

Abstract
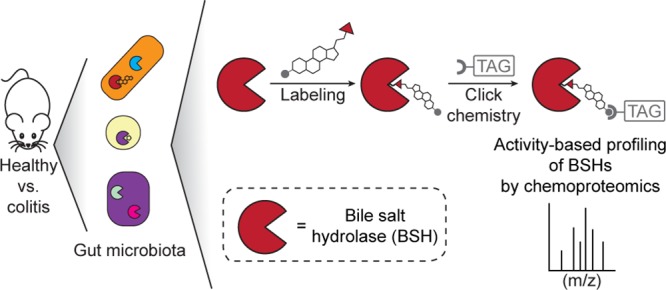
The metagenome of the gut microbiome encodes tremendous potential for biosynthesizing and transforming small-molecule metabolites through the activities of enzymes expressed by intestinal bacteria. Accordingly, elucidating this metabolic network is critical for understanding how the gut microbiota contributes to health and disease. Bile acids, which are first biosynthesized in the liver, are modified in the gut by enzymes expressed by commensal bacteria into secondary bile acids, which regulate myriad host processes, including lipid metabolism, glucose metabolism, and immune homeostasis. The gateway reaction of secondary bile acid biosynthesis is mediated by bile salt hydrolases (BSHs), bacterial cysteine hydrolases whose action precedes other bile acid modifications within the gut. To assess how changes in bile acid metabolism mediated by certain intestinal microbiota impact gut physiology and pathobiology, methods are needed to directly examine the activities of BSHs because they are master regulators of intestinal bile acid metabolism. Here, we developed chemoproteomic tools to profile changes in gut microbiome-associated BSH activity. We showed that these probes can label active BSHs in model microorganisms, including relevant gut anaerobes, and in mouse gut microbiomes. Using these tools, we identified altered BSH activities in a murine model of inflammatory bowel disease, in this case, colitis induced by dextran sodium sulfate, leading to changes in bile acid metabolism that could impact host metabolism and immunity. Importantly, our findings reveal that alterations in BSH enzymatic activities within the gut microbiome do not correlate with changes in gene abundance as determined by metagenomic sequencing, highlighting the utility of chemoproteomic approaches for interrogating the metabolic activities of the gut microbiota.
Short abstract
Activity-based profiling of bile salt hydrolase activity using click-chemistry-based chemoproteomics reveals that enzymatic activity increases in a mouse model of colitis.
Introduction
The human microbiome is a vast and diverse consortium of microorganisms that has numerous effects on our health and physiology.1,2 It comprises an estimated 100 trillion microbes, including bacteria, viruses, archaea, and fungi, that colonize many anatomical sites within our bodies. Among these microbiomes, the densest microbial population resides in the intestines due to the exposure of this organ to microorganisms from our diet and external environment via the gastrointestinal tract.
The gut microbiome contains approximately 100 times the number of genes in the human genome, and this metagenome encodes numerous biosynthetic enzymes that have enormous potential for the biotransformation of small-molecule metabolites.3 The metabolic activity of this gut bioreactor provides many important functions for the host, including breaking down indigestible components of our diet, biosynthesizing essential vitamins and nutrients, and regulation of immunity.2 Accordingly, elucidating the metabolic potential of the many enzymatic reactions occurring within the intestines is critical for understanding how the activities of the gut microbiota contribute to human health and disease.4
Bile acids (BAs) are important metabolites that are initially produced by the host and are subsequently chemically diversified by the gut microbiota.5,6 First, so-called primary BAs are synthesized from cholesterol by hepatocytes in the liver to produce saturated, hydroxylated C24 cyclopentanephenanthrene sterols such as cholic acid and chenodeoxycholic acid. These free BAs are further modified in the liver to increase water solubility through conjugation of the carboxylic acid to glycine or taurine. The conjugated BAs are then actively secreted into bile and stored in the gall bladder. During digestion, bile is released into the small intestine, where the conjugated BAs act as detergents to solubilize dietary lipids and lipid-soluble vitamins.
In the small intestine, conjugated BAs are metabolized by bile salt hydrolase (BSH) enzymes expressed by the gut microbiota via hydrolysis at the C24 amide bond to release unconjugated BAs (Figure 1).7 The BSH-catalyzed step is considered the “gateway reaction” of microbiota-mediated bile salt metabolism because deconjugation must occur before all other transformations affected by the gut microbiome. These include dehydroxylation, dehydrogenation, and sulfation, leading to a large collection of so-called secondary BAs, which have direct effects on the microbiota and also mediate many important biological processes, including host metabolism and immune regulation.8 Thus, BSHs are an important bacterial enzyme class that produces critical metabolites necessary for the proper physiological function of the gut. Despite the significance of these enzymes, their functions in the gut are not well-understood due in part to a lack of tools to assess their activities.
