p-MIR-reporter plasmid (Ambion, ThermoFisher Sci-entific, Waltham, MA, USA). 293T cells were seeded into 24-well plates and cotransfected with firefly luciferase reporter wild type (WT) plasmid, firefly luciferase reporter Mut plasmid, miR-193a mimics, inhibitors, or controls by Lipofectamine 2000. After 24 h of transfection, the cell lysates were collected and the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) was used to detect the firefly luciferase activities.
Western blotting
The protein was extracted as previously described.16 Antibodies against EGFR27 and p5328 (Abcam Biotechnology, Cambridge, UK), GAPDH21 (Santa Cruz, CA, USA) were used for blotting. The band intensity was quantified using ImageJ v1.50e (National Institutes of Health (NIH), Bethesda, MD, USA).
Assessment of cell proliferation, migration, and apoptosis
For the Cell Counting Kit-8 (CCK-8), 5-Ethynyl-2′-deoxyuridine (EdU), Transwell, and apoptosis assays, A549 cells under different treatments were measured at different time points using a CCK-8 kit (CK04-500, Dojindo, Japan), an EdU assay kit (RiBoBio, Guangzhou, China), a transwell Boyden Chamber (6.5 mm, Costar, Corning, NY, USA), and an Annexin V-FITC/PI staining kit (BD Biosciences, San Diego, CA, USA), respectively, according to the manufacturers’ instructions as previously described.16
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed using an EZ-ChIP assay kit (Upstate Biotechnology, Inc., Lake Placid, NY, USA) according to the manufacturer’s instructions as previously described.24 Soluble chromatin was prepared from A549 cells, and incubated with anti-EGFR and anti-p53 antibody (Abcam, ab1101) or mouse IgG as a negative control. The primer pairs used for the PCR analysis were as follows: for EGFR site 1: 5′-ATTCTTGTCTTTGAGTTGGCTCAC-3′ (forward) and 5′-ACCAGCGTTTCCCAAAAGGT-3′ (reverse); for EGFR site 2: 5′-TCAAGAAAGTATGCTGCCCG-3′ (forward) and 5′-TTCCCCATCTAGAACCTGGAG-3′ (reverse); for p53 site: 5′-TATGCTGCCCGGTACTATGCT-3′ (forward) and 5′- TCAACTTCCCCATCTAGAACCT-3′ (reverse). All data were normalized to the input.
Immunohistochemistry staining
Immunohistochemistry staining was performed as described.16 Immunohistochemical staining of paraffin sections was performed by a microwave-based antigen retrieval technique, and the specimen slides were incubated with primary anti-bodies against Ki67 (Cell Signaling Technology, Danvers, MA, USA) and EGFR (Abcam) overnight at 4°C. The expression of Ki67 and EGFR was quantitated using Image-Pro Plus 6.0 software.
In vivo studies
A total of 18 mice (male, nu/nu, 6-week-old, 18–20 g) were purchased from the Laboratory Animal Center of Oncogene Biotechnology Company (No. OTech-2017-01082, Yangzhou, PR China), and maintained under specific-pathogen-free conditions according to the NIH guide for the care and use of mice. The mice were housed with an inverse 12 h day–night cycle (free access to water and a maintenance diet) with lights on at 8:30 pm in a temperature- (22 ± 1°C) and humidity-(55 ± 5%) controlled room. All cages contained wood shavings, bedding, and a cardboard tube for environmental enrichment. The animals’ health status was monitored throughout the experiments by a health surveillance programme according to Federation of European Laboratory Animal Science Associations (FELASA) guidelines. The mice were free of all viral, bacterial, and parasitic pathogens listed in the FELASA recommendations, except for Helicobacter species.
All 18 mice were divided randomly into six groups (3 mice/group). A549 cells were infected with miR-193a overexpression lentivirus (miR-193a) or p53-shRNA lentivirus (p53-shRNA), or transfected with EGFR overexpression plasmids (EGFR) and cotransfected with miR-193a overexpression lentivirus plus EGFR overexpression plasmids (miR-193a+EGFR) or miR-193a overexpression lentivirus plus p53-shRNA lentivirus (miR-193a+ p53-shRNA). Then, the cells were implanted subcutaneously into nude mice (1 × 106 cells per mouse). The needle was inserted into the left side of the armpit, midway down, 5 mm deep at a 45° angle. After A549 cells injection, the mice were housed under the same conditions as previously.
Thereafter, the weights of mice were measured on day 0, 3, 6, 9, 12, 15, 18, 21, and 24. Mice were sacrificed by cervical dislocation 24 days after injection to remove the xenografted tumors, and the volumes and weights of the tumors were measured (Supplementary Figure 1). The animal experiments were carried out in accordance with the NIH guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978) and the guidelines of the First Affiliated Hospital of USTC.
