

Abstract
YcaO enzymes are known to catalyze the ATP-dependent formation of azoline heterocycles, thioamides, and (macro)lactamidines on peptide substrates. These enzymes are found in multiple biosynthetic pathways, including those for several different classes of ribosomally synthesized and post-translationally modified peptides (RiPPs). However, there are major knowledge gaps in the mechanistic and structural underpinnings that govern each of the known YcaO-mediated modifications. Here, we present the first structure of any YcaO enzyme bound to its peptide substrate in the active site, specifically that from Methanocaldococcus jannaschii which is involved in the thioamidation of the α-subunit of methyl-coenzyme M reductase (McrA). The structural data are leveraged to identify and test the residues involved in substrate binding and catalysis by site-directed mutagenesis. We also show that thioamide-forming YcaOs can carry out the cyclodehydration of a related peptide substrate, which underscores the mechanistic conservation across the YcaO family and allows for the extrapolation of mechanistic details to azoline-forming YcaOs involved in RiPP biosynthesis. A bioinformatic survey of all YcaOs highlights the diverse sequence space in azoline-forming YcaOs and suggests their early divergence from a common ancestor. The data presented within provide a detailed molecular framework for understanding this family of enzymes, which reconcile several decades of prior data on RiPP cyclodehydratases. These studies also provide the foundational knowledge to impact our mechanistic understanding of additional RiPP biosynthetic classes.
Short abstract
YcaO domain enzymes modify peptide backbones. The first structure of a YcaO bound to a peptide substrate, and associated biochemical studies demonstrate the mechanistic conservation across all YcaOs.
Introduction
Proteins that contain the YcaO domain (domain of unknown function DUF181, protein family PF02624, TIGR families TIGR00702 and TIGR03549, and InterPro families IPR003776 and IPR019938) are prevalent in bacteria and archaea, and over 17 000 sequences containing this domain have been annotated in GenBank (as of mid-2018).1 The most widely studied YcaO-domain-containing proteins are those that carry out post-translational modification on protein or peptide substrates, although the founding member of this superfamily, the ycaO gene product from Escherichia coli, has yet to be functionally characterized.2 The function of YcaO domains is currently best understood in the context of biosynthesis of various classes of ribosomally synthesized and post-translationally modified peptides (RiPPs),3 including the linear azol(in)e-containing peptides (LAPs),4 thiopeptides,5 and a subset of cyanobactins.6 The prototypical member of the extended YcaO family is one of two catalytic components of the tripartite microcin B17 synthetase, which specifically carries out the ATP-dependent installation of azoline heterocycles on the backbone of a precursor peptide.7 The biosynthetic gene cluster (BGC) for microcin B17, a DNA gyrase-inhibiting LAP, was originally identified on a plasmid that encodes for a 69-residue precursor peptide, three genes for peptide backbone heterocyclization, as well as additional genes for self-immunity and cellular export.8
Initial in vitro reconstitution studies of microcin B17 demonstrated that the trimeric heterocycle synthetase consisted of a YcaO protein, a member of the E1 ubiquitin-activating (E1-like) family of enzymes, and a flavin-dependent dehydrogenase.7,9 Subsequent studies established that the YcaO and the E1-like protein work in concert to carry out ATP-dependent cyclodehydration.10 Specifically, the YcaO facilitates the attack of the side chain from Cys/Ser/Thr onto the preceding amide bond, and carries out ATP-dependent O-phosphorylation, amide nitrogen deprotonation, and phosphate elimination to yield the azoline heterocycle. The E1-like protein potentiates cyclodehydration activity by binding to the leader sequence of the precursor peptide through a RiPP precursor peptide recognition element (RRE).11 The crystal structure of the E. coli YcaO (EcYcaO) in complex with ATP, along with structure- and sequence-guided mutational studies on the RiPP cyclodehydratase (BalhD) from Bacillus sp. Al Hakam, firmly established the functionality of the YcaO as the catalytic component of the cyclodehydratase.2 More recent studies on the structure of a fused E1-like/YcaO from a cyanobactin biosynthetic cluster showed that binding of the leader sequence by the E1-like domain stabilizes active site loops of the YcaO, and accelerates the rate of azoline installation.12
YcaO domains are known to catalyze peptide backbone modifications other than cyclodehydration, which include installation of lactamidine rings13 and thioamides.14 Two RiPP classes exhibit characteristic lactamidines installed by YcaO enzymes, the bottromycins15,16 and the klebsazolicins (Figure 1a).17 Both of these natural products contain azoline modifications as well as lactamidine rings, and in vitro studies from each biosynthetic pathway demonstrate notable differences in their constituent biosynthetic strategies. For instance, the precursor peptides for the bottromycins contain a follower sequence, as opposed to a leader sequence. Additionally, the YcaO enzymes do not utilize an RRE, while those for the klebsazolicins contain a leader sequence that is necessary for the RRE-dependent post-translational modifications. Lastly, and perhaps most notably, two distinct, stand-alone YcaO enzymes are required for azoline and macrolactamidine formation during bottromycin biosynthesis,15,16 while a single microcin B17 synthetase-like heterotrimeric complex catalyzes the installation of both the azoline and lactamidine modifications found in klebsazolicin.17
Figure 1.

Comparison of post-translational modification installed on peptide substrates by YcaO enzymes and representative natural products. (A) Example modifications include cyclodehydration of peptide substrates with Cys or Ser to form thiazoles (blue) or oxazoles (green), respectively, thioamidation (red) of the backbone amide, and installation of macrolactamidine (pink). (B) Each of the YcaO-mediated transformations occurs through the formation of a common O-phosphorylated hemiorthoamide intermediate. Installation of azoline moieties necessitates an intramolecular attack from an adjacent β-nucleophilic side chain whereas thioamidation utilizes an external sulfide.
The potential involvement of a YcaO in thioamide formation was first proposed for the thioviridamide biosynthetic pathway (Figure 1a), wherein the mature RiPP contains multiple peptide backbone thioamides.18,19 Further, a conserved thioglycine is present in the α-subunit of all methyl-coenzyme M reductase (McrA) proteins analyzed to date, which was proposed to be installed in a manner similar to that for thioviridamide.20 Subsequently, phylogenetic analysis demonstrated that all methanogens contain at least one YcaO, nearly all of which are encoded adjacently to a gene annotated as encoding a “TfuA-like” protein (PF07812, IPR012924).21 Mass spectral analysis of McrA from cell lysates of Methanosarcina acetivorans bearing genetic deletions of ycaO, tfuA, or both genes demonstrated the essentiality of the YcaO–TfuA pair in thioamide installation.14 More recently, we have demonstrated the ATP-dependent in vitro reconstitution of thioamidation on a synthetic peptide corresponding to the McrA sequence flanking the site of thioglycine modification, using purified YcaO and TfuA proteins from M. acetivorans and either a chemical or enzymatic source of sulfide.22 Although less represented in GenBank, there are a few methanogen genomes that lack a discernible tfuA, including Methanopyrus kandleri and Methanocaldococcus jannaschii. We have demonstrated that the YcaO proteins from these two hyperthermophiles can catalyze thioamidation on the cognate McrA peptide in a TfuA-independent fashion.22
While the various post-translational modifications catalyzed by YcaOs result in different chemical outcomes, the reactions are believed to share a common mechanism (Figure 1b). Specifically, YcaOs catalyze the attack of a nucleophilic species at the amide carbon to form a (presumptive) hemiorthoamide intermediate. The identity of the nucleophile varies depending on the modification; in azoline formation, the nucleophile is the side chain of an adjacent Cys/Ser/Thr residue. For (macro)lactamidine formation, the nucleophile is either the N-terminus of the peptide or theoretically any other amine nucleophile present in the peptide. The nucleophile for thioamidation derives from a yet unidentified exogenous sulfur source, which may vary depending on whether or not the organism encodes a TfuA protein.
While discovery efforts have been seeking new roles for YcaO proteins beyond the examples highlighted here, structure–function studies of the canonical backbone modification reaction have yet to be fully elucidated. Structural studies of the cryptic EcYcaO2 and the cyanobactin synthetase LynD12 afforded insights into how this new protein fold engages the nucleotide, and how a RiPP cyclodehydratase binds to the leader sequence, respectively. However, there are currently no structural data for any YcaO with its substrate peptide bound at the active site. More generally, the lack of core peptide-bound structure for nearly any RiPP biosynthetic enzyme likely reflects the underlying promiscuity and low substrate affinity of the cognate catalytic domains. Here, we present the crystal structures of the thioamide-forming YcaO from M. jannaschii (MjYcaO) along with the cocrystal structure with the cognate McrA-derived peptide (MjMcrA) containing the site of thioglycine modification. Structure-based mutational analysis affords a detailed description of residues involved in both peptide binding and catalysis. Notably, we show that MjYcaO and MkYcaO are competent McrA peptide cyclodehydratases for substrate variants containing a Cys residue adjacent to the modification site, supporting a common mechanism among different classes of modifying enzymes. Structure-based modeling studies yielded insights into the mechanistic difference among the YcaOs involved in the biosynthesis of LAPs, azoline-containing cyanobactins, thiopeptides, and thioamide-containing polypeptides such as thioviridamide and McrA. The data presented within provide an improved molecular context for interpreting decades of prior biochemical observations.
Results
Biochemical and Biophysical Studies of Thioglycine Formation
Difficulties with the production and stability of the full-length MCR alpha subunit that lacked the thioamide modification precluded using the protein for biochemical studies. We previously demonstrated using an end-point mass spectrometry assay that recombinantly expressed and purified M. acetivorans YcaO (MaYcaO) and TfuA proteins can carry out peptide backbone thioamidation. Those studies employed a synthetic 13-mer peptide derived from the α-subunit of the MCR (McrA), bearing five N- and C-terminal residues that flank the Gly465 site of modification as well as a double Gly linker (GG–460RLGFFGFDLQD470, Figure 2a).22 Moreover, recombinant M. jannaschii YcaO (MjYcaO) carried out the thioamidation of a similar peptide derived from MjMcrA with the natural site of modification at Gly448 (GG–443RLGFYGYDLQD453, Figure 2a) in a TfuA-independent manner. For simplicity, we hereafter refer to the thioamidation site as position 0 with the N- and C-terminal flanking positions given negative and positive integer values, respectively. For both the MaMcrA- and MjMcrA-derived peptides, thioamidation was dependent on ATP and sulfide. To characterize the thioamidation kinetics, we employed a high-performance-liquid-chromatography-based (HPLC-based) assay to follow the initial rate of product formation as a function of substrate concentration. The second-order rate constant (kcat/KM) for MjYcaO was 8.9 × 102 M–1 s–1 with a KM of 59.9 μM and kcat of 5.3 × 10–2 s–1 for the 11-mer peptide substrate (Figure 2b). Notably, the measured catalytic efficiency was an order of magnitude lower than that measured by monitoring phosphate production,22 which may partially arise because of the technical issues with the assay conditions, unproductive attack of water instead of sulfide, and/or the ATPase activity from MjYcaO in the absence of peptide substrate (SI, Figure S1).
Figure 2.
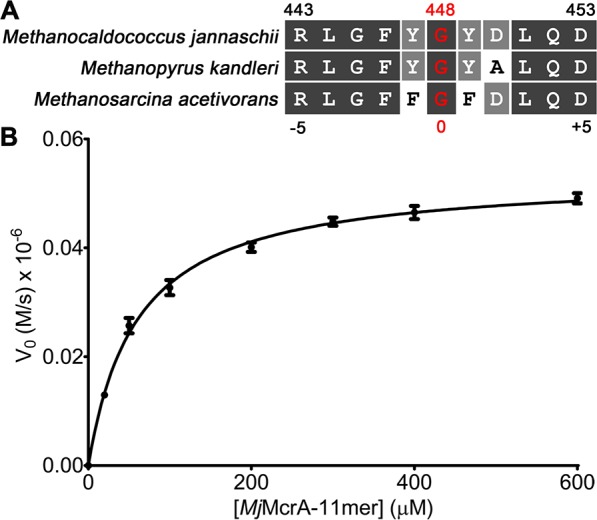
Sequence alignment of McrA peptides and HPLC-based kinetic analysis of the MjYcaO thioamidation reaction. (A) Alignment of McrA peptide sequences (11-mer) flanking the site of thioamidation, including sequences of Mj, Mk, and MaMcrA with the MjMcrA numbering code labeled on both the top and bottom. (B) HPLC-based kinetic analysis on the production of thioamidated MjMcrA 11-mer using varied concentrations of MjMcrA 11-mer peptide substrate.
As previously reported, MjYcaO binds to a fluorescein-isothiocyanate-labeled (FITC-labeled) 13-mer peptide (GG–RLGFYGYDLQD), derived from MjMcrA, with a KD of 1.1 μM as determined by fluorescence polarization (FP).22 We also observed binding toward the same FITC-labeled peptide that lacked the double Gly linker (443RLGFYGYDLQD453). Using the 13-mer peptide, we established a competitive binding assay with the competitor ligands being supplied as recombinant peptides fused the C-terminus of maltose-binding protein (MBP) (SI, Figure S2). The wild-type McrA peptide gave a binding constant (Ki) of ∼1.3 μM. Systematic Ala-substitution was conducted on the recombinant MBP-tagged peptide to reveal residues governing MjYcaO binding. The competitive assay identified the region from Leu(−4)–Leu(+3) to be important (SI, Figure S3 and Table S1) which was largely consistent with end-point enzymatic activity assays monitored by matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry (MALDI-TOF-MS).
Crystal Structures of MjYcaO–ATP and MjYcaO–ATP–Peptide Complexes
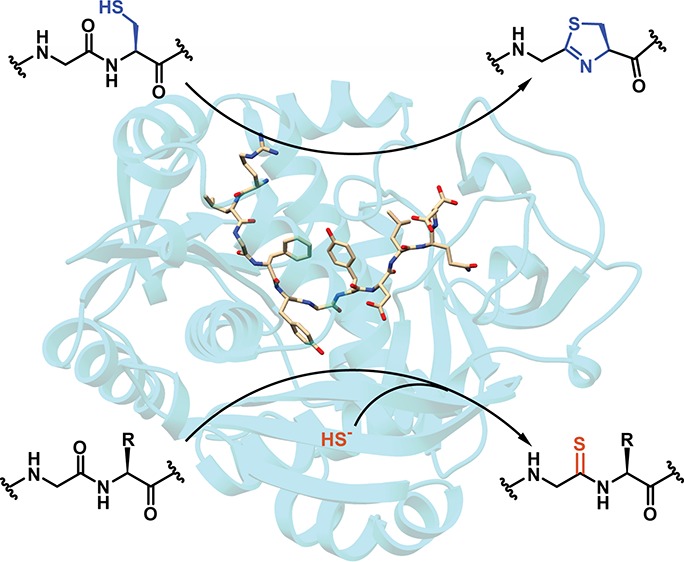
We next sought to obtain structural data for MjYcaO in the presence and absence of the 11-mer MjMcrA peptide (SI, Table S2). Crystals of MjYcaO–ATP diffracted to 2.3 Å resolution and the resultant structure recapitulates the overall architecture observed previously in the structure of M. kandleri YcaO (MkYcaO) [Protein Data Bank (PDB) code 6CI7] (Figure 3).22 As predicted by previous sequence comparisons,2 the active site features of MjYcaO are essentially identical to other YcaO enzymes, such as TruD (PDB code 4BS9), EcYcaO (PDB code 4Q86), and MkYcaO and include a nearly invariant constellation of residues that are involved in interactions with the bound nucleotide. Notably, residues spanning Gly276–Lys290, which are located within a segment between helices α9 and α11, are disordered in the structure. The equivalent region is similarly disordered in all eight crystallographically independent copies in the structure of MkYcaO,22 highlighting the mobility of this region as a common feature of the methanogenic YcaO enzymes associated with MCR modification. Our prior postulation that this region undergoes an ordering upon binding the substrate is borne out by our cocrystal studies described below.
Figure 3.

Crystal structures of MjYcaO in the presence and absence of the MjMcrA 11-mer peptide substrate. (A) Overall structure of MjYcaO bound to the MjMcrA peptide, ATP, and Mg2+. Regions of the polypeptide that become ordered upon substrate binding are colored in blue, and the peptide is shown in yellow sticks. (B) Superposition of the structures of MjYcaO in the absence (in gray) and presence (pink/blue/green) of the peptide substrate showing the ordering and movement that occur upon peptide binding. (C) Simulated annealing difference Fourier map (1.95 Å resolution) calculated with coefficients |Fobs| – |Fcalc| and contoured at 2.5σ above background, with the coordinates of the peptide omitted prior to refinement. The site of thioamidation is indicated with a black arrow. (D) Surface representation of the active site showing the binding pockets that engage the peptide substrate. Pocket 1 is colored in blue, pocket 2 in red, and pocket 3 in purple.
While numerous attempts to obtain crystals of MjYcaO in complex with the 13-mer MjMcrA peptide (GG–RLGFYGYDLQD) described previously were unsuccessful,22 equilibration of the enzyme with ATP and the aforementioned 11-mer peptide (RLGFYGYDLQD) yielded crystals that diffracted to 1.95 Å resolution (Figure 3a). Following the determination of crystallographic phases using the coordinates of MjYcaO without the substrate peptide, inspection of the resultant electron density map revealed significant conformational changes in the vicinity of the ATP-binding site. Specifically, the loops encompassing residues Lys4–Tyr10 (preceding helix α1), Asp236–Leu245 (between strands β10 and β11), strands β1 and β2 (between Leu50 and Ala63), and helix α8 (consisting of residues Pro292–Lys302) all move inward toward the nucleotide. Lastly, residues Gly276–Lys290, which are disordered in the structures of both MjYcaO and MkYcaO in the absence of the peptide, form a well-ordered α-helix at the upper edge of the active site cleft (Figure 3b). These combined secondary structural movements and ordering result in the formation of a well-defined cradle adjacent to the ATP. Clear and unambiguous density corresponding to the bound McrA peptide is observed within this cavity.
The MjMcrA peptide binds in a U-shaped manner across the surface of the cradle formed at the MjYcaO-binding site, with the position corresponding to the site of modification [McrA–Gly(0)] situated at the base (Figure 3c). The bound nucleotide engages two Mg2+ ions, as previously observed in the structures of the YcaO proteins from E. coli and Lyngbya aestuarii (LynD, PDB code 4V1T). Although Gly(0) is positioned proximal to ATP–Mg2+ with the γ-phosphate 5.7 Å away from the carbonyl oxygen, prior studies indicated that the position of the reactive γ-phosphate varies among different structures of LynD,12 consistent with its role in catalysis. Likewise, the position of the γ-phosphate varies in structures of MjYcaO with and without bound peptide. The orientation of the Gly(0) carbonyl is fixed by interactions of the preceding Phe(−2)/Tyr(−1), and the following Tyr(+1), which would position the sulfur nucleophile for attack on the si face of the carbonyl carbon, followed by attack of the hemiorthoamide intermediate to the γ-phosphate of ATP. This observation is corroborated by the reactivity deficiency on McrA peptide variants with Phe(−2), Tyr(−1), or Tyr(+1) substituted with Ala (SI, Table S1). The α-helix that becomes ordered upon binding of the McrA peptide is positioned along the flanking side of the peptide and may help to eliminate solvent from the active site.
Residues Involved in Peptide Binding
Numerous interactions between MjYcaO and the MjMcrA peptide define the specificity of the interaction, most notably a series of hydrophobic packing contacts that position the substrate at the active site (Figures 3d and 4a). Specifically, Leu(−4) of the peptide is positioned in a pocket (pocket 1) formed by MjYcaO residues Ile234, Leu245, Val247, Leu298, His300, Trp303, and Phe304. Notably, among sequenced methanogens (197 complete genomes in GenBank as of mid-2018), the Trp303–Phe304 pair is conserved in 189 (96%) of the YcaO sequences. Methanogens that contain the Trp303–Phe304 YcaO motif display either Leu or Met at the (−4) position in the cognate McrA sequences (SI, Figure S4). For the remaining genomes (n = 8), the equivalent residue in McrA is Phe, and the cognate YcaOs compensate for this larger residue by displaying a smaller Tyr–Leu pair in place of Trp–Phe. A second hydrophobic pocket (pocket 2) defines the binding site for Tyr(−1), and consists of MjYcaO residues Tyr66 and Gln268, as well as Glu82 that is within hydrogen bonding distance. Lastly, Tyr(+1) from the MjMcrA peptide is docked into a third hydrophobic pocket (pocket 3) created by MjYcaO residues Trp180, Thr366, and Ile367, as well as Phe(−2) from the MjMcrA peptide. On the C-terminal portion of the MjMcrA peptide, notable interactions include a salt bridge formed between Asp(+2) in MjMcrA and Arg278 of MjYcaO, as well as a hydrogen bonding interaction between the amide of Asp(+2) and Thr154 of MjYcaO.
Figure 4.
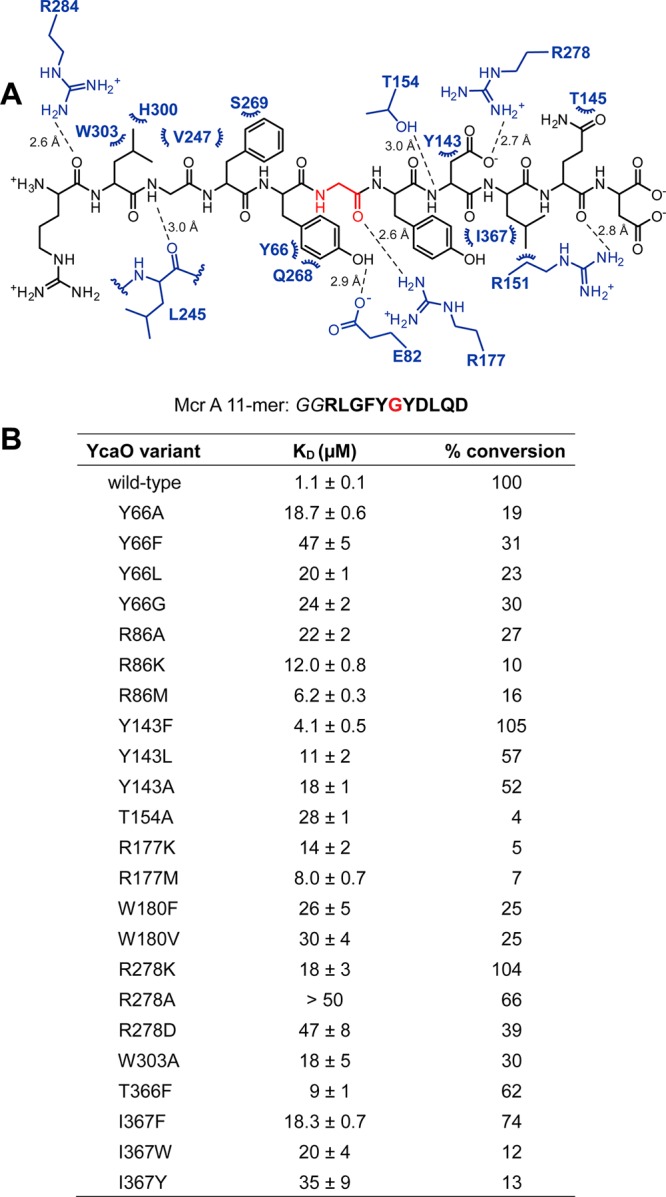
Residues important for the interactions between MjYcaO and MjMcrA 11-mer. (A) The coordinates obtained for the MjYcaO–MjMcrA 11-mer complex were processed by LigPlot Plus35 (Dimplot function) to extract the putative interactions between the protein and peptide with the default runtime parameters. Residues from the MjYcaO are in blue, and those from the MjMcrA 11-mer peptide are in black with the modified Gly highlighted in red. Hydrogen bonds are denoted by dashed lines (distances indicated next to the lines), and hydrophobic contacts are indicated by arcs. Stereochemistry is omitted for clarity. (B) Summary of the binding constants and conversion of the MjMcrA 11-mer substrate by MjYcaO variants. The synthetic peptide derived from MjMcrA [Arg(−5)–Asp(+5)] is shown with the residue naturally thioamidated Gly(0) in red. Binding constants were determined by fluorescence polarization assay with the FITC-labeled MjMcrA 11-mer peptide with Gly–Gly as a linker. Error is represented as SEM (n = 3). Conversion was measured by HPLC (n = 2) and normalized to wild-type MjYcaO.
To elucidate the relevance of the crystallographically observed interactions, we carried out a mutational analysis of MjYcaO and measured the binding affinities of the variants using the aforementioned FP-based assay (Figure 4b and SI, Figures S2 and S5). Ala-substitution at individual residues within pocket 1 (W303A), pocket 2 (W180F/V), or pocket 3 (Y66A/F/L/G) of MjYcaO significantly increased the KD to the wild-type MjMcrA peptide (18–25-fold, Figure 4b, SI, Figure S5). Decreasing the size of the binding pockets in MjYcaO via the T366F or I367F/Y/W variants in pocket 2 similarly resulted in KD increases (7–35-fold, respectively). Notably, Ile367 contacts another key residue, MjMcrA–Leu(+3), which contributes to the larger KD increase for the I367F/Y/W variants. Combined, variants generated at these positions define the importance of the interactions with each of the three pockets for binding of the MjMcrA peptide substrate. The observed interactions are also supported by alterations conducted on the MjMcrA peptide: Leu(−4) and Gly(−3) are held by pocket 1, and Ala-substitution resulted in at least 6-fold increases in Ki. Likewise, the MjMcrA–F(−2)A variant resulted in a 3-fold increase in Ki (SI, Table S1).
The importance of the salt bridge formed between MjMcrA–Asp(+2) and MjYcaO–Arg278 was evaluated by the MjYcaO–R278 K variant with an 18-fold increase in KD. More deleterious effects (nearly 50-fold increases in KD) were observed upon removal of the charge (R278A) or reversing the polarity (R278D) (Figure 4b, SI, Figure S5). Notably, Arg278 is located in the α-helix spanning Gly276–Lys290, which is disordered in the structure of MjYcaO and MkYcaO in the absence of bound peptide.22 Similarly, the MjMcrA–D(+2)A variant has a Ki 4-fold higher than wild-type. Hence, the interactions between MjYcaO–Arg278 and MjMcrA–Asp(+2) likely play a crucial role in binding specificity. Other additional interaction with MjMcrA–Asp(+2) occurs with Thr154 and Tyr143, and the T154A MjYcaO variant resulted in a 25-fold increase in KD, while the Y143F MjYcaO variant showed a 4-fold increase in KD. Decreasing the size of Tyr143 to Leu or Ala resulted in further increases in the KD up to 18-fold (Figure 4b, SI, Figure S5). These combined data highlight the importance of each of the three pockets, as well as electrostatic interactions in establishing the specificity of MjYcaO for binding of the MjMcrA peptide substrate.
Residues Involved in Catalysis
The availability of the first crystal structure of a YcaO enzyme with a bound core peptide affords an opportunity to identify residues that may be directly involved in catalysis and to test putative functions of these target residues using a mutational approach. The proposed mechanism for thioamidation involves the formation of a tetrahedral intermediate from the attack of an external sulfide on the target amide carbonyl, followed by the attack of the resultant backbone oxyanion onto the γ-phosphate of ATP.22 A general base would be required to deprotonate the thiol group of the tetrahedral intermediate to achieve phosphate elimination, which leads to the final product (Figure 1b). In addition, enzyme residues may also be necessary to enhance the electrophilicity of the amide carbon, to stabilize the presumed oxyanion of the hemiorthoamide and/or the phosphorylated intermediate. In the cocrystal structure of MjYcaO, Arg177 is poised to stabilize the oxyanion and enhance the electrophilicity at the carbon. Consequently, the R177 K/M/A variants had a moderate effect on substrate binding (increases in KD of ∼10-fold, Figure 4b, SI, Figure S5)22 but significantly reduced activity (<10% of wild-type). Likewise, Arg86 likely plays a role in stabilization of the phosphorylated tetrahedral intermediate, while Thr154 is poised to aid phosphate elimination. Substitutions at these positions have a larger effect on peptide binding but again show minimal activity (R86A/K/M had <30% of the wild-type activity while T154A exhibited only 4%). In addition, residues Arg86, Arg177, and Thr/Ser154 are highly conserved (SI, Figure S6), suggesting that these residues may play the same role across all YcaOs. Notably, biochemical and crystallographic studies of an unrelated class of RiPP biosynthetic enzyme, the LanMs, which are involved in the biosynthesis of class II lanthipeptides, showed that they similarly utilize a Thr/Arg pair for phosphate elimination to yield an α,β-unsaturated product,23,24 although proceeding through a β-elimination mechanism.25
Mechanistic Conservation across YcaOs for Thioamidation and Cyclodehydration
Structure-based multiple sequence alignments of YcaOs, including members that catalyze the formation of thioamides, azoline heterocycles, and macrolactamidines, reveal that many of the active site residues identified in MjYcaO are highly conserved across these divergent classes (SI, Figure S6 and Table S3). On the basis of this conservation, the chemical outcome from different classes of YcaOs may largely be due to the identity of the proximal nucleophile that attacks the amide carbonyl carbon. Therefore, we tested the ability of MjYcaO to catalyze cyclodehydration of a peptide in which the residue following Gly(0) is replaced with a Cys [RLGFYGCDLQD, Y(+1)C 11-mer]. Treatment of this peptide with wild-type MjYcaO and ATP at pH 7.5 yielded almost no change (SI, Figure S7). Since the Y(+1)C 11-mer still effectively binds to the MjYcaO (Ki is 2-fold higher than that of wild-type, SI, Figure S8), we reasoned that the lack of a catalytic general base to deprotonate the Cys thiol may account for the absence of cyclodehydration activity. To that end, we tested activity at pH 7.5 using either 5 mM phosphate (H2Na2PO4) or phosphite (HNa2PO3) as a surrogate for the missing general base. Reactions carried out on the Y(+1)C 11-mer with MjYcaO, ATP, and either of these anions resulted in a mass decrease consistent with the loss of water (m/z calculated, 1268.5728 Da; observed, 1268.5731 Da; error, 0.25 ppm) while no new peak was observed when MjYcaO was omitted from the reaction (Figure 5a,b). On the basis of our hypotheses that the thioamide-forming YcaO enzyme lacks the catalytic base to deprotonate the Cys thiol, we reasoned that elevated pH would improve its ability to catalyze cyclodehydration. Reactions on the Y(+1)C 11-mer were carried out with MjYcaO and ATP in the absence of these anions but at an elevated pH (9.0) sufficient for deprotonation of the Cys thiol (pKa ∼ 8, SI, Figure S7). Notably, at this pH, ∼50% of the starting material (based on ion intensity) was converted to the dehydrated species. In addition, by supplying the same Y(+1)C 11-mer substrate to MkYcaO we observed enzyme-dependent conversion to the dehydrated species, highlighting the cyclodehydratase activity from another thioamide-forming methanogenic YcaO enzyme (SI, Figure S7).
Figure 5.

MALDI-TOF-MS analysis of MjMcrA 11-mer peptide thioamidation and MjMcrA–Y(+1)C 11-mer cyclodehydration. The sequences of the McrA 11-mer variants with the site for thioamidation (Gly(0)) in red, and the Cys(+1) replacement in blue. (A) Thioamidation catalyzed by MjYcaO. Top: mass spectrum of the unmodified MjMcrA 11-mer peptide, m/z 1346.6 Da. Bottom: mass spectrum of the MjMcrA 11-mer peptide after reacting with MjYcaO, Na2S, and ATP, showing the thioamidated product, m/z 1362.6 Da. (B) Cyclodehydration catalyzed by MjYcaO. From top to bottom: mass spectrum of the unmodified MjMcrA–Y(+1)C 11-mer peptide; MjMcrA–Y(+1)C 11-mer treated with MjYcaO and ATP in the presence of phosphate; acid hydrolysis of cyclodehydrated MjMcrA–Y(+1)C 11-mer peptide in [18O]-labeled water; control where MjYcaO is omitted. (C) View of the MjYcaO (in cyan) bound to MjMcrA (shown as tan sticks). The site of nucleophilic attack by the sulfur source is shown with a purple arrow. (D) Superposition of the computational structure of PatD (green) and TruD crystal structure (purple) with the MjMcrA 11-mer peptide structure (shown as tan sticks) modeled in the active site. In the peptide, Tyr(+1) has been modified to a Ser for presentation.
The site of dehydration was located to between Gly(0) and Cys(+1) by MS/MS experiment, and no fragmentation was observed at the amide bond connecting them (SI, Figure S9). For a further evaluation of the formation of a thiazoline at this position, the reaction mixture containing the dehydrated species was lyophilized and subsequently reconstituted in [18O]-labeled H2O with 1% (v/v) formic acid. This treatment resulted in the conversion of the dehydrated species to a new product 2 Da heavier than the starting peptide (m/z calculated, 1288.5876 Da; observed, 1288.5896 Da; error, 1.55 ppm; SI, Figure S10). The [18O] is located at the amide bond between Gly(0) and Cys(+1) (SI, Figure S10), consistent with the incorporation of [18O] from solvent into the peptide via the mild acid hydrolysis of a thiazoline, as observed previously for experiments using YcaO cyclodehydratases.10 To confirm that both the thioamidation and cyclodehydration reactions catalyzed by MjYcaO proceed via a phosphorylation mechanism, we carried out HPLC analysis for each reaction using the appropriate substrate. In each case, the consumption of ATP and generation of ADP were observed, consistent with a phosphorylation mechanism for both reactions (SI, Figure S11). These data further suggest that all YcaO enzymes modify their substrates through the same mechanism, and the identity of the nucleophile determines the outcome.
Insights into the Mechanism of Peptide Backbone Cyclodehydration
Although MjYcaO was capable of catalyzing cyclodehydration on an appropriate substrate, the reaction only occurred upon addition of an anion, which may act as a general base surrogate, or at an elevated pH to ensure deprotonation of the Cys thiol to the more reactive thiolate. In addition, MjYcaO was not able to catalyze cyclodehydration on substrates whose (+1) residue was substituted with Ser or Thr despite their wild-type-like binding affinity [Y(+1)S 11-mer Ki is ∼2-fold higher than that of wild-type, SI, Figure S8]. These observations suggest that YcaO cyclodehydratases contain an active site general base that can deprotonate the side chain β-hydroxyl or thiol of Cys/Ser/Thr of the substrate peptide to facilitate intramolecular nucleophilic attack. To identify this active site base, we superimposed the crystal structures of the cyanobactin cyclodehydratases PatD and TruD with the MjYcaO cocrystal structure looking for potential bases in the vicinity of the β-carbon atom of Tyr(+1). While essentially no polar side chains from the cyclodehydratases are found near Tyr(+1) in the MjYcaO structure, the C-terminal carboxylate of PatD is superimposed directly adjacent to Tyr(+1) (Figure 5c,d). This observation suggests a catalytic role for the C-terminus of azoline-forming YcaO enzymes. We had previously demonstrated the importance of this Pro-rich C-terminus for cyclodehydration.2 Intriguingly, among the 1890 bioinformatically identified putative azoline-forming YcaOs (i.e., peptide backbone cyclodehydratases), ∼90% of them contain Pro-rich C-termini, often Pro–X–Pro–X–Pro as the last five residues. Enrichment of Pro is not observed in the C-termini of YcaO enzymes involved in thioamide or macrolactamidine formation (SI, Figures S6 and S12 and the SI data set 1). The C-terminus of MjYcaO does not contain the Pro-rich motif and is not located near the active site.
Phylogenetic Analysis of the YcaO Protein Family
We next sought to analyze sequence relationships and functