

Abstract
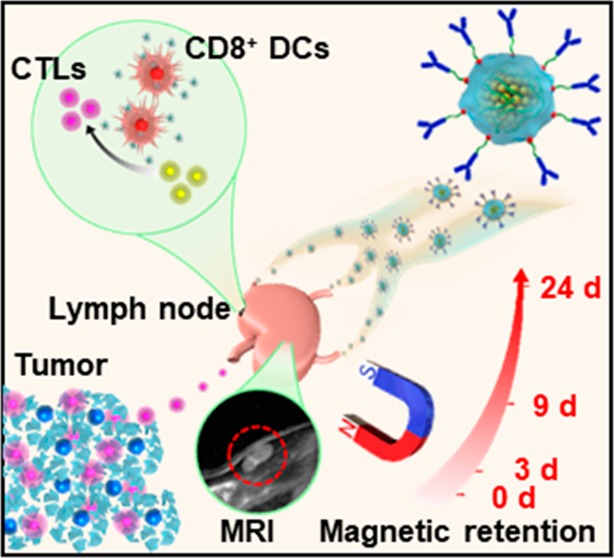
A novel cancer vaccine is developed by using Fe3O4 magnetic nanoclusters (MNCs) as the core and cancer cell membranes decorated with anti-CD205 as the cloak. Because of the superparamagnetism and magnetization of MNCs, it is first achieved for the magnetic retention of vaccine in the lymph nodes with a magnetic resonance imaging (MRI) guide, which opened the time window for antigen uptake by dendritic cells (DCs). Meanwhile, the camouflaged cancer cell membranes serve as a reservoir of various antigens, enabling subsequent multiantigenic response. Additionally, the decorated anti-CD205 direct more vaccine into CD8+ DCs, facilitating the major histocompatibility complex (MHC) I cross-presentation. These unique advantages together lead to a great proliferation of T cells with superior clonal diversity and cytotoxic activity. As a result, potent prophylactic and therapeutic effects with few abnormalities are observed on five different tumor models. Therefore, such a cancer-derived magnetosome with the integration of various recent nanotechnologies successfully demonstrates its promise for safe and high-performance cancer vaccination.
Short abstract
A magnetosome was engineered for magnetic retention in lymph nodes and efficient activation of cellular immunity, which could serve as a high-performance cancer vaccine.
After years of clinical practices, traditional anticancer strategies such as chemotherapy and radiotherapy still provide unsatisfactory results. The most critical problem associated with traditional anticancer therapies is untargeted cytotoxicity that impacts both cancerous and healthy tissues, resulting in serious side effects.1−3 This situation has led to an urgent call for developing new strategies for anticancer therapy with safe and efficient performance. In recent years, increased knowledge of immunology and oncology has significantly catalyzed the development of novel cancer vaccines. This treatment modality can, in principle, specifically eliminate cancer cells by inducing a host immune response.4,5 On one hand, numerous antibodies generated via humoral immunity can be utilized to neutralize cancer cells, thus playing an important role in cancer prevention.6 On the other hand, cytotoxic T lymphocytes (CTLs) sourced from the cellular immunity have the capability to directly lyse cancer cells, which is a requirement for effective therapeutic cancer vaccines.7,8 However, therapeutic cancer vaccines tend to be intractable as the immune system has previously tolerated the cancer cells, which have been disguised with self-antigens.9
Unfortunately, exposure to cancer-derived antigens alone fails to elicit strong immune responses due to poor immunogenicity. To address this, efforts in cancer vaccine development have revolved around the design of adjuvants that improve the immune responses.10 For example, researchers explored a series of Toll-like receptor (TLR) agonists including CpG oligodeoxynucleotide (CpG-ODN),8,11,12 flagellin,13−15 and monophosphoryl lipid A (MPLA),16−19 which have proven useful for antigen-presenting cell (APC) maturation. Considering that antigen uptake by APCs is prerequisite for an immune response, recent efforts have focused on the construction of efficient antigen delivery systems.5,8,19,20 As numerous APCs and T cells reside in the lymph nodes, directly delivering antigens into the lymph nodes seems an efficient strategy.21,22 Correspondingly, many nanodelivery systems, such as liposomes, dendrimers,23 and micelles,24 have attracted considerable interest due to their ability to efficiently drain the lymph nodes. In addition, TLR agonists can be codelivered to the lymph nodes, which should further improve the immune responses.25,26 Although promising, a series of problems have been associated with these nanovaccines. For example, efferent lymphatic vessels promoted the outflow of vaccines in the lymph nodes, thus reducing the possibility of antigen capture by APCs.27 Even if the nanovaccines are intercepted, the antigens released in APCs are always processed and presented with the major histocompatibility complex (MHC) II, which mainly activate humoral immunity rather than cellular immunity.28 As a result, nanovaccine performance falls short of expectations.
Herein, we developed a novel cancer-derived magnetosome as a high-performance cancer vaccine (Scheme 1). Briefly, Fe3O4 magnetic nanoclusters (MNCs) with uniform size, high superparamagnetism, and magnetization were synthesized as the core of the magnetosome. Their packed properties enabled us to magnetically retain them in the lymph nodes with a magnetic resonance imaging (MRI) guide. The MNC core was then adsorbed with the TLR agonist CpG-ODN (C) and camouflaged with cancer cell membranes (M) via electrostatic interactions, which served as the APC activator and multiple antigen reservoir. Via intrinsic biosynthesis and metabolic incorporation of phospholipids, cancer cell membranes could be engineered to contain azides. This technique facilitated subsequent decoration with dibenzocyclooctyne-modified (DBCO-modified) anti-CD205 (A) through mild and efficient click chemistry (Scheme 1A).29,30 In this case, the formed cancer-cell-membrane-coated CpG-ODN-loaded MNC with anti-CD205 decoration (A/M/C-MNC) could be preferentially recognized by CD8+ dendritic cells (DCs), which mainly reside in the lymph nodes and have the capability for MHC I cross-presentation and CD8+ T cell stimulation (Scheme 1B).31,32 As a result, a large amount of T cells proliferated with great clonal diversity and superior cytotoxic activity. Using five different tumor models, potent anticancer efficacy and safety were systematically and integrally demonstrated, showing the great promise of utilizing this novel platform for cancer vaccination.
Scheme 1. Schematic Illustration of A/M/C-MNC for Immunotherapy.

(A) Fabrication process of A/M/C-MNC. (B) Illustration of A/M/C-MNC-mediated cellular immune responses eliciting cytotoxicity T lymphocytes (CTLs) and memory T cells (TM cells) for cancer immunotherapy.