Statistical analyses
Every experiment was performed at least three times. The data were presented as the mean ± SEM of at least three independent experiments. The differences between the groups were analyzed by performing Student’s t-tests. The differences among more than two groups were analyzed by performing ANOVA. Two-tailed Pearson’s correlation coefficient analysis and graphs were made using GraphPad Prism 6 (GraphPad Software). Throughout the text, figures, and figure legends, the following terminology was used to denote statistical significance: *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
EGFR expression was frequently upregulated in NSCLC specimens
As an important player in tumorigenesis, EGFR expression is up-regulated in various cancerous tissues. First, we searched the TCGA database and compared the EGFR expression levels between cancerous (NSCLC) and matched normal adjacent tissues (NAT). The results showed that EGFR mRNA expression level in NSCLC was significantly higher than that in NAT (Figure 1a). Next, we determined the association between EGFR expression and the malignancy of lung cancers. The data of TCGA database showed that malignancy grade rose with increasing EGFR expression levels in NSCLC (Figure 1b), indicating that NSCLC malignancy of was correlated with EGFR overexpression. In support of this correlation, patients with high levels of EGFR had significantly shorter overall survival time than those with low levels of EGFR (Figure 1c). We confirmed the observation by detecting EGFR expression in human NSCLC specimens. Tumor tissues and normal adjacent tissues obtained from 27 patients (Table 1) with NSCLC, and four tumor tissues and nontumorous tissues obtained from 4 patients with NSCLC (Supplementary Table S1) were analyzed (Figure 1d and Supplementary Figure S2). Compared with the NATs, the protein levels of EGFR were significantly increased in NSCLC tissues (Figure 1d and e). These above results demonstrated that EGFR was upregulated in lung cancers, associated with tumor malignancy and poor prognosis.
Figure 1.

Upregulation of epidermal growth factor receptor (EGFR) in non-small cell lung carcinoma (NSCLC) tissues compared with normal tissues. (a) EGFR expression analysis based on TCGA dataset showed that EGFR mRNA levels were higher in lung cancerous tissues (NSCLC) than in matched normal adjacent tissues (NAT). (b) EGFR expression was correlated with the clinical stage of lung cancer. (c) Kaplan-Meier analysis of survival of patients with NSCLC. (d, e) Western blot analysis of the EGFR protein levels in 27 paired NSCLC and NAT samples. (d) Representative images. (e) Quantitative analysis; **p < 0.01.
Identification of EGFR as a target of miR-193a
MiRNA has been demonstrated to play crucial roles in cancer development by directing post-transcriptionally regulating target gene expression.11 Thus, we supposed that EGFR might be regulated by miRNA. Three computational algorithms, TargetScan, miRanda, and PicTar, were used in combination to identify the miRNAs that potentially target EGFR. The candidate miRNAs targeting the 3′-UTR of EGFR are shown in Figure 2a. Due to the inverse expression pattern between the miRNAs and their targets in tumor development and the upregulation of EGFR protein level in NSCLC, we aimed to identify the miRNAs that were downregulated in NSCLC. We then designed a biotinylated anti-EGFR mRNA probe. This modification did not affect the suppression of EGFR by miRNAs. After transfecting EGFR mRNA probe into A549 for 48 h, we pulled down the biotinylated anti-EGFR mRNA probe by using streptavidin-coated magnetic beads, and measured the coprecipitated miRNAs. Among the seven predicted miRNAs, miR-193a displayed the highest level (Figure 2b). Therefore, we selected miR-193a for further analysis. Then, we explored the correlation between miR-193a and EGFR expression in NSCLC tissues. By measuring the miR-193a levels in the same 27 pairs of NSCLC tissues and NAT, we found that miR-193a was consistently downregulated in NSCLC tissues (Figure 2c). Importantly, Spearman correlation analysis revealed the significantly inverse correlation between miR-193a and EGFR protein levels in NSCLC tissues (Figure 2d). Based on these findings, we reasoned that miR-193a might be a potential regulator of EGFR.
Figure 2.

Identification of epidermal growth factor receptor (EGFR) as miR-193a target. (a) Schematic overview regarding 10 candidate microRNAs (miRNAs) targeting EGFR 3′-untranslated region (UTR). (b) Quantitative reverse transcriptase-PCR (RT-PCR) analysis of miRNA levels in A549 after pulling down by control probe or EGFR mRNA probe. (c) Quantitative RT-PCR analysis of the miR-193a expression levels in the same 27 pairs of non-small cell lung carcinoma (NSCLC) and normal tissue samples. (d) Pearson’s correlation scatter plot of the fold changes of miR-193a and EGFR protein in human NSCLC tissue pairs. *p < 0.05, **p < 0.01.
Validation that EGFR was post-transcriptionally regulated by miR-193a
To further verify that miR-193a directly targets the presumed binding sites in EGFR 3′-UTR, we performed luciferase reporter assays. The full-length 3′-UTR of EGFR containing presumed binding site for miR-193a was placed downstream of the firefly luciferase gene in a reporter plasmid (Figure 3a), and the resulting plasmids were then transfected into human 293T cells with control plasmid (β-gal) and miR-193a mimics, inhibitors or negative control RNAs. Expectedly, the luciferase reporter assays demonstrated that, compared with the cells transfected with control mimics, overexpressing miR-193a inhibited approximately 60% of the luciferase activity of the reporters; however, inhibition of miR-193a resulted in an increase in reporter activity. To further validate the phenomena, point mutations were introduced into sites complementary to miR-193a within the EGFR mRNA 3ʹ-UTR. This altered luciferase reporter was unaffected by miR-193a overexpression or knockdown (Figure 3b).
Figure 3.

MiR-193a directly regulated epidermal growth factor receptor (EGFR) expression at the post-transcriptional level. (a) Schematic depicting the binding site for miR-193a in the 3′-untranslated region (UTR) of the EGFR mRNA. (b) The relative luciferase activities in 293T cells transfected with wild type or mutant EGFR 3′-UTR. (c) Quantitative reverse transcriptase-PCR (RT-PCR) analysis of miR-193a levels in A549, H322, and H1299 cells transfected with control mimic (miR-NC), miR-193a mimic (miR-193a), control inhibitor (anti-miR-NC), or miR-193a inhibitor (anti-miR-193a). (d)Western blot analysis of EGFR protein levels in A549, H322, and H1299 cells after transfection. (e) Quantitative RT-PCR analysis of the EGFR mRNA levels in A549, H322, and H1299 cells after transfection. *p < 0.05, **p < 0.01, ***p < 0.001.
Next, we assessed the effects of miR-193a on EGFR protein expression levels. We modulated miR-193a level in NSCLC cells to determine whether EGFR is inversely correlated with miR-193a in vitro. We efficiently overexpressed and inhibited miR-193a in A549 cells with miR-193a mimics and inhibitors, respectively (Figure 3c). Expectedly, EGFR protein expression in the A549 cells was significantly inhibited by miR-193a overexpression, whereas treatment with the miR-193a inhibitors increased EGFR protein levels (Figure 3d). However, the miR-193a alterations did not significantly affect EGFR mRNA levels (Figure 3e). Then, we repeated the above transfection experiments in two additional lung cancer cell lines (i.e., H322 and H1299) to evaluate the robustness of the effect and observed consistent results (Figure 3c–e). Overall, we concluded that the miR-193a specifically regulated EGFR protein expression post-transcriptionally.
MiR-193a attenuated the effects of EGFR on cell proliferation, invasion, and apoptosis in lung cancer cells
EGFR is known to promote cell proliferation and invasion and suppress apoptosis.2 To determine whether miR-193a inhibits EGFR expression and thus affects cell functions, we performed EdU and CCK-8 experiments to study the effect of miR-193a on lung cell proliferation. A549 cells overexpressing miR-193a showed decreased proliferation; however, inhibiting miR-193a showed the opposite effect on cell proliferation (Figure 4a and d). Subsequently, we performed cell invasion and apoptosis assays and found that miR-193a overexpression decreased cell invasion and increased cell apoptosis (Figure 4b and c). Then, we validated that the inhibition of cell proliferation and invasion and the promotion of apoptosis by miR-193a was a result of the inhibition of EGFR. A549 cells transfected with the EGFR overexpression plasmids showed increased cell proliferation and migration and decreased apoptosis. In contrast, transfection with the EGFR siRNA had the opposite effect on cell functions (Supplementary Figure S3a–g). Moreover, compared with the cells transfected with the miR-193a mimics alone, the cells transfected with both the miR-193a mimics and EGFR overexpression plasmids exhibited significantly higher proliferation and invasion rates and a lower apoptosis rate (Figure 4e–h, Supplementary Figure S4). Altogether, these data revealed that miR-193a inhibited NSCLC cell proliferation and invasion and promotes apoptosis by suppressing EGFR.
Figure 4.

MiR-193a inhibited non-small cell lung carcinoma (NSCLC) cell proliferation and invasion and promotes NSCLC cell apoptosis by targeting epidermal growth factor receptor (EGFR). A549 cells were transfected with miR-NC, miR-193a mimic (miR-193a), anti-miR-NC, miR-193a inhibitor (anti-miR-193a), miR-NC plus control plasmid (control), miR-193a mimic plus control plasmid (miR-193a), miR-NC plus EGFR overexpression plasmid (EGFR), or miR-193a mimic plus EGFR overexpression plasmid (miR-193a+EGFR). (a, e) Representative images and histogram statistics from cell proliferation assays (EdU). (b, f) Transwell analysis of invaded A549 cells. (c, g) Cell apoptosis assays were performed after the transfection. (d, h) Cell proliferation assays (CCK-8) were performed 12, 24, 48, 72, and 96 h after the transfection. *p < 0.05, **p < 0.01, ***p < 0.001.
MiR-193a was regulated by p53 and miR-193a formed a feedback loop with EGFR
The mechanism by which miR-193a expression is downregulated in NSCLC remains unknown. We predicted the putative transcription factor binding sites in miR-193a promoter. AT-rich minimal consensus sequences (ATRSs) have been shown to be crucial for the function of nuclear EGFR,29–32 Thus, we predicted that EGFR and p53 could bind to the miR-193a promoter. The tumor suppressor, p53, is a transcription factor that induces or represses many genes and miRNAs.33 Previous studies have shown that p53 deletions or mutations frequently occur in cancer development, and that p53 is correlated with tumor metastasis and tumor progression.34,35 Interestingly, genomic analysis predicted that there were two putative EGFR binding sites, and one p53 binding site in the promoter regions of miR-193a (Figure 5a). Hence, we investigated whether p53 and EGFR, which are both transcriptional factors, can recognize and bind the miR-193a promoter and regulate miR-193a expression. Thus, we performed ChIP assays in A549 cells. Expectedly, the assays using p53 antibody showed robust PCR product enrichment indicative of p53 binding in miR-193a promoter (Figure 5b). The ChIP assays using the EGFR antibody showed robust PCR product enrichment indicative of EGFR binding at sites 1 and 2 in miR-193a promoter (Figure 5b). Then, we cloned p53-binding sites and EGFR-binding sites 1 and 2 sequences into a firefly luciferase reporter plasmid. The luciferase reporter assays showed that p53 overexpression promoted the firefly luciferase in the plasmids in which the p53 binding site sequences had been inserted into the promoter region (Figure 5c). However, after mutating the binding sequences, luciferase activity was unaffected (Figure 5c). In contrast, overexpression of EGFR inhibited the firefly luciferase in the plasmids in which the EGFR binding sites 1 and 2 sequences had been inserted into the promoter region (Figure 5c). Furthermore, we overexpressed and knocked down p53 (Supplementary Figure S5) or EGFR in A549 cells, and measured the miR-193a response by qRT-PCR. The ectopic expression of p53 in A549 cells resulted in a higher miR-193a expression than the cells transfected with a control vector, whereas the p53 knockdown resulted in a lower miR-193a expression level (Figure 5d). In contrast, EGFR overexpression inhibited the expression of miR-193a, and the EGFR knockdown resulted in an increase in miR-193a level. Similar alterations were observed in the levels of the precursors of miR-193a, suggesting that the alteration of miR-193a was correlated with transcriptional changes (Supplementary Figure S6).
Figure 5.

p53 directly induced miR-193a and epidermal growth factor receptor (EGFR) to form a double-negative feedback loop. (a) Schematic illustrating the putative p53/EGFR-binding sites in the miR-193a promoter. (b) Direct binding of p53 and EGFR to promoter regions of miR-193a as indicated by PCR-based chromatin immunoprecipitation (ChIP) assays. Binding was confirmed by semi-quantitative PCR, followed by gel electrophoresis. (c) Luciferase reporter assays confirmed the suppression of the miR-193a promoter by EGFR, and the promotion of the miR-193a promoter by p53 through the potential binding. (d) qRT-PCR analysis of the miR-193a levels in A549 cells after the knockdown or overexpression of p53 or EGFR. (e) Quantitative RT-PCR showing the miR-193a levels in non-small cell lung carcinoma (NSCLC) cell lines, i.e., A549: p53 WT; H1299: p53 Null. (f, g) Western blot analysis of p53 and EGFR protein levels in A549 and H1299 cells. (f) Representative images, (g) quantitative analysis. (h) Western blot analysis of p53 and EGFR levels in A549 cells transfected with control siRNA, p53 siRNA, p53 siRNA plus miR-193a mimic, control vector, p53 vector, or p53 vector plus miR-193a inhibitor. *p < 0.05; **p < 0.01; ***p < 0.001.
Subsequently, we measured the miR-193a levels in the NSCLC cell lines A549 (p53 WT) and H1299 (p53 null). Expectedly, the A549 cell line exhibited the highest miR-193a level (Figure 5e). Meanwhile, the A549 cells showed a higher p53 protein level and lower EGFR level than the H1299 cells (Figure 5f and g). Moreover, we performed overexpression and knockdown assays to validate that the upregulation of miR-193a caused by p53 can directly regulate the expression of EGFR in A549 cells. Expectedly, compared with the treatment with the p53 siRNA alone, EGFR protein level was inhibited by cotreatment with the miR-193a mimics and p53 siRNA. Compared with the transfection with the p53 vector alone, the cotransfection with the miR-193a inhibitor and p53 vector increased EGFR expression (Figure 5h, Supplementary Figure S7). Altogether, these results demonstrated that p53 positively regulated miR-193a transcription via specific binding in the promoter regions, whereas EGFR transcriptionally suppressed miR-193a expression; therefore, miR-193a and EGFR formed a double-negative feedback loop (see Discussion).
p53 regulated-miR-193a and EGFR feedback loop influenced NSCLC tumorigenesis
To explore the effect of p53 regulated-miR-193a and EGFR double-negative feedback loop on NSCLC cells in vivo, we established a subcutaneous xenografted model of NSCLC in nude mice. A549 cells were infected with miR-193a overexpression lentivirus (miR-193a) or p53-shRNA lentivirus (p53-shRNA). A549 cells were transfected with EGFR overexpression plasmids (EGFR) and cotransfected with miR-193a overexpression lentivirus plus EGFR overexpression plasmids (miR-193a+EGFR), or miR-193a overexpression lentivirus plus p53-shRNA lentivirus (miR-193a+ p53-shRNA). Then, the cells were implanted subcutaneously into nude mice, and tumor growth was evaluated 24 days after the cell implantation. Compared with the control group, a significant increase in the sizes and weights of the tumors was observed in the EGFR group and the p53-shRNA group, whereas the tumors in the miR-193a group grew dramatically slower (Figure 6a and b). In addition, miR-193a overexpression attenuated the growth-promoting effect of EGFR, suggesting that miR-193a suppresses tumor growth by silencing EGFR. Moreover, the miR-193a+p53-shRNA group grew dramatically slower than the p53-shRNA group (Figure 6a and b). Moreover, the levels of miRNAs and proteins in tumors were confirmed by qRT-PCR and western blot, respectively. A significant upregulation of miR-193a and EGFR was found in miR-193a and EGFR groups, respectively, and overexpressing EGFR could reverse the inhibitory effect of miR-193a on EGFR. In addition, a significant downregulation of miR-193a was observed in the p53-shRNA group, and overexpressing miR-193a could weaken the inhibitory effect of p53-shRNA on miR-193a (Figure 6c and d, Supplementary Figure 8). Consistent with this finding, the immunohistochemical studies also determined the lower levels of EGFR in the group implanted with miR-193a-overexpressing cells (Figure 6e).
Figure 6.

Effects of the p53-induced miR-193a and epidermal growth factor receptor (EGFR) double-negative feedback loop on the growth of non-small cell lung carcinoma (NSCLC) xenografts in mice. (a) Representative images of tumors from mice implanted with control A549 cells, miR-193a-overexpressing A549 cells, EGFR-overexpressing A549 cells, miR-193a plus EGFR co-overexpressing A549 cells, p53-inhibiting A549 cells, or miR-193a-overexpressing plus p53-inhibiting A549 cells. A549 cells (1 × 106 cells per 0.1 ml) with different treatments were implanted subcutaneously into 6-week-old mice (3 mice per group). (b) The time course of tumor growth in the implanted mice. Tumor volume was measured every 3 days for 24 days after the inoculation. (c) Quantitative RT-PCR analysis of miR-193a levels in tumors from the implanted mice. (d) Western blotting analysis of EGFR and p53 protein levels in tumors from the implanted mice. (e) Representative images and quantitative analysis from hematoxylin and eosin (H&E), Ki67, EGFR and TUNEL staining of tumor sections obtained from the six mouse groups. All data were shown as the mean ± SEM of three separate experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
The hematoxylin and eosin (H&E) results showed decreased cell mitosis and increased cell apoptosis in miR-193a group and increased mitosis in the EGFR and p53-shRNA groups, whereas the miR-193a+EGFR and miR-193a+ p53-shRNA groups showed more cell mitosis than the miR-193a group, suggesting that upregulation of miR-193a could attenuate the proinvasive and antiapoptotic effect of EGFR (Figure 6e). Ki67 and TUNEL staining showed that EGFR group and p53-shRNA group contained more Ki67-positive and fewer TUNEL-positive cells than the control group. The miR-193a group had fewer Ki67-positive and more TUNEL-positive cells than the control group (Figure 6e). The miR-193a+EGFR and miR-193a+p53-shRNA groups had fewer Ki67-positive and more TUNEL-positive cells than the EGFR and p53-shRNA groups. Altogether, the results determined the biological role of the p53-induced-miR-193a and EGFR feedback loop in NSCLC progression.
Discussion
Currently, identifying a cure for NSCLC is challenging because the poor understanding of NSCLC pathogenesis.36–38 Therefore, exploring the mechanisms underlying NSCLC progression is critical for the further developing new approaches for NSCLC.
The activation of EGFR, which is a member of the EGFR family of receptor tyrosine kinases (RTKs), initiates signaling via various pathways that ultimately results in cell proliferation, survival, angiogenesis, invasion, and metastasis.2,32,39 Deregulation or activity of EGFR has been strongly correlated with the etiology of human cancers.1,2 Since its discovery, many studies have reported the nuclear localization of EGFR and its role as a transcription factor in regulating genes and miRNA expression in tumor.29,32,40 Previous studies have suggested that the EGFR signaling pathway is important for NSCLC tumorigenesis, proliferation, maintenance, angiogenesis, migration, metastasis, and survival.5,7,41 Consistently, we demonstrated that miR-193a inhibited NSCLC proliferation and invasion and promoted NSCLC apoptosis by directly regulating EGFR in vitro and in vivo. Interestingly, we further found that EGFR could, in turn, be recruited to the miR-193a promoter and repress miR-193a expression. Thus, miR-193a and EGFR form a double negative feedback loop that will amplify EGFR and minimize miR-193a expression in NSCLC cells. The feedback regulation may indicate the overexpression of EGFR and downregulation of miR-193a in NSCLC (Figure 7). How does this feedback loop involve in or promote NSCLC progression? Because double-negative feedback equals positive feedback and will amplify a response into a self-sustained mode independent of the original stimulus, the feedback loop composed of EGFR and miR-193a may allow NSCLC cells to become more malignant. For instance, this feedback loop may enable NSCLC cells to reproduce more rapidly and metastasize to new microenvironments. Thus, we uncovered a new mechanism to explain ectopic expression of EGFR in NSCLC.
Figure 7.
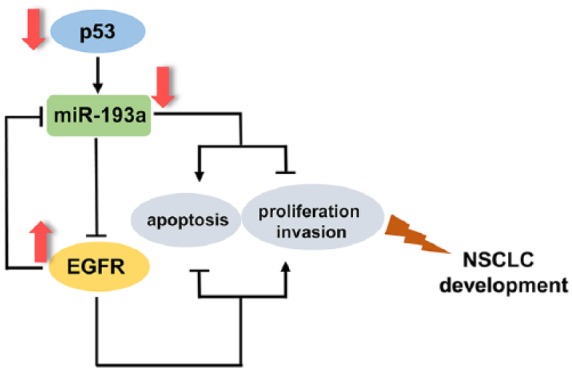
Schematic for the p53/miR-193a/ epidermal growth factor receptor (EGFR) feedback loop in non-small cell lung carcinoma (NSCLC).
The p53 tumor suppressor protein is one of the most commonly altered proteins in human cancers.42,43 p53 regulates vital functions within cells, including differentiation, senescence, and cell death.33,44 Previous reports have shown that p53 deletions or mutations are frequently found in cancers, and that p53 function loss is involved in tumor metastasis and progression.35,45–47 EGFR is an important cancer-driving gene that is frequently overexpressed in various cancers, thereby promoting cancer initiation and progression.2 However, the correlation between the transcription factor p53 and EGFR is poorly understood. In this study, we reported the following link between p53 and EGFR through miR-193a: p53-induced miR-193a could further inhibit EGFR expression. The EGFR accumulation induced by the p53 loss represents a new mechanism driving NSCLC progression. In addition, the link among p53, miR-193a, and EGFR indicates that the dysregulation of p53 is an initial factor in NSCLC progression.
Our findings may provide new approaches for NSCLC therapies. In principle, restoration of p53 expression in cancer cells would promote miR-193a expression, further inhibiting the expression of oncogenic EGFR, which, in turn, could relieve the suppression of EGFR on miR-193a, thereby accelerating the accumulation of miR-193a in NSCLC cells and inhibiting tumor growth. Consistent with this view, our results determined that the restoration of p53/miR-193a attenuated the antiapoptotic, proproliferative and proinvasive effect of EGFR in NSCLC cells and inhibited tumor growth in vivo. Thus, the feedback loop comprising p53, miR-193a, and EGFR might serve as a target for NSCLC treatment. Considerable further studies should be performed to explore feasible strategies to inhibit EGFR expression.
This study has some limitations that have to be pointed out. The effects of p53-miR-193a-EGFR regulatory loop were just validated in NSCLC. However, lung cancer contains different types, such as smallcell lung carcinoma, more validation should be performed in various lung cancers. Besides, there is still a long way for the clinical application, more research should be conducted on p53, miR-193a, and EGFR. And we would like to further elucidate the molecular mechanisms of NSCLC and develop new approaches for molecular therapeutics for this disease.
Conclusion
In conclusion, this paper finds a feedback loop comprising EGFR and p53-regulated miR-193a in NSCLC. This finding is in line with the opinion that miRNAs form regulatory motifs with protein regulators to confer robustness to biological processes, and that their dysregulation can lead to dysfunction.
Supplemental Material
Supplemental material, Supplementary_Data for The p53/miR-193a/EGFR feedback loop function as a driving force for non-small cell lung carcinoma tumorigenesis by Wei Wang, Xia-Bo Shen, Wei Jia, Da-Bing Huang, Yong Wang and Yue-Yin Pan in Therapeutic Advances in Medical Oncology
Acknowledgments
We gratefully thank Dr Hang-Cheng Zhou and Dr Ao Xu (pathologists in the Department of Pathology, The First Affiliated Hospital of USTC) for their help in patients’ samples collection of NSCLC.
Footnotes
Author contributions: YYP conceived and designed the experiments. WW, XBS, WJ, DBH, and YW participated in the experiments and drafted the manuscript. All authors read and approved the final manuscript.
Funding: This work was partly supported by the Natural Science Foundation of Anhui Province (No.1708085QH177 and 1908085MH260), Hefei Mu-nicipal Independent Innovation Policy “Borrowing and Transferring” Project (J2018Y01) and National Natural Science Foundation of China (No.81201906).
Conflict of interest statement: The authors declare no potential conflicts of interest.
Supplemental material: Supplemental material for this article is available online.
ORCID iD: Wei Wang  https://orcid.org/0000-0001-7656-0782
https://orcid.org/0000-0001-7656-0782
Contributor Information
Wei Wang, Department of Medical Oncology, The First Affiliated Hospital of USTC, Hefei, PR China.
Xia-Bo Shen, Department of Medical Oncology, Anhui Provincial Hospital, Hefei, PR China.
Wei Jia, Department of Medical Oncology, The First Affiliated Hospital of USTC, Hefei, PR China.
Da-Bing Huang, Department of Medical Oncology, The First Affiliated Hospital of USTC, Hefei, PR China.
Yong Wang, Department of Medical Oncology, The First Affiliated Hospital of USTC, Hefei, PR China.
Yue-Yin Pan, Department of Medical Oncology, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, No.17 Lujiang Road, Luyang District, Hefei 230001, Anhui Province, PR China.
References
- 1. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 2005; 5: 341–354. [DOI] [PubMed] [Google Scholar]
- 2. Forrester SJ, Kawai T, O’Brien S, et al. Epidermal growth factor receptor transactivation: mechanisms, pathophysiology, and potential therapies in the cardiovascular system. Annu Rev Pharmacol Toxicol 2016; 56: 627–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sharma SV, Bell DW, Settleman J, et al. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007; 7: 169–181. [DOI] [PubMed] [Google Scholar]
- 4. Collisson EA, Campbell JD, Brooks AN, et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014; 511: 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cappuzzo F, Hirsch FR, Rossi E, et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst 2005; 97: 643–655. [DOI] [PubMed] [Google Scholar]
- 6. Llovet JM, Villanueva A, Lachenmayer A, et al. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol 2015; 12: 408–424. [DOI] [PubMed] [Google Scholar]
- 7. Seshacharyulu P, Ponnusamy MP, Haridas D, et al. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets 2012; 16: 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Llovet JM, Hernandez-Gea V. Hepatocellular carcinoma: reasons for phase III failure and novel perspectives on trial design. Clin Cancer Res 2014; 20: 2072–2079. [DOI] [PubMed] [Google Scholar]
- 9. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–297. [DOI] [PubMed] [Google Scholar]
- 10. Kloosterman WP, Plasterk RHA. The diverse functions of MicroRNAs in animal development and disease. Dev Cell 2006; 11: 441–450. [DOI] [PubMed] [Google Scholar]
- 11. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136: 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jin F, Wang Y, Li M, et al. MiR-26 enhances chemosensitivity and promotes apoptosis of hepatocellular carcinoma cells through inhibiting autophagy. Cell Death Dis 2017; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kumar MS, Erkeland SJ, Pester RE, et al. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci USA 2008; 105: 3903–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu ZB, Chen X, Zhao Y, et al. Serum microRNA signatures identified in a genome-wide serum microRNA expression profiling predict survival of non-small-cell lung cancer. J Clin Oncol 2010; 28: 1721–1726. [DOI] [PubMed] [Google Scholar]
- 15. Raponi M, Dossey L, Jatkoe T, et al. MicroRNA Classifiers for Predicting Prognosis of Squamous Cell Lung Cancer. Cancer Res 2009; 69: 5776–5783. [DOI] [PubMed] [Google Scholar]
- 16. Yue J, Lv D, Wang C, et al. Epigenetic silencing of miR-483-3p promotes acquired gefitinib resistance and EMT in EGFR-mutant NSCLC by targeting integrin beta3. Oncogene 2018; 37: 4300–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu J, Cheng Y, Li Y, et al. microRNA-128 plays a critical role in human non-small cell lung cancer tumourigenesis, angiogenesis and lymphangiogenesis by directly targeting vascular endothelial growth factor-C. Eur J Cancer 2014; 50: 2336–2350. [DOI] [PubMed] [Google Scholar]
- 18. Liu F, Cai Y, Rong X, et al. MiR-661 promotes tumor invasion and metastasis by directly inhibiting RB1 in non small cell lung cancer. Mol Cancer 2017; 16: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chang T-H, Tsai M-F, Gow C-H, et al. Upregulation of microRNA-137 expression by Slug promotes tumor invasion and metastasis of non-small cell lung cancer cells through suppression of TFAP2C. Cancer Lett 2017; 402: 190–202. [DOI] [PubMed] [Google Scholar]
- 20. Yu T, Li J, Yan M, et al. MicroRNA-193a-3p and –5p suppress the metastasis of human non-small-cell lung cancer by downregulating the ERBB4/PIK3R3/mTOR/S6K2 signaling pathway. Oncogene 2015; 34: 413–423. [DOI] [PubMed] [Google Scholar]
- 21. Liang H, Liu M, Yan X, et al. miR-193a-3p functions as a tumor suppressor in lung cancer by down-regulating ERBB4. J Biol Chem 2015; 290: 926–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fisher JN, Terao M, Fratelli M, et al. MicroRNA networks regulated by all-trans retinoic acid and Lapatinib control the growth, survival and motility of breast cancer cells. Oncotarget 2015; 6: 13176–13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ozsolak F, Poling LL, Wang Z, et al. Chromatin structure analyses identify miRNA promoters. Genes Dev 2008; 22: 3172–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y, Liang H, Zhou G, et al. HIC1 and miR-23 similar to 27 similar to 24 clusters form a double-negative feedback loop in breast cancer. Cell Death Differ 2017; 24: 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martinez NJ, Walhout AJM. The interplay between transcription factors and microRNAs in genome-scale regulatory networks. Bioessays 2009; 31: 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu Y, Liu R, Yang F, et al. miR-19a promotes colorectal cancer proliferation and migration by targeting TIA1. Mol Cancer 2017; 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang C, Xu H, Zhou Z, et al. Blocking of the EGFR-STAT3 signaling pathway through afatinib treatment inhibited the intrahepatic cholangiocarcinoma. Exp Ther Med 2018; 15: 4995–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Z, Ding Y, Wang X, et al. Pseudolaric acid B triggers ferroptosis in glioma cells via activation of Nox4 and inhibition of xCT. Cancer Lett 2018; 428: 21–33. [DOI] [PubMed] [Google Scholar]
- 29. Lin SY, Makino K, Xia WY, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol 2001; 3: 802–808. [DOI] [PubMed] [Google Scholar]
- 30. Tsai Y-C, Chen W-Y, Siu MK, et al. Epidermal growth factor receptor signaling promotes metastatic prostate cancer through microRNA-96-mediated downregulation of the tumor suppressor ETV6. Cancer Lett 2017; 384: 1–8. [DOI] [PubMed] [Google Scholar]
- 31. Chang T-C, Wentzel EA, Kent OA, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 2007; 26: 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brand TM, Iida M, Li C, et al. The nuclear epidermal growth factor receptor signaling network and its role in cancer. Discov Med 2011; 12: 419–432. [PMC free article] [PubMed] [Google Scholar]
- 33. Soussi T. p53 alterations in human cancer: more questions than answers. Oncogene 2007; 26: 2145–2156. [DOI] [PubMed] [Google Scholar]
- 34. Blandino G, Levine AJ, Oren M. Mutant p53 gain of function: differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 1999; 18: 477–485. [DOI] [PubMed] [Google Scholar]
- 35. Muller PAJ, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 2014; 25: 304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer 2010; 10: 760–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liao ZX, Lin SH, Cox JD. Status of particle therapy for lung cancer. Acta Oncol 2011; 50: 745–756. [DOI] [PubMed] [Google Scholar]
- 38. Wozniak A. Challenges in the current antiangiogenic treatment paradigm for patients with non-small cell lung cancer. Crit Rev Oncol Hematol 2012; 82: 200–212. [DOI] [PubMed] [Google Scholar]
- 39. Carpenter G, Lembach KJ, Morrison MM, et al. Characterization of binding of I-125-labeled epidermal growth-factor to human fibroblasts. J Biol Chem 1975; 250: 4297–4304. [PubMed] [Google Scholar]
- 40. Chang Y-S, Chen W-Y, Yin JJ, et al. EGF receptor promotes prostate cancer bone metastasis by downregulating miR-1 and activating TWIST1. Cancer Res 2015; 75: 3077–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005; 97: 339–346. [DOI] [PubMed] [Google Scholar]
- 42. Chari NS, Pinaire NL, Thorpe L, et al. The p53 tumor suppressor network in cancer and the therapeutic modulation of cell death. Apoptosis 2009; 14: 336–347. [DOI] [PubMed] [Google Scholar]
- 43. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 2014; 14: 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu J, Zhang C, Feng Z. Tumor suppressor p53 and its gain-of-function mutants in cancer. Acta Biochim Biophys Sin (Shanghai) 2014; 46: 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhu J, Sammons MA, Donahue G, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015; 525: 199–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lane DP, Cheok CF, Lain S. p53-based cancer therapy. Cold Spring Harb Perspect Biol 2010; 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hsu IC, Tokiwa T, Bennett W, et al. P53 gene mutation and integrated hepatitis-B viral-DNA sequences in human liver-cancer cell-lines. Carcinogenesis 1993; 14: 987–992. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Supplementary_Data for The p53/miR-193a/EGFR feedback loop function as a driving force for non-small cell lung carcinoma tumorigenesis by Wei Wang, Xia-Bo Shen, Wei Jia, Da-Bing Huang, Yong Wang and Yue-Yin Pan in Therapeutic Advances in Medical Oncology