Figure 1.

Chemoproteomic, activity-based approach for profiling bile salt hydrolase (BSH) activity within the gut microbiome during health and disease (e.g., colitis). (a) Scheme of overall chemical strategy to covalently label active BSH enzymes via their active-site cysteine. (b) Structure of the activity-based probe Ch-AOMK used to identify BSH activity in the gut microbiome. (c) Cu-catalyzed azide–alkyne cycloaddition (CuAAC) click chemistry reaction to tag labeled enzymes with an affinity handle or contrast agent (e.g., TAG) for pull-down or imaging of BSH activity. (d) BSH carries out the deconjugation reaction of glyco- and tauro-conjugated bile acids, which is the first major step of bile acid metabolism in the intestines.
Traditional biochemical assays that measure starting material consumption or product formation are less well-suited to identifying and characterizing enzymatic activities from complex biological mixtures such as whole proteomes. Alternatively, functional metagenomics can enable the discovery of new enzymatic activities encoded by the gut microbiome by ectopic expression of metagenomic fragments in model organisms. Using this approach, for example, Jones et al. determined that the human gut microbiome possesses BSH activity within at least three different phyla.9 Whereas this work represents a major advance because it demonstrates the distribution of BSH activity within the intestines, limitations of genomics-based strategies include incomplete coverage arising from potential toxicity of overexpressing certain clones in heterologous systems or incomplete expression of biosynthetic gene clusters, which both lead to unintended elimination of potential biosynthetic enzymes. In addition, artificial expression of exogenous biosynthetic enzymes in a heterologous microorganism in vitro, or even when reconstituted in vivo, may result in nonphysiological enzyme levels or localization within the tissue of interest and therefore may not reflect physiologically relevant enzymatic activities.10
Chemoproteomic tools such as activity-based probes (ABPs) have the unique capacity to target desired enzymatic activities within complex biological systems and have thus revolutionized our ability to discover and characterize enzymes without the need for purification or heterologous expression of the target enzymes.11 ABPs comprise a targeting group to direct the probe to enzymes of interest and a selective, electrophilic chemical warhead to covalently label an active-site nucleophilic residue once the enzyme has bound the targeting group. The enzymatic activity can be detected by inclusion within the ABP of a bioorthogonal handle that can subsequently undergo a click chemistry tagging reaction, such as the copper-catalyzed azide–alkyne cycloaddition (CuAAC), to endow the target protein with an imaging agent or affinity probe. ABPs have been applied to detect enzymatic activity from complex biological samples by either visualization using imaging modalities or protein identification by pull-down using affinity-based reagents, followed by mass spectrometry (MS)-based proteomics.
Inflammatory bowel diseases (IBDs), including colitis and Crohn’s disease, are serious conditions that afflict millions worldwide.12 Symptoms of IBDs include persistent diarrhea, abdominal pain, rectal bleeding, weight loss, and fatigue. These conditions greatly affect patient quality of life and are typically characterized by chronic inflammation in the intestines due to misregulated immune responses.13 Although the exact causes of IBDs remain unknown, dysbiosis of a healthy gut microbiome correlates with increased IBD incidence, suggesting that the microbes play a major role in regulating disease pathogenesis.14
As important metabolites that regulate microbial composition, host immunity, and other aspects of gut physiology, BAs are prime candidates for factors that may be impacted in IBD dysbiosis. In fact, BA metabolism has been examined in IBDs; however, these studies were indirect because they were only able to assess the total levels of conjugated and free BAs, rather than the enzymatic activity that underpins this important biochemical transformation.15 Although photo-cross-linking probes hold promise for identifying cholic-acid-binding proteins, by design they do not inform on BA-metabolizing enzymatic activities and therefore cannot be applied to track changes in BA metabolism.16 Recently, a continuous fluorescent assay was developed to monitor BSH activity within intact bacteria, though the fluorogenic ABP was not used to detect endogenous enzyme within the gut microbiome and cannot inform on the identities of BSH-active bacteria.17 Interestingly, a proteomics study used a general cysteine-reactive ABP to discover an increase in several classes of microbial hydrolases in a mouse model of IBD; however, the impact on bile acid metabolism was not directly addressed.18 Due to the potential importance of certain cysteine hydrolases, notably BSHs, in modulating bile acid metabolism in this disease, we were motivated to develop a direct method for detecting and identifying changes in BSH enzymatic activity that could be applied to assess alterations to global BA metabolism in mouse models of IBD. To this end, we designed and synthesized a trifunctional ABP to profile BSH activity within the gut microbiome in healthy and colitis samples.
Results and Discussion
Because BSH is a cysteine hydrolase, our probe (Figure 1a–c, Scheme S1) contains an acyloxymethylketone warhead, which selectively labels the active-site nucleophile in this enzyme class.19 To selectively target the probe to BSHs, we endowed it with a cholic acid moiety, because conjugated cholic acids are substrates of BSHs also known as choloylglycine hydrolases (CGHs).20,21 Finally, the probe (named Ch-AOMK for cholic acid–acyloxymethylketone) also contains an azido functional group to enable visualization or enrichment of CGH activity from complex biological samples using CuAAC tagging with either fluorophore- or biotin-conjugated alkynes (Figure 1, Scheme S2).22
We first demonstrated that Ch-AOMK can covalently label active CGH in vitro. In this assay, we incubated Clostridium perfringens CGH with Ch-AOMK, followed by CuAAC tagging with a rhodamine 110-alkyne derivative (Fluor 488-alkyne). Analysis of the samples at various times by gel electrophoresis and in-gel fluorescence imaging showed selective labeling of active CGH, but not heat-killed enzyme, with increasing signal over time (Figure 2a, Figure S1a). The enzymatic labeling was detectable after 1 h and reached a maximum at 8 h. To determine that the enzyme retains activity over this period of time in vitro, we performed a biochemical activity assay under similar reaction conditions with its natural substrate, glycocholate, that measures the release of glycine after enzymatic hydrolysis (Figure S1b).23 We also demonstrated that increasing the concentration of Ch-AOMK led to a dose-dependent increase in CGH labeling (Figure 2b, Figure S1c).
Figure 2.

Ch-AOMK labels Clostridium perfringens BSH in vitro in a time- and dose-dependent manner. Active or heat-killed (HK) BSH was treated with Ch-AOMK for various amounts of time using (a) Ch-AOMK at 500 μM or (b) varying concentrations of Ch-AOMK at 37 °C for 24 h, after which the samples were tagged using the copper-catalyzed azide–alkyne cycloaddition (CuAAC) with Fluor 488-alkyne. The samples were analyzed by SDS-PAGE and visualized by in-gel fluorescence. A.U. = arbitrary unit. The bands were quantified by densitometry using ImageJ (bottom panels). Error bars represent standard deviation from the mean. * p < 0.05, ** p < 0.01, *** p < 0.001, n.s. = not significant, n = (a) 8, (b) 6.
To assess whether Ch-AOMK can selectively label active CGH within bacterial lysates, we overexpressed C. perfringens CGH in Escherichia coli. We found that Ch-AOMK efficiently labeled wildtype (WT) CGH but not the catalytically inactive Cys2Ser (C2S) mutant, using CuAAC tagging with Fluor-488, gel electrophoresis, and in-gel fluorescence imaging (Figure 3a, Figure S2a,b). We also verified that active CGH labeled with Ch-AOMK could be enriched following CuAAC tagging with biotin-alkyne and pull-down using streptavidin-conjugated agarose (Figure 3b, Figures S3 and S4d). Here, we verified WT and C2S mutant expression by probing for their C-terminal FLAG peptide epitope tag, and importantly, the labeling was dependent on the amount of bacterial lysate (Figure S2).
Figure 3.

Ch-AOMK labels C. perfringens BSH expressed in Escherichia coli. Ch-AOMK (500 μM) was incubated with lysates from C. perfringens wildtype (WT) BSH or C2S mutant expressed in E. coli at 37 °C for 24 h. Following Ch-AOMK labeling, CuAAC tagging was carried out with (a) Fluor 488-alkyne or (b) biotin-alkyne. (a) Samples were subjected to SDS-PAGE, and the gel was visualized using fluorescence, followed by Coomassie staining. (b) Samples were analyzed either by Western blot with streptavidin-HRP or by silver staining. Input is 2% of the elution. The arrowhead indicates the expected size of BSH (37 kDa). A.U. = arbitrary unit. The bands were quantified by densitometry using ImageJ (bottom panels). Error bars represent standard deviation from the mean. * p < 0.05, ** p < 0.01, *** p < 0.001, n.s. = not significant, n = (a) 6, (b) 5.
We next determined that Ch-AOMK can label active BSH within model anaerobic bacterial strains from the gut microbiome whose BSH activities have been biochemically characterized.24 Lysates generated from Bifidobacterium bifidum and Bifidobacterium longum were incubated with Ch-AOMK, followed by CuAAC tagging with Fluor 488-alkyne, and samples were analyzed by gel electrophoresis and in-gel fluorescence imaging (Figure 4a,b, Figure S4b,c). Gratifyingly, Ch-AOMK could facilitate selective visualization of a band with an approximate molecular weight of 35 kDa, the expected size of BSH, from both of these bacteria.24 As a control, Ch-AOMK labeling of the 35 kDa species was eliminated in the presence of iodoacetamide (IA), which alkylates cysteine residues and therefore prevents the AOMK warhead from targeting the BSH active-site nucleophile. We unequivocally identified the B. bifidum and B. longum BSHs as the targets of Ch-AOMK by following the ABP labeling step with CuAAC tagging with biotin-alkyne, streptavidin-agarose enrichment, and identification by mass spectrometry, which verified that the 35 kDa species were indeed BSHs expressed by these bacteria (Figure 4c,d, Figure S4e,f and Table S1).
Figure 4.

Ch-AOMK labels BSH from gut anaerobes. Ch-AOMK (500 μM) was incubated with lysates from (a, c) Bifidobacterium bifidum or (b, d) Bifidobacterium longum at 37 °C for 24 h. (a, b) Lysates were treated with iodoacetamide (IA, 20 mM) prior to incubation with Ch-AOMK as a negative control. Following Ch-AOMK labeling, CuAAC tagging was carried out with (a, b) Fluor 488-alkyne or (c, d) biotin-alkyne. (a, b) Samples were subjected to SDS-PAGE, and the gel was visualized using fluorescence, followed by Coomassie staining. (c, d) Samples were analyzed either by Western blot with streptavidin-HRP or by silver staining. Input is 2% of the elution. The arrowhead indicates the expected size of BSHs (35 kDa). A.U. = arbitrary unit. The bands were quantified by densitometry using ImageJ (bottom panels). Error bars represent standard deviation from the mean. * p < 0.05, ** p < 0.01, *** p < 0.001, n.s. = not significant, n = (a) 5, (b) 4, (c) 4, (d) 5.
To demonstrate that Ch-AOMK can label active BSH within complex biological samples, we then tested its ability to target BSH activity within the murine gut microbiome. Lysates from gut bacteria isolated from mouse fecal samples were incubated with Ch-AOMK, followed by CuAAC tagging with Fluor 488-alkyne (Figure 5a, Figure S5a). We found substantial and selective Ch-AOMK labeling in these samples of proteins with approximate molecular weights of 35 kDa, the expected mass of most known gut bacterial BSH enzymes.24 Ch-AOMK labeling of these species was abrogated by pretreatment with IA, providing evidence that the labeling is due to covalent reaction with cysteines.
Figure 5.

Changes in BSH activity in the gut microbiome are identified using Ch-AOMK in healthy and colitic mice. (a) Bacterial lysates (100 μg) isolated from healthy mouse gut microbiomes were incubated with Ch-AOMK (100 μM) at 37 °C for 24 h. After CuAAC tagging with Fluor 488-alkyne, samples were analyzed by SDS-PAGE, followed by visualization using fluorescence. As a negative control for cysteine labeling, iodoacetamide (IA, 20 mM) was added prior to Ch-AOMK. Coomassie staining served as the loading control. Alternatively, (b) lysates (2.5 mg) were incubated with Ch-AOMK (100 μM) at 37 °C for 12 h. After CuAAC tagging with biotin-alkyne, labeled proteins were enriched by streptavidin-agarose pull-down and analyzed either by Western blot with streptavidin-HRP or by silver staining. Input is 2% of the elution. (c–e, g, h) Mice were treated with dextran sodium sulfate (DSS, 3% w/v, ad libitum) for 8 days, and (c, d) bacterial populations from the gut microbiome were lysed and analyzed as in parts a and b. The arrowhead indicates the expected mass of BSHs (35 kDa). The bands were quantified by densitometry using ImageJ (a–d, bottom panels). A.U. = arbitrary unit. (e) Phylogenetic tree of BSHs identified within the mouse metagenomic assemblies. Bootstrap confidence levels reflect 100 phylogenetic tree reconstructions and are indicated by the blue circles. Green indicates Firmicutes, magenta indicates Bacteroidetes, and bold indicates active BSHs identified by chemoproteomics using Ch-AOMK labeling, followed by CuAAC-based tagging, enrichment, and protein identification by mass spectrometry (MS)-based proteomics, using a 2-fold enrichment cutoff (see also Table S2). The heatmap corresponds to samples from three independent experiments (1–3) from mice treated with DSS, which were also analyzed by MS-based proteomics to identify changes in BSH activity during disease (see also Table S5). Red indicates higher BSH activity in DSS compared to control mice, blue indicates lower BSH activity in DSS compared to control mice (fold change according to heatmap), and gray indicates that the BSH was not identified in the indicated mass spectrometry experiment. (f) Fold change (log2) of enrichment of BSH from healthy mouse microbiomes comparing Ch-AOMK treatment to no Ch-AOMK treatment (y-axis) versus bsh gene abundance using reads per kilobase million (RPKM) in the mice (x-axis). (g) Fold change (log2) of enrichment of BSH from microbiomes of mice treated with DSS compared to vehicle controls (y-axis) versus ratio of bsh gene abundance (RPKM) in mice treated with DSS compared to controls (x-axis). The data in part g correspond to experiment 2 in part e. (h) Quantification by MS-based metabolomics of fecal bile acid levels in DSS colitis versus control mice. CA, cholic acid; TCA, taurocholic acid; DCA, deoxycholic acid; TDCA, taurodeoxycholic acid; CDCA, chenodeoxycholic acid; TCDCA, taurochenodeoxycholic acid; LCA, lithocholic acid; TLCA, taurolithocholic acid. Each plot indicates the ratio of a corresponding unconjugated to conjugated BA pair. Error bars represent standard deviation from the mean. Interquartile ranges (IQRs, boxes), median values (line within box), whiskers (lowest and highest values within 1.5 times IQR from the first and third quartiles), and outliers beyond whiskers (dots) are indicated. * p < 0.05, ** p < 0.01, *** p < 0.001, n.s. = not significant, n = (a) 5, (b) 6, (c) 6, (d) 5, (e) 3, (h) 20.
We next identified these labeled proteins using MS-based proteomics, following CuAAC with biotin-alkyne and pull-down using streptavidin-agarose, which verified their identities as BSHs (Figure 5b, Figure S5b and Table S2). Although bsh genes are common across phyla, according to our assemblies of shotgun metagenomic sequences from the mouse gut microbiome, the majority of active BSHs within the healthy gut microbiome was derived from several bacteria within the phylum Bacteroidetes, which are Gram-negative bacteria that constitute one of the dominant phyla within the gut microbiome (e.g., 20–40% of healthy individuals)25 and whose BSH activities have recently been biochemically characterized (Figure 5e).26 To determine whether Ch-AOMK can label BSHs from phylogenetically distant bacteria, we calculated the phylogenetic distances between protein sequences of the active BSHs from the gut microbiome and bacterial BSHs used in the in vitro studies (Figure S6 and Table S3). From these results, we conclude that Ch-AOMK does not have a bias for bacteria from the same taxonomic classification. In addition, Ch-AOMK labeling was not solely dependent on gene abundance because several bacteria within the Bacteroidetes phylum whose bsh genes were at lower abundance exhibited similar enrichment of BSH activity using our chemoproteomics approach to others with more abundant bsh genes (Figure 5f, Table S4).
Then, we applied our strategy to profile the BSH activity in IBDs within mice. We used a well-established mouse model of colitis that is induced by treatment with dextran sodium sulfate (DSS), which induces intestinal inflammation resembling the human disease (Figure 1a, Figure S7a).27 In these studies, global BSH activity detected by Ch-AOMK labeling increased significantly during DSS treatment, as indicated by CuAAC tagging with Fluor 488-alkyne and in-gel fluorescence (Figure 5c, Figure S7c). Additionally, Ch-AOMK labeling, followed by CuAAC with biotin-alkyne and streptavidin-agarose pull-down, led to significantly increased enrichment of BSHs from DSS-treated mice (Figure 5d, Figure S7d). We verified that BSH activity increases during DSS colitis using the biochemical activity assay described above (Figure S7b).23 Together, these results suggest that BSH activity increases during DSS colitis in mice.
To examine the individual BSH enzymes contributing to this increase, we analyzed the enriched samples by MS-based proteomics. Unexpectedly, we found that the overall increase in BSH activity is not due to the activities of specific bacteria because individual bacterial BSHs had altered activities during independent DSS experiments (1–3, Figure 5e). These data suggest that some bacterial BSH activities increase or decrease, while others remain the same (Figure 5e), but the global level of BSH activity consistently increases during DSS treatment (Figure 5c, Figure S7c). Again, we found that these changes in BSH activity do not correlate with bsh gene abundance within the gut microbiome metagenomic assemblies (Figure 5g, Table S4). These results highlight the dynamic nature of the gut microbiome and the stochastic nature of perturbations to the microbial composition that accompany DSS colitis (Figure S7e–g).27
Finally, we turned to MS-based metabolomics to determine whether the increase in global BSH activity leads to changes in levels of relevant primary and secondary BA metabolites. Critically, we found that DSS treatment led to increased ratios of deconjugated to conjugated BAs across four major primary and secondary BAs in the gut, consistent with our Ch-AOMK data indicating increased BSH activity in DSS colitis (Figure 5h). Thus, we conclude that global BSH activity increases in a mouse model of DSS colitis and that this increase is due to the collective BSH activities of the gut microbiome, rather than the activities of specific, individual BSH enzymes.
Our findings synergize with a recent report from Wright and co-workers, who developed an ABP for profiling the activity of β-glucuronidases, xenobiotic metabolizing enzymes, within healthy mouse gut microbiomes.28 In that study, the authors found that the individual bacteria contributing to global β-glucuronidase activity differed in replicate groups of mice. As well, they found that antibiotic treatment decreased global β-glucuronidase activity but that the remaining activity was again due to distinct types of bacteria in replicate experiments, suggesting interindividual variability at the level of microbiota-derived enzymatic activity. Notably, our examination of BSH activity using the ABP Ch-AOMK revealed similar interindividual variability wherein distinct bacteria contributed to changes in global BSH activity in different replicate experimental groups in DSS-treated compared to healthy mice. We envision that our chemoproteomic strategy for labeling active BSHs could be combined with orthogonally functionalized ABPs to simultaneously profile multiple activities of different enzyme families, including β-glucuronidases, within the gut microbiome.
An interesting future direction will be the extension of these approaches to profile enzymatic activities within human gut microbiomes, which could identify changes in activity that accompany different physiological states. In addition, chemoproteomic strategies using ABPs could also be applied to healthy and diseased human samples to identify changes in enzymatic activities that arise from various pathological conditions. These findings could enable the discovery of enzymes or their products as potential biomarkers for these diseases. As such, our findings that BSH activity increases in a mouse model of colitis suggest the potential use of BSH activity as a biomarker for human IBDs, whose currently available biomarkers have limited clinical utility because they are not sufficiently specific to enable an accurate diagnosis.29
Conclusion
In summary, we have developed a targeted chemoproteomic strategy for profiling BSH activity in complex biological settings, such as the gut microbiome. We synthesized an ABP that targets the active-site cysteine residue within BSH enzymes and can be applied to detect enzymatic activity in vitro using purified BSH. We demonstrated that this approach can label active BSHs within model microorganisms and gut anaerobes. Moreover, we applied our technology to identify and characterize changes in BSH activity that occur within the gut microbiome during IBD in a mouse model of colitis. Using the ABP, we discovered that colitis-associated gut dysbiosis led to increases in global BSH activity, a finding that was confirmed by metabolomic analysis of bile acid metabolites. Interestingly, chemoproteomic and metagenomic analyses revealed that the relative contributions of individual BSH enzymes varied and did not correlate with gene abundance. Thus, our findings highlight the utility of activity-based, chemoproteomic approaches to directly identify disease-associated perturbations in enzymatic activities that may not depend on changes in gene abundance. In the future, we envision that this approach may prove useful to understand variations in BSH activity during other important physiological processes and diseases impacted by bile acid metabolism, such as metabolic syndrome.
Acknowledgments
This work was supported by the Arnold and Mabel Beckman Foundation (Beckman Young Investigator Award to P.V.C.) and a President’s Council of Cornell Women Affinito-Stewart Grant (P.V.C.). I.L.B. is supported by a Sloan Research Fellowship, a Packard Fellowship for Science and Engineering, and a Pew Biomedical Scholarship. We thank Gael Nicolas for technical assistance, Samantha Scott for animal husbandry, and Nicole Spiegelman and Albert Vill for technical advice. We also thank the staff in the Proteomics and Genomics Facilities at the Biotechnology Resource Center of Cornell University and NIH SIG 1S10 OD017992-01 grant support for the Orbitrap Fusion mass spectrometer.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.9b00147.
Table S1. B. bifidum and B. longum data (XLSX)
General materials and methods, synthetic schemes, additional data and figures, including Ch-AOMK labeling of BSHs, phylogenetic tree of putative BSHs, and 1H and 13C NMR of Ch-AOMK (PDF)
Table S2. Mouse microbiome data with Ch-AOMK (XLSX)
Table S3. All BSH similarity matrix (XLSX)
Table S4. RPKM fold change (XLSX)
Table S5. DSS 1, 2, and 3 (XLSX)
The authors declare no competing financial interest.
Supplementary Material
References
- Tremaroli V.; Bäckhed F. Functional Interactions between the Gut Microbiota and Host Metabolism. Nature 2012, 489 (7415), 242–249. 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- Belkaid Y.; Hand T. W. Role of the Microbiota in Immunity and Inflammation. Cell 2014, 157 (1), 121–141. 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel N.; Balskus E. P. Exploring and Understanding the Biochemical Diversity of the Human Microbiota. Cell Chem. Biol. 2016, 23 (1), 18–30. 10.1016/j.chembiol.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Dorrestein P. C.; Mazmanian S. K.; Knight R. Finding the Missing Links among Metabolites, Microbes, and the Host. Immunity 2014, 40 (6), 824–832. 10.1016/j.immuni.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlström A.; Sayin S. I.; Marschall H.-U.; Bäckhed F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24 (1), 41–50. 10.1016/j.cmet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- Ridlon J. M.; Kang D.-J.; Hylemon P. B. Bile Salt Biotransformations by Human Intestinal Bacteria. J. Lipid Res. 2006, 47 (2), 241–259. 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- Begley M.; Hill C.; Gahan C. G. M. Bile Salt Hydrolase Activity in Probiotics. Appl. Environ. Microbiol. 2006, 72 (3), 1729–1738. 10.1128/AEM.72.3.1729-1738.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin A. S.; Fischbach M. A. A Biosynthetic Pathway for a Prominent Class of Microbiota-Derived Bile Acids. Nat. Chem. Biol. 2015, 11 (9), 685–690. 10.1038/nchembio.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B. V.; Begley M.; Hill C.; Gahan C. G. M.; Marchesi J. R. Functional and Comparative Metagenomic Analysis of Bile Salt Hydrolase Activity in the Human Gut Microbiome. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (36), 13580–13585. 10.1073/pnas.0804437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niphakis M. J.; Cravatt B. F. Enzyme Inhibitor Discovery by Activity-Based Protein Profiling. Annu. Rev. Biochem. 2014, 83 (1), 341–377. 10.1146/annurev-biochem-060713-035708. [DOI] [PubMed] [Google Scholar]
- Shannon D. A.; Weerapana E. Covalent Protein Modification: The Current Landscape of Residue-Specific Electrophiles. Curr. Opin. Chem. Biol. 2015, 24, 18–26. 10.1016/j.cbpa.2014.10.021. [DOI] [PubMed] [Google Scholar]
- Khor B.; Gardet A.; Xavier R. J. Genetics and Pathogenesis of Inflammatory Bowel Disease. Nature 2011, 474 (7351), 307–317. 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloy K. J.; Powrie F. Intestinal Homeostasis and Its Breakdown in Inflammatory Bowel Disease. Nature 2011, 474 (7351), 298–306. 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- Palm N. W.; de Zoete M. R.; Cullen T. W.; Barry N. A.; Stefanowski J.; Hao L.; Degnan P. H.; Hu J.; Peter I.; Zhang W.; et al. Immunoglobulin A Coating Identifies Colitogenic Bacteria in Inflammatory Bowel Disease. Cell 2014, 158 (5), 1000–1010. 10.1016/j.cell.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duboc H.; Rajca S.; Rainteau D.; Benarous D.; Maubert M. A.; Quervain E.; Thomas G.; Barbu V.; Humbert L.; Despras G.; et al. Connecting Dysbiosis, Bile-Acid Dysmetabolism and Gut Inflammation in Inflammatory Bowel Diseases. Gut 2013, 62 (4), 531–539. 10.1136/gutjnl-2012-302578. [DOI] [PubMed] [Google Scholar]
- Zhuang S.; Li Q.; Cai L.; Wang C.; Lei X. Chemoproteomic Profiling of Bile Acid Interacting Proteins. ACS Cent. Sci. 2017, 3 (5), 501–509. 10.1021/acscentsci.7b00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandvold K. R.; Weaver J. M.; Whidbey C.; Wright A. T. A Continuous Fluorescence Assay for Simple Quantification of Bile Salt Hydrolase Activity in the Gut Microbiome. Sci. Rep. 2019, 9 (1), 1359. 10.1038/s41598-018-37656-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayers M. D.; Moon C.; Stupp G. S.; Su A. I.; Wolan D. W. Quantitative Metaproteomics and Activity-Based Probe Enrichment Reveals Significant Alterations in Protein Expression from a Mouse Model of Inflammatory Bowel Disease. J. Proteome Res. 2017, 16 (2), 1014–1026. 10.1021/acs.jproteome.6b00938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato D.; Boatright K. M.; Berger A. B.; Nazif T.; Blum G.; Ryan C.; Chehade K. A. H.; Salvesen G. S.; Bogyo M. Activity-Based Probes That Target Diverse Cysteine Protease Families. Nat. Chem. Biol. 2005, 1 (1), 33–38. 10.1038/nchembio707. [DOI] [PubMed] [Google Scholar]
- Batta A. K.; Salen G.; Shefer S. Substrate Specificity of Cholylglycine Hydrolase for the Hydrolysis of Bile Acid Conjugates. J. Biol. Chem. 1984, 259 (24), 15035–15039. [PubMed] [Google Scholar]
- Huijghebaert S. M.; Hofmann A. F. Influence of the Amino Acid Moiety on Deconjugation of Bile Acid Amidates by Cholylglycine Hydrolase or Human Fecal Cultures. J. Lipid Res. 1986, 27 (7), 742–752. [PubMed] [Google Scholar]
- Patterson D. M.; Nazarova L. A.; Prescher J. A. Finding the Right (Bioorthogonal) Chemistry. ACS Chem. Biol. 2014, 9 (3), 592–605. 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]
- Bond C.; Nair P. P.; Gordon M.; Reback J. The Enzymatic Cleavage of the Carbon-Nitrogen Bond in 3α,7α,12α-Trihydroxy-5β-Cholan-24-Oylglycine. J. Biol. Chem. 1967, 242 (1), 7–11. [PubMed] [Google Scholar]
- Kumar R. S.; Brannigan J. A.; Prabhune A. A.; Pundle A. V.; Dodson G. G.; Dodson E. J.; Suresh C. G. Structural and Functional Analysis of a Conjugated Bile Salt Hydrolase from Bifidobacterium Longum Reveals an Evolutionary Relationship with Penicillin V Acylase. J. Biol. Chem. 2006, 281 (43), 32516–32525. 10.1074/jbc.M604172200. [DOI] [PubMed] [Google Scholar]
- Arumugam M.; Raes J.; Pelletier E.; Paslier D. Le; Yamada T.; Mende D. R.; Fernandes G. R.; Tap J.; Bruls T.; Batto J.-M.; et al. Enterotypes of the Human Gut Microbiome. Nature 2011, 473 (7346), 174. 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L.; Seaton S. C.; Ndousse-Fetter S.; Adhikari A. A.; DiBenedetto N.; Mina A. I.; Banks A. S.; Bry L.; Devlin A. S. A Selective Gut Bacterial Bile Salt Hydrolase Alters Host Metabolism. eLife 2018, 7, e37182. 10.7554/eLife.37182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munyaka P. M.; Rabbi M. F.; Khafipour E.; Ghia J. E. Acute Dextran Sulfate Sodium (DSS)-Induced Colitis Promotes Gut Microbial Dysbiosis in Mice. J. Basic Microbiol. 2016, 56 (9), 986–998. 10.1002/jobm.201500726. [DOI] [PubMed] [Google Scholar]
- Whidbey C.; Sadler N. C.; Nair R. N.; Volk R. F.; DeLeon A. J.; Bramer L. M.; Fansler S. J.; Hansen J. R.; Shukla A. K.; Jansson J. K.; et al. A Probe-Enabled Approach for the Selective Isolation and Characterization of Functionally Active Subpopulations in the Gut Microbiome. J. Am. Chem. Soc. 2019, 141 (1), 42–47. 10.1021/jacs.8b09668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norouzinia M.; Chaleshi V.; Alizadeh A. H. M.; Zali M. R. Biomarkers in Inflammatory Bowel Diseases: Insight into Diagnosis, Prognosis and Treatment. Gastroenterol. Hepatol. from Bed to Bench 2017, 10 (3), 155–167. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.