For long-term retention in the lymph nodes, the magnetosome core should have high magnetization for magnetic control. Meanwhile, the combination of high superparamagnetism is desirable, as it will help us to noninvasively monitor the dynamic distribution in the lymph nodes via MRI. Unfortunately, these two properties are contradictory in traditional magnetic nanoparticles. To circumvent this challenge, we previously developed a hydrothermal treatment method to synthesize MNC with uniform size, which consisted of stacked ∼10 nm particle building units (Figure 1A). This unique architecture endowed the MNC with both superior magnetization and superparamagnetism. The positive charge sourced from doped polyethylenimine (PEI) also facilitated the adsorption of CpG-ODN (Figure S1A) and subsequent camouflaging of 4T1 cancer cell membrane fragments (MFs), which were pre-engineered to contain azides (Figure S1B–D) and enriched by ultracentrifugation (Figure S1E). Correspondingly, we observed the formation of a uniform layer around the MNC (Figure 1A and Figure S1F,G) containing mostly membrane protein components (Figure 1B and Figure S1H), and the sandwiched CpG-ODN could thus be well-retained during electrophoresis (Figure S1A). Finally, DBCO-modified anti-CD205 (Figure S1I–K) could be mildly and efficiently anchored around the magnetosome (M/C-MNC) via click chemistry, which was confirmed by super-resolution imaging as shown in Figure 1C.
Figure 1.

Construction and characterization of A/M/C-MNC. (A) TEM imaging of MNC and M/C-MNC. The space between the red lines indicates the cancer cell membrane. (B) SDS-PAGE analysis of proteins in whole cell (WC), M/C-MNC, and A/M/C-MNC. (C) Stochastic optical reconstruction microscopic (STORM) imaging of A/M/C-MNC labeled with a fluorescent secondary antibody. (D) Fluorescence imaging of A/M/C-MNC. The anti-CD205 (pink), membrane (green), and CpG-ODN (blue) were labeled with a fluorescent secondary antibody, DiD, and FAM, respectively. (E) Size number curves and surface ζ potentials of MNC, C-MNC, M/C-MNC, and A/M/C-MNC measured by dynamic light scattering (DLS). (F) Stability of A/M/C-MNC over a 2 week period in PBS. (G) Magnetic hysteresis loop and rapid response of A/M/C-MNC measured at room temperature. The saturation magnetization σs of A/M/C-MNC was determined to be 63.56 emu g–1. (H) T2 relaxation rate (1/T2, s–1) versus Fe concentration (mM) at 7 T. Inset: T2-weighted MRI images of A/M/C-MNC in 0.1% molten agar gel at different Fe concentrations. All data represent the mean ± SD (n = 3).
For further verification and in vivo utilization, the resulting A/M/C-MNC was systematically characterized. As shown in Figure 1D, MNC, CpG-ODN, membrane, and anti-CD205 perfectly colocalized with each other, indicating the successful assembly of these four components. The gradual increase in the particle size and decreased ζ potential also demonstrated successful CpG-ODN adsorption, membrane camouflaging, and anti-CD205 decoration, and their amounts could be tuned up to 10, 73.68, and 6.8 μg for every 100 μg of MNC (Figure 1E and Figure S1L–N). During 2 weeks of storage, little change was observed on the size and ζ potential of A/M/C-MNC (Figure 1F and Figure S2). Meanwhile, the high saturation magnetization (∼70 emug–1) enabled rapid enrichment by a commercial magnetic scaffold (Figure 1G), and the MRI signal intensity was proportional to the particle concentration (Figure 1H). These packed features thus ensured the integrated structure during in vivo transportation, paved the way for lymph node retention, and shed light on the T2 contrast imaging.
To examine the proposed use of magnetic control to extend the retention time in the lymph nodes, we labeled the membrane component with 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyaine iodide (DiR) and tracked its fate in vivo. After optimization, the magnetic field should be applied immediately after injection at the draining lymph node (Figures S3–S5). As shown in Figure 2A,B, the signal of the free MF in the lymph nodes was very weak because of its rapid clearance. Once formulated into the nanovaccine, A/M/C-MNC drained into the lymph nodes within 24 h, as indicated by the brighter signal. However, because of the efferent lymphatic vessels, attenuation was observed, and the signal had mostly disappeared by day 9. Notably, we observed a significantly improved signal in the lymph nodes upon applying a magnetic field (m), which could be preserved more than 3 weeks. Compared to the MF group, the A/M/C-MNC with magnetic retention (A/M/C-MNC(m)) group demonstrated a 5.99-fold extension of the retention half-life (Figure 2C), and the area under the curve was also shown to increase up to 48.1-fold (Figure 2D). The great improvement in the lymph node delivery was further verified by histological examination (Figure 2E). Specifically, more than 50% of the cells were surrounded with the DiR signal in the A/M/C-MNC(m) group, while the signal clearly decreased to 16.07% and 4.1% for A/M/C-MNC and MF, respectively. To gain deeper insight into the systemic distribution, we excised organs in the A/M/C-MNC(m) group (Figure 2F). Compared to the fluorescence observed in the lymph nodes, other organs exhibited a faint signal. This indicated that most of the A/M/C-MNC had been magnetically retained in the lymph nodes rather than distributed elsewhere, which also caused no changes of A/M/C-MNC accumulation in other lymph nodes (Figure S3). Because of the superparamagnetism of the MNC core, similar results could also be obtained by in vivo MRI (Figure 2G). The A/M/C-MNC(m) group achieved a significantly higher T2 signal in the lymph nodes than in the other organs, which again demonstrated the success of our proposed use of magnetic control to improve lymph node delivery. Considering that the magnetic retention effect was cumulative (Figure S6), and the fluorescence signals became negligible longer than 21 days in the A/M/C-MNC(m) group (Figure 2B), we continuously applied the magnetic field for 21 days in the latter experiments.
Figure 2.

Magnetic field-induced lymph node (LN) retention and anti-CD205-mediated CD8+ DCs targeting of A/M/C-MNC in vivo. (A) Delivery visualization of DiR-labeled cell membrane fragments (MFs), A/M/C-MNC, and A/M/C-MNC under magnetic field retention in the lymph node (A/M/C-MNC(m)) after vaccination. (B) Mean fluorescence intensity (MFI) statistics at the lymph nodes after vaccination. (C) Retention half-life of different formulations in the lymph nodes. Bars a–c in parts C–E and G were MF, A/M/C-MNC, and A/M/C-MNC(m), respectively. (D) Relative bioavailability statistics of different formulations in the lymph nodes. (E) Fluorescence imaging of lymph node sections 2 days after vaccination with different formulations. The nuclei were counterstained with DAPI (blue), and all formulations were labeled with DiD (red). (F) Fluorescence imaging of excised lymph nodes and visceral organs 2 days after vaccination with A/M/C-MNC(m). I–VI were lung, liver, kidneys, heart, spleen, and lymph nodes, respectively. (G) In vivoT2-weighted MR imaging of mice vaccinated with different formulations. The red circles outline the lymph nodes. (H) Representative fluorescence images of lymph node sections after vaccination with DiD-labeled M/C-MNC and A/M/C-MNC. (I) MFI of DCs in the lymph nodes after vaccination with different DiD-labeled formulations. Ab, anti-CD205 conjugation; m, magnetic field retention in the lymph nodes. (J) Analysis of DC maturation markers CD40 and CD86, MHC-II, and cross-presentation marker MHC-I in the lymph nodes after vaccination with different formulations. All data represent the mean ± SD (n = 3).
Although the improved lymph node retention might facilitate DC uptake, we still had to face the hurdle of antigen cross-presentation. Considering the abundance of CD8+ DCs with high CD205 expression and unique cross-presentation ability, we further proposed to utilize anchored anti-CD205 to direct the magnetosomes to CD8+ DCs. To validate this approach, we investigated CD8+ DC targeting and presentation after vaccination with different magnetosome formulations. Immunostaining of lymph node sections revealed only a few M/C-MNCs scattered with CD8+ DCs (indicated in white in the merged image) due to their random distribution in the lymph nodes (Figure 2H). In contrast, significantly higher colocalization was observed between A/M/C-MNC and CD8+ DCs. The good targeting performance of A/M/C-MNC was verified using another quantitative method, wherein anti-CD205 decoration improved the internalization in CD8+ DCs compared to that in CD8– DCs and other immune cells (Figure S7). As this benefit could be further enhanced upon application of a magnetic field, we observed the highest magnetosome uptake by CD8+ DCs in the A/M/C-MNC(m) group (Figure 2I).
Having demonstrated the preferential uptake of A/M/C-MNC by CD8+ DCs, we next sought to evaluate DC maturation and presentation (Figure 2J). As indicators of DC maturation, the costimulators CD40 and CD86 demonstrated similar expression profiles after treatment with different magnetosome formulations. Compared with the phosphatic buffer solution (PBS) group, the MF and MNC with membranes camouflaging (M/MNC) groups showed a very small improvement in both CD8+ DCs and CD8– DCs, which could be ameliorated by loading with CpG-ODN (M/C-MNC). Once anti-CD205 was anchored (A/M/C-MNC) for targeting delivery, the expression of these costimulators by CD8+ DCs significantly increased, while little change was observed for CD8– DCs. Such a difference could be further enhanced under a magnetic field, leading to a much superior maturation of CD8+ DCs in the A/M/C-MNC(m) group. In addition to the costimulators, we determined MHC expression to assess antigen presentation. As expected, MHC I expression in CD8+ DCs was consistently higher than that in CD8– DCs, indicating satisfactory cross-presentation in CD8+ DCs. Because of the promotion effect of anti-CD205 antibodies,33 MHC II and MHC I expression both gradually increased in the order of PBS, MF, M/MNC, M/C-MNC, A/M/C-MNC, and A/M/C-MNC(m). The efficient cross-presentation of cancer-associated antigens could also be found using OVA-expressed cancer cell (EG7) membrane as the cloak (Figure S8). All the results illustrated effective humoral and cellular responses after A/M/C-MNC(m) treatment.
The significantly improved expression of MHCs and costimulators prompted us to evaluate T cell proliferation and activation. As shown in Figure 3A, lymph node growth followed the same order as the MHC expression: PBS, MF, M/MNC, M/C-MNC, A/M/C-MNC, and A/M/C-MNC(m). Specifically, the A/M/C-MNC(m) group showed a ∼12.5-fold increase in the lymph node weight compared with the PBS group. This significant increase in weight was attributed to the notable increase in the proliferation of stimulated T cells, which was verified by immunofluorescence staining of Ki67, a biomarker of cell proliferation (Figure 3B).34 As the cell membranes utilized is composed of many cancer-derived antigens, the proliferating T cells showed great clonal diversity (indicated by complementarity-determining region (CDR) 3 high-throughput sequencing, as shown in Figure 3C and Figure S9). This multiantigenic response could overcome the limitations of immune evasion during tumor evolution.35,36
Figure 3.

CTL proliferation in the lymph nodes in vivo. (A) Weight and size of the lymph nodes after vaccination with different formulations. (B) Immunohistochemical analysis of Ki67 expression. The nuclei were counterstained with DAPI (blue), and the Ki67 was labeled with fluorescent secondary antibody (red). (C) Frequencies of 200 representative CDR3 sequences in the lymph nodes before (left) and after (right) vaccination. (D) CFSE-based flow cytometry of CD8+ T cell proliferation in vivo after different treatments. (E) Normalized expression of intracellular IFN-γ and granzyme B in CD8+ T cells after vaccination with different formulations. (F) CTL proliferation percent 4 weeks after vaccination with different formulations. All data represent the mean ± SD (n = 3).
Considering the superior maturation and presentation of CD8+ DCs, we next considered the CD8+ T cells, which were indispensable for cancer cell elimination. As shown in Figure 3D, different treatments led to very different CD8+ T cell proliferation profiles. As expected, the most intensive proliferation was achieved in the A/M/C-MNC(m) group, with 83.7% of the CD8+ T cells having undergone division by day 5. In contrast, cell division was significantly reduced in the other groups. To further examine CD8+ T cell proliferation, we determined the levels of interferon γ (IFN-γ) and granzyme B (Figure 3E and Figure S10). Again, the A/M/C-MNC(m) group exhibited the highest levels of these two indicators, demonstrating a good transformation to CTLs. Given the distinct lymph node retention times of the different magnetosome formulations, the expansion dynamics of the CTLs also exhibited significant differences (Figure 3F). In the treatments without application of a magnetic field, the expansions returned to baseline within 2 weeks due to magnetosome elimination from the efferent lymphatic vessels. In contrast, the lymph node residence time was remarkably extended in the A/M/C-MNC(m) group through magnetic control. Thus, robust expansion at day 5 and a prolonged duration in the lymph nodes (4 weeks) were observed, which could further enhance the therapeutic effect against cancer.
The encouraging results described above prompted us to systematically evaluate the anticancer performance of the engineered magnetosomes on different 4T1 breast tumor models. In addition to the remarkable cancer prophylactic effects (details in the Supporting Information and Figure S11), we also tested the utility of our vaccine formulation in a more clinically relevant therapeutic setting. The mice in this study were challenged with 4T1 cells at the mammary fat pad and then subsequently immunized with the different magnetosome formulations (Figure 4A). Like the prophylactic results, the tumor growth rates again decreased in the order of PBS, MF, M/MNC, M/C-MNC, A/M/C-MNC, and A/M/C-MNC(m) (Figure 4B). Correspondingly, significantly prolonged survival was achieved in the A/M/C-MNC(m) group (Figure 4C). This superior therapeutic efficacy was also found to correlate with improved CTL infiltration (Figure 4D), which plays an important role in direct tumor lysis via secretion of IFN-γ and granzyme B. Furthermore, we tested the feasibility of combining anti-PD-1 (aP) therapy with the engineered magnetosomes (A/M/C-MNC(m)+aP). With the assistance of this immune checkpoint inhibitor, the aggressive tumor development could be well-controlled, and all the mice remained alive after 80 days (Figure 4C). As metastasis to distant lungs and bone is typically observed in breast cancer patients in the clinic, the antimetastasis performance of the magnetosomes was also evaluated. Metastatic foci in the lung and metastasis-induced erosion in the tibia were found to different extents for most groups, while this was not observed in the mice treated with A/M/C-MNC(m) and A/M/C-MNC(m)+aP (Figure 4 E,F and Figure S12).
Figure 4.

A/M/C-MNC-mediated inhibition of the growth and spontaneous metastasis of 4T1 primary tumors in vivo. (A) Treatment schedule for the therapeutic performance. (B) Individual tumor growth kinetics of primary tumors receiving different treatments. (C) Survival of mice in part B. (D) Immunohistochemical analysis of the intratumoral infiltration of CTLs in primary tumors. (E) Representative photographs of spontaneous lung metastasis nodules. (F) Computed tomography (CT) imaging of spontaneous tibia metastasis.
To mimic a more malignant metastasis process, we established hematogenous metastasis by intravenous inoculation of luciferase-expressing 4T1 (Luc-4T1) cells (Figure 5A). The mice were pretreated with different magnetosome formulations, and then, lung metastasis was detected in vivo via bioluminescence. As shown in Figure 5B, metastasis to the lung was inhibited in the M/C-MNC group as the bioluminescence signal was significantly reduced. When the magnetosomes were decorated with anti-CD205 (A/M/C-MNC) and thereby targeted to the CD8+ DCs, a more potent immune response was observed, leading to a 25.8-fold reduction in the bioluminescence signal compared with the PBS group (Figure 5C). With further magnetic retention at the lymph nodes (A/M/C-MNC(m)), the bioluminescence signal completely disappeared, and no metastatic foci were observed histologically, indicating complete inhibition of lung metastasis (Figure 5D). Finally, the addition of aP allowed us to achieve a 100% survival rate after 80 days, which is an inspiring result given the malignant nature of this metastasis model (Figure 5E).
Figure 5.

A/M/C-MNC-mediated remarkable prophylactic effects in hematogenous metastasis of 4T1 tumor cells. (A) Hematogenous metastasis experimental design. (B) Representative bioluminescence images of hematogenous metastasis in lungs with different pretreatments. (C) MFI statistics of lung metastasis in part B. (D) Survival of mice in part B. (E) H&E-stained lung sections of different groups. All data represent the mean ± SD (n = 6).
The residual tumor cells after surgical excision always result in tumor recurrence, which has been a common problem in the clinic.7,37,38 The results described above gave us hope and prompted us to explore utilizing our vaccine formulation to prevent postsurgical tumor recurrence. To this end, a tumor recurrence model was established by surgically resecting the majority of the primary Luc-4T1 tumor in the mammary fat pad (Figure 6A). Correspondingly, we could observe almost the same intensity of the bioluminescence in each group after the surgical operation (Figure 6B). After immunization with the different magnetosome formulations, we observed distinct tumor recurrence in the various treatment groups (Figure 6B,C). Compared with the PBS group, which demonstrated aggressive recurrence throughout the body, vaccination with A/M/C-MNC inhibited the growth of the recurrent tumor at the primary site, while no metastasis signal was observed elsewhere. A better result could be observed in the A/M/C-MNC(m) group, wherein the bioluminescence signal and tumor size at the primary site showed a more significant decrease. Like the aforementioned data show, treatment with A/M/C-MNC(m)+aP again outperformed the other treatment regimens. Tumor recurrence was completely suppressed, and a 100% survival rate was achieved during a long-term observation period (Figure 6C,D).
Figure 6.

A/M/C-MNC induced significant suppression of the postoperative recurrence and long-term immune memory effects. (A) Treatment schedule. (B) Left: Representative bioluminescence images of the primary tumor before and after tumor resection and the recurrent tumor. Right: Individual tumor growth kinetics of primary tumors and recurrent tumors receiving different treatments. (C) Left: MFI of recurrent tumors in part B. Right: MFI of lung metastasis in part B. (D) Survival percentage of the recurrent tumor-bearing mice receiving different treatments. (E) Therapeutic schedule. (F) Representative bioluminescence imaging of the primary tumors before and after tumor resection and the rechallenged tumors. (G) Flow cytometry plots and the proportions of effector memory T cells in the spleen examined on the same day as rechallenging with 4T1 cells. (H) Average tumor growth curves of the rechallenged distant tumors receiving different treatments. (I) Percentage of the tumor-free mice in part H. All data represent the mean ± SD (n = 6).
Another important feature of cancer vaccine is its durable memory effect, which is critical for a favorable cancer prognosis.39 To explore this, we evaluated the immunological memory induced by our engineered magnetosome formulations against cancer. To establish an appropriate animal model (Figure 6E), we immunized the Luc-4T1 tumor-bearing mice with the different magnetosome formulations, which was then followed by a complete tumor resection (indicated by the disappearance of the tumor signal at the primary site as shown in Figure 6F). Two weeks later, Luc-4T1 tumor cells were inoculated at a distant mammary fat pad, and the effect of immunological memory on the growth of this distant tumor was investigated. Compared to the PBS group, the effector memory T cells in the spleen increased in the order of A/M/C-MNC, A/M/C-MNC(m), and A/M/C-MNC(m)+aP (Figure 6G). Correspondingly, distinct tumor growth inhibition efficiencies were observed at the distant tumor (Figure 6F,H). Compared with the PBS group, the growth of the tumor at the distant mammary fat pad was significantly inhibited with a dramatically reduced bioluminescence signal in the A/M/C-MNC group (Figure 6F). Because of further magnetic retention at the lymph nodes, we could almost eliminate the distant tumor, and only one mouse was found with a very small tumor at day 80 (Figure 6I). When combined with aP, neither bioluminescence nor the tumor was detected at the distant tumor site. As a result, all mice remained tumor-free after 80 days, thus demonstrating an effective immunological memory effect after treatment with the A/M/C-MNC(m)+aP regimen.
In addition to the prophylactic and therapeutic investigations described above, we carried out a series of safety evaluations of the magnetosome-containing formulations. For example, the levels of both tumor necrosis factor α (TNF-α) and interleukin-6 (IL-6) remained similar to those of the PBS group (Figure S13), thus eliminating any concerns that our magnetosome treatment would elicit a cytokine storm. Likewise, biochemical markers in the blood including aspartate aminotransferase (AST), alanine aminotransferase (ALT), blood urea nitrogen (BUN), lactic dehydrogenase (LDH), and alkaline phosphatase (ALP) were all in the normal range (Figure S14), indicating a healthy condition. In addition, few abnormalities were observed in the body weight, temperature, and organ histology (Figures S15 and S16), further confirming the safe use of the A/M/C-MNC(m) formulations.
In summary, we have developed a novel class of magnetosomes using MNC as the core and cancer cell membranes decorated with anti-CD205 as the cloak. This versatile design incorporates recent nanoparticle technology for safe and high-performance anticancer vaccination. The superparamagnetism and magnetization of the MNC core enabled magnetic retention of the magnetosomes in the lymph nodes as detected by MRI, which increased the time window for antigen uptake by DCs. Meanwhile, the camouflaged cancer cell membrane served as a reservoir of various membrane antigens and paved the way for a subsequent multiantigenic responses. In addition, the decorated anti-CD205 directed more magnetosomes into CD8+ DCs, thus facilitating MHC I cross-presentation. With these unique advantages, a large number of T cells proliferated with great clonal diversity and superior cytotoxic activity. Consequently, potent prophylactic and therapeutic effects with few abnormalities were systematically demonstrated on five different tumor models, which could fit various requirements in the clinic.
The above satisfactory results not only showed the great promise of engineered magnetosomes as a new cancer vaccine modality but also opened a new area of research for future studies. Using the current generalizable strategy, magnetosomes can be prepared with various cancer cell membranes, which can then be used to evaluate different cancers in terms of specificity and protection. As magnetosome formulations can work together with other immunotherapies such as CpG-ODN and anti PD-1, their combination with other adjuvants or checkpoint blockades can also be explored to identify an optimal regimen. In addition, simultaneous efforts should be made to use the own excised primary tumor to construct the magnetosome-based vaccine, therefore taking a further step toward personalized treatment in the clinic.
Experimental Section
Reagents and Materials
The magnetic MNC and azide-choline (AECho) were prepared as described previously.29,30,40 DBCO-Alexa Fluor 488 was purchased from ClickChemistryTools (Scottsdale, AZ). Antimouse CD205 monoclonal antibody and antimouse CD44 monoclonal antibody were purchased from Abcam (Shanghai, China). FITC-conjugated antimouse CD11c, APC/Cy7-conjugated antimouse CD3, BV510-conjugated antimouse CD3, eFlour 450-conjugated antimouse CD4, eFlour 450-conjugated antimouse CD8a, PE-conjugated antimouse CD8a, PE-conjugated antimouse CD40, APC-conjugated antimouse CD80, APC/Cy7-conjugated antimouse CD86, PerCP/eFlour 710-conjugated antimouse MHC I, eFlour 450-conjugated antimouse CD44, PerCP Cy5.5-conjugated antimouse CD62L, BV605-conjugated antimouse MHC II, PE-conjugated antimouse IFN-γ, and eFluor 660-conjugated Granzyme B were purchased from eBiosicience (Beijing, China). Advanced RPMI 1640 medium and fetal bovine serum (FBS) were obtained from Gibco (Beijing, China). DiD and DiR dye were purchased from Fanbo Biochemicals (Beijing, China). CFSE was purchased from ThermoFisher (Shanghai, China).
Preparation of Azide-Modified Cell Membrane Fragments
4T1 cells were cultured at 37 °C for 24 h, and then, the medium was replaced with fresh medium containing 0.2 mM AECho and cultured for another 24 h. For verification of the N3 groups on cells, these cells were stained with DBCO-Alexa Fluor 488 and detected by confocal laser scanning microscope (CLSM, Leica). Then, the cells pretreated with AECho were washed with HEPES buffer solution (HBS) and degraded using IKAT18 basic ULTRA-TURRAX (IKA). The resulting cellular membrane fragments were applied to a discontinuous sucrose density gradient consisting of 55% (w/v), 40% (w/v), and 30% (w/v) of sucrose, and then purified by ultracentrifugation. Three bands were clearly observed and were collected and analyzed for the extracellular CD44 membrane biomarker via Western blot analysis. Western blot analysis indicated that most membrane fractions were enriched in band 1, which was selected to coat C-MNC.
Binding of CpG-ODN to MNC
The MNC dispersed in H2O was incubated with CpG-ODN for 1 h at 4 °C and purified by magnetic separation to remove free CpG-ODN. The optimum loading efficiency of CpG-ODN was determined by the infinite M200 microplate spectrophotometer (TECAN). Gel retardation assay was performed on a 1% (w/v) agarose gel containing SUPER Green II (Fanbo Biochemicals) at 80 V for 30 min in TAE buffer. A bioimaging system (MF-Chemi BIS 3.2, DNR) was used for the final imaging.
Coating CpG-ODN and MNC Complex with Cell Membrane Fragments
Cancer cell membrane and C-MNC were coextruded through a porous membrane to form M/C-MNC. Subsequently, M/C-MNC was washed with PBS through magnetic separation. The coated membrane layer was visualized by transmission electron microscope (Zeiss Libra 120 PLUS EFTEM) and CLSM. For TEM imaging, samples were deposited on a copper grid and negatively stained with uranyl acetate before imaging. For CLSM imaging, M/C-MNC were sequentially reacted with anti-CD44 antibody and Alexa Fluor 488-labeled secondary antibody. The colocalization of MNC and the fluorescent secondary antibody could confirm the successful coating of the membrane. Moreover, C-MNC was incubated with unmodified cell membrane (N3–) or azide-modified cell membrane (N3+), followed by incubation with DBCO-Alexa Fluor 488. The successful coating could be confirmed by the colocalization of DBCO-Alexa Fluor 488 and MNC in the N3+ group.
Conjugation of DBCO-Ab to M/C-MNC
Membrane-coated C-MNC (M/C-MNC) was incubated with excessive DBCO-Ab for 1 h at 37 °C. Then, the surplus DBCO-Ab was removed through washing with PBS, and the production was collected by magnetic separation. For an estimation of the saturation of DBCO-Ab conjugation, a series of diluted DBCO-Ab were individually incubated with the same amount of M/C-MNC (50 μg of MNC). After washing with PBS, they were incubated with excessive fluorescence-labeled secondary antibody, and then, the fluorescence intensity was measured by microplate reader (TECAN). The total protein amount on M/C-MNC was measured using a BCA kit.
Magnetic Field Administration
The lymph nodes were pinpointed through anatomy experiments. For all experiments concerning magnetic field application, we fixed a small flake magnet at the draining lymph nodes. First, the lymph node region was depilated. Then, the magnet was fixed at the lymph node through a specific medical double-sided adhesive tapes, which made the magnet fit better to the skin and was beneficial for the magnetic field to focus at the lymph node. The magnet is cylindrical with 5 mm in diameter and 1.5 mm in height, which was suitable for adhesive tape adhesion and magnetic field application. In addition, the magnetic force of the magnet mainly concentrates in a region of 10 mm × 10 mm × 10 mm, which can effectively penetrate the skin and focus to the lymph node with little impact on the other area.
In Vivo Delivery of Nanovaccine to Draining Lymph Nodes
BALB/c mice were individually injected subcutaneously at the tail base with 100 μg of A/M/C-MNC, and then divided into two groups randomly. One group was administrated with magnetic field at the area of inguinal lymph nodes while the other group was not. For the mice of the group with magnetic field administration, a flake magnet with enough magnetic force to enrich A/M/C-MNC was fixed at the closest lymph node through an efficient fixation system. Then, another group injected with 4T1 cell membrane fragments was used as control. MRI analysis was taken using a 7 T Bruker pharmascan animal instrument (Bruker Optics, Tsukuba, Japan). MRI images of each phantom were obtained on a 7 T superconducting magnet with T2-weighted (3000/13.2) spin–echo sequences. For in vivo fluorescence imaging, DiR-labeled A/M/C-MNC and 4T1 cell membrane fragments were used. Imaging analysis was taken at different time intervals using the ex/in vivo imaging system (FX Pro, Kodak) with a 730 nm excitation and a 790 nm filter to collect the fluorescence signal of DiR. For frozen section fluorescence imaging, DiD-labeled A/M/C-MNC and 4T1 cell membrane fragments were used. Two days after the subcutaneous injection, the inguinal lymph nodes of each mouse were collected and frozen in optimum cutting temperature (OCT) tissue compound (Sakura, Tokyo, Japan) on dry ice, and then sectioned into 10 μm slices. After staining with 0.1 μg mL–1 DAPI solution, the slices were imaged by an automatic multispectral imaging system (PerkinElmer).
In Vivo Uptake by CD8+ Dendritic Cells
BALB/c mice were randomly assigned to four groups: A–m–, A–m+, A+m–, and A+m+. Groups A–m– and A–m+ were injected subcutaneously at the tail base with DiD-labeled M/C-MNC, while the other two groups were injected with DiD-labeled A/M/C-MNC. Groups A–m+ and A+m+ were administered a magnetic field at their inguinal lymph nodes, while the other two groups were not. Two days later, the inguinal lymph nodes of each mouse were collected and triturated into single cell suspensions. The cells were stained with FITC-conjugated antimouse CD11c and PE-conjugated antimouse CD8a at 4 °C for 30 min. Then, the cells were dispersed in 500 μL of PBS and analyzed by flow cytometry.
In Vivo Dendritic Cell Maturation
BALB/c mice were randomly assigned into six groups and subcutaneously injected at the tail base with PBS, MF, M/MNC, M/C-MNC, A/M/C-MNC, or A/M/C-MNC, respectively. A magnetic field was administrated at inguinal lymph nodes of the last group (A/M/C-MNC(m)). Two days later, inguinal lymph nodes of each mouse were individually collected and triturated into single cell suspension. Then, the cells were stained with FITC-conjugated antimouse CD11c, eFlour 450-conjugated antimouse CD8a, PE-conjugated antimouse CD40, APC-conjugated antimouse CD80, APC/Cy7-conjugated antimouse CD86, PerCP/eFlour 710-conjugated antimouse MHC I, and BV605-conjugated antimouse MHC II. After being washed, the cells were dispersed in 500 μL of PBS and analyzed by flow cytometry.
In Vivo CTL Proliferation
For frozen section fluorescence imaging, lymph node segments collected from the mice vaccinated with PBS, MF, M/MNC, M/C-MNC, A/M/C-MNC, or A/M/C-MNC(m) were prepared as described above. Cells with high proliferation activity were successively labeled with antimouse Ki67 antibody followed by a fluorescence-labeled secondary antibody and were then analyzed via an automatic multispectral imaging system (PerkinElmer). For carboxyfluorescein-succinimidyl-amino-ester-based (CFSE-based) flow cytometry analysis, T cells from spleens and lymph nodes of PBS-, MF-, M/MNC-, M/C-MNC-, A/M/C-MNC-, or A/M/C-MNC(m)-treated groups were labeled with CFSE and then intravenously injected into the mice that had been vaccinated with the corresponding formulations. Three days after the injection, the inguinal lymph nodes and spleens of each mouse were collected, triturated into single cell suspensions, and then labeled with APC/Cy7-conjugated antimouse CD3 antibody and PE-conjugated antimouse CD8a antibody. After being washed, the cells were dispersed in 500 μL of PBS and analyzed by flow cytometry. Similarly, CD8+ T cell proliferation was detected in mice that had been vaccinated for 3, 5, 7, 14, and 21 days. For the determination of the activity of cytotoxic T lymphocytes (CTLs), the T cells from the spleens and lymph nodes of the PBS, MF, M/MNC, M/C-MNC, A/M/C-MNC, and A/M/C-MNC(m) groups were fixed, permeabilized, and then labeled with APC/Cy7-conjugated antimouse CD3 antibody, eFluor 450-conjugated antimouse CD8a antibody, PE-conjugated antimouse IFN-γ antibody, and eFluor 660-conjugated Granzyme B antibody. The expression of IFN-γ and Granzyme B by CD8+ T cells was detected by flow cytometry.
CDR3 High-Throughput Sequencing
RNA extraction of lymph node resided cells was performed following RNeasy Plus mini kit (Qiagen). Samples were analyzed by high-throughput sequencing of TCR using the ImmuHubTM TCR profiling system at the deep level (ImmuQuad Biotech, Hangzhou, China). Briefly, a 5′ RACE unbiased amplification protocol was used, and the sequencing was performed on an Illumina HiSeq X10 system with PE150 mode (Illumina). A postsequencing algorithm was applied to raw sequencing data for PCR and sequencing errors correction and V, D, J, C gene segments mapping with IMGT. The resulting nucleotide and amino acid sequences of CDR3 of TCRβ were determined, and those with out-of-frame and stop codon sequences were removed from the identified TCRβ repertoire. We further defined the amounts of each TCRβ clonotype by adding numbers of TCRβ clones sharing the same nucleotide sequence of CDR3.
Statistical Analysis
All the data were presented as mean ± SD. Unpaired student’s t test (two-tailed) was used for comparison between two groups. Statistical significance was set at **p < 0.05, ***p < 0.01.
Safety statement
No unexpected or unusually high safety hazards were encountered in this line of research.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.9b00060.
Additional experimental section; supplementary text about prophylactic effects; additional experimental figures including vaccine construction, vaccine retention in different lymph nodes with or without magnetic field application, study of time-dependent magnetic field application, uptake by immune cells, antigen cross-presentation, CDR3 repertoire, cytokine secretion, prophylactic effect, lung and tibia metastasis, and safe estimation (PDF)
Author Contributions
W.W. and H.-Y.X. designed and supervised the whole project. F.L., W.N., F.Z., G.L., C.L., Y.L., W.B., L.Z., S.W., and X.G. performed the research and experiments. F.L., W.W., and H.-Y.X. analyzed the data and wrote the manuscript. All the authors revised the manuscript and approved the submission.
The work was supported by the National Natural Science Foundation of China (81571813, 21874011, 21622608 and 21821005), National Key R&D Program of China (2017YFA0207900), and Strategic Priority Research Program of the Chinese Academy of Sciences (XDB29040303).
The authors declare no competing financial interest.
Supplementary Material
References
- Dantzer R.; Meagher M. W.; Cleeland C. S. Translational approaches to treatment-induced symptoms in cancer patients. Nat. Rev. Clin. Oncol. 2012, 9 (7), 414–426. 10.1038/nrclinonc.2012.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleeland C. S.; Allen J. D.; Roberts S. A.; Brell J. M.; Giralt S. A.; Khakoo A. Y.; Kirch R. A.; Kwitkowski V. E.; Liao Z.; Skillings J. Reducing the toxicity of cancer therapy: recognizing needs, taking action. Nat. Rev. Clin. Oncol. 2012, 9 (8), 471–478. 10.1038/nrclinonc.2012.99. [DOI] [PubMed] [Google Scholar]
- Blumenfeld C. M.; Schulz M. D.; Aboian M. S.; Wilson M. W.; Moore T.; Hetts S. W.; Grubbs R. H. Drug capture materials based on genomic DNA-functionalized magnetic nanoparticles. Nat. Commun. 2018, 9 (1), 2870. 10.1038/s41467-018-05305-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Mooney D. J. Biomaterial-assisted targeted modulation of immune cells in cancer treatment. Nat. Mater. 2018, 17 (8), 761–772. 10.1038/s41563-018-0147-9. [DOI] [PubMed] [Google Scholar]
- Yang R.; Xu J.; Xu L.; Sun X.; Chen Q.; Zhao Y.; Peng R.; Liu Z. Cancer cell membrane-coated adjuvant nanoparticles with mannose modification for effective anticancer vaccination. ACS Nano 2018, 12 (6), 5121–5129. 10.1021/acsnano.7b09041. [DOI] [PubMed] [Google Scholar]
- Lollini P.-L.; Cavallo F.; Nanni P.; Forni G. Vaccines for tumour prevention. Nat. Rev. Cancer 2006, 6 (3), 204–216. 10.1038/nrc1815. [DOI] [PubMed] [Google Scholar]
- Wang T.; Wang D.; Yu H.; Feng B.; Zhou F.; Zhang H.; Zhou L.; Jiao S.; Li Y. A cancer vaccine-mediated postoperative immunotherapy for recurrent and metastatic tumors. Nat. Commun. 2018, 9 (1), 1532. 10.1038/s41467-018-03915-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuai R.; Ochyl L. J.; Bahjat K. S.; Schwendeman A.; Moon J. J. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 2017, 16 (4), 489–496. 10.1038/nmat4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burg S. H.; Arens R.; Ossendorp F.; van Hall T.; Melief C. J. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16 (4), 219–233. 10.1038/nrc.2016.16. [DOI] [PubMed] [Google Scholar]
- Coffman R. L.; Sher A.; Seder R. A. Vaccine adjuvants: putting innate immunity to work. Immunity 2010, 33 (4), 492–503. 10.1016/j.immuni.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugovillarino G.; Ito S.; Klinman D. M.; Glimcher L. H. The adjuvant activity of CpG DNA requires T-bet expression in dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (37), 13248–13253. 10.1073/pnas.0506638102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann S.; Heeg K.; Dalpke A. Immunostimulatory DNA as adjuvant: efficacy of phosphodiester CpG oligonucleotides is enhanced by 3′ sequence modifications. Vaccine 2003, 21 (9), 990–995. 10.1016/S0264-410X(02)00550-9. [DOI] [PubMed] [Google Scholar]
- Skountzou I.; Martin M. P.; Wang B.; Ye L.; Koutsonanos D.; Weldon W.; Jacob J.; Compans R. W. Salmonella flagellins are potent adjuvants for intranasally administered whole inactivated influenza vaccine. Vaccine 2010, 28 (24), 4103–4112. 10.1016/j.vaccine.2009.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salman H. H.; Irache J. M.; Gamazo C. Immunoadjuvant capacity of flagellin and mannosamine-coated poly(anhydride) nanoparticles in oral vaccination. Vaccine 2009, 27 (35), 4784–4790. 10.1016/j.vaccine.2009.05.091. [DOI] [PubMed] [Google Scholar]
- Didierlaurent A.; Snelgrove R.; Edwards L.; Williams A.; Sirard J. C.; Hussell T. TLR5 signalling alters subsequent immune responses in the lung. Immunology 2005, 116, 26–26. [Google Scholar]
- Hollingshead S.; Jongerius I.; Exley R. M.; Johnson S.; Lea S. M.; Tang C. M. Structure-based design of chimeric antigens for multivalent protein vaccines. Nat. Commun. 2018, 9 (1), 1051. 10.1038/s41467-018-03146-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouliot K.; Buglione-Corbett R.; Marty-Roix R.; Montminy-Paquette S.; West K.; Wang S.; Lu S.; Lien E. Contribution of TLR4 and MyD88 for adjuvant monophosphoryl lipid A (MPLA) activity in a DNA prime-protein boost HIV-1 vaccine. Vaccine 2014, 32 (39), 5049–5056. 10.1016/j.vaccine.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Liu R.; Zhu N. Enhancement of humoral and cellular immune responses by monophosphoryl lipid A (MPLA) as an adjuvant to the rabies vaccine in BALB/c mice. Immunobiology 2013, 218 (12), 1524–1528. 10.1016/j.imbio.2013.05.006. [DOI] [PubMed] [Google Scholar]
- Moon J. J.; Suh H.; Bershteyn A.; Stephan M. T.; Liu H.; Huang B.; Sohail M.; Luo S.; Um S. H.; Khant H. Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses. Nat. Mater. 2011, 10 (3), 243–251. 10.1038/nmat2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho N. H.; Cheong T. C.; Min J. H.; Wu J. H.; Lee S. J.; Kim D.; Yang J. S.; Kim S.; Kim Y. K.; Seong S. Y. A multifunctional core-shell nanoparticle for dendritic cell-based cancer immunotherapy. Nat. Nanotechnol. 2011, 6 (10), 675–682. 10.1038/nnano.2011.149. [DOI] [PubMed] [Google Scholar]
- Guan J.; Shen Q.; Zhang Z.; Jiang Z.; Yang Y.; Lou M.; Qian J.; Lu W.; Zhan C. Enhanced immunocompatibility of ligand-targeted liposomes by attenuating natural IgM absorption. Nat. Commun. 2018, 9 (1), 2982. 10.1038/s41467-018-05384-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll S.; Delon J.; Brotz T. M.; Germain R. N. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science 2002, 296 (5574), 1873–1876. 10.1126/science.1071065. [DOI] [PubMed] [Google Scholar]
- Gonzalez S. F.; Lukacskornek V.; Kuligowski M. P.; Pitcher L. A.; Degn S. E.; Kim Y.; Cloninger M. J.; Martinezpomares L.; Gordon S.; Turley S. J. Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat. Immunol. 2010, 11 (5), 427–434. 10.1038/ni.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karabin N. B.; Allen S.; Kwon H. K.; Bobbala S.; Firlar E.; Shokuhfar T.; Shull K. R.; Scott E. A. Sustained micellar delivery via inducible transitions in nanostructure morphology. Nat. Commun. 2018, 9 (1), 624. 10.1038/s41467-018-03001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn G. M.; Laga R.; Darrah P. A.; Ishizuka A. S.; Balaci A. J.; Dulcey A. E.; Pechar M.; Pola R.; Gerner M. Y.; Yamamoto A. In vivo characterization of the physicochemical properties of polymer-linked TLR agonists that enhance vaccine immunogenicity. Nat. Biotechnol. 2015, 33 (11), 1201–1210. 10.1038/nbt.3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitani G.; Rinaldi A.; Bertoni F.; Sallusto F.; Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1|[ndash]|polarizing program in dendritic cells. Nat. Immunol. 2005, 6 (8), 769–776. 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine D. J.; Swartz M. A.; Szeto G. L. Engineering synthetic vaccines using cues from natural immunity. Nat. Mater. 2013, 12 (11), 978–990. 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudziak D.; Kamphorst A. O.; Heidkamp G. F.; Buchholz V. R.; Trumpfheller C.; Yamazaki S.; Cheong C.; Liu K.; Lee H. W.; Park C. G. Differential antigen processing by dendritic cell subsets in vivo. Science 2007, 315 (5808), 107–111. 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- Xiong K.; Wei W.; Jin Y.; Wang S.; Zhao D.; Wang S.; Gao X.; Qiao C.; Yue H.; Ma G. Biomimetic immuno-magnetosomes for high-performance enrichment of circulating tumor cells. Adv. Mater. 2016, 28 (36), 7929–7935. 10.1002/adma.201601643. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Wei W.; Wang P.; Zuo L.; Li F.; Xu J.; Xi X.; Gao X.; Ma G.; Xie H. Y. Biomimetic magnetosomes as versatile artificial antigen-presenting cells to potentiate t-cell-based anticancer therapy. ACS Nano 2017, 11 (11), 10724–10732. 10.1021/acsnano.7b04955. [DOI] [PubMed] [Google Scholar]
- Stern J. N.; Keskin D. B.; Kato Z.; Waldner H.; Schallenberg S.; Anderson A.; Von H. B.; Kretschmer K.; Strominger J. L. Promoting tolerance to proteolipid protein-induced experimental autoimmune encephalomyelitis through targeting dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (40), 17280–17285. 10.1073/pnas.1010263107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granellipiperno A.; Pritsker A.; Pack M.; Shimeliovich I.; Arrighi J. F.; Park C. G.; Trumpfheller C.; Piguet V.; Moran T. M.; Steinman R. M. Dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin/CD209 is abundant on macrophages in the normal human lymph node and is not required for dendritic cell stimulation of the mixed leukocyte reaction. J. Immunol. 2005, 175 (7), 4265–4273. 10.4049/jimmunol.175.7.4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkholz K.; Schwenkert M.; Kellner C.; Gross S.; Fey G.; Thurner B. S.; Schuler G.; Schaft N.; Dörrie J. Targeting of DEC-205 on human dendritic cells results in efficient MHC classII–restricted antigen presentation. Blood 2010, 116 (13), 2277–2285. 10.1182/blood-2010-02-268425. [DOI] [PubMed] [Google Scholar]
- Huang A. C.; Postow M. A.; Orlowski R. J.; Mick R.; Bengsch B.; Manne S.; Xu W.; Harmon S.; Giles J. R.; Wenz B. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545 (7652), 60–65. 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A. W.; Sobral M. C.; Badrinath S.; Choi Y.; Graveline A.; Stafford A. G.; Weaver J. C.; Dellacherie M. O.; Shih T. Y.; Ali O. A. A facile approach to enhance antigen response for personalized cancer vaccination. Nat. Mater. 2018, 17 (6), 528–534. 10.1038/s41563-018-0028-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Türeci Ö.; Löwer M.; Schrörs B.; Lang M.; Tadmor A.; Sahin U. Challenges towards the realization of individualized cancer vaccines. Nat. Biomed. Eng. 2018, 2 (8), 566–569. 10.1038/s41551-018-0266-2. [DOI] [PubMed] [Google Scholar]
- Paik S.; Shak S.; Tang G.; Kim C.; Baker J.; Cronin M.; Baehner F. L.; Walker M. G.; Watson D.; Park T. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004, 351 (27), 2817–2826. 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- Baker D. G.; Masterson T. M.; Pace R.; Constable W. C.; Wanebo H. The influence of the surgical wound on local tumor recurrence. Surgery 1989, 106 (3), 525–532. [PubMed] [Google Scholar]
- Finn O. J. The dawn of vaccines for cancer prevention. Nat. Rev. Immunol. 2017, 18 (3), 183–194. 10.1038/nri.2017.140. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Zhao L.; Wang S.; Yang J.; Lu G.; Luo N.; Gao X.; Ma G.; Xie H. Y.; Wei W. Construction of a biomimetic magnetosome and its application as a sirna carrier for high-performance anticancer therapy. Adv. Funct. Mater. 2018, 28 (1), 1703326. 10.1002/adfm.201703326. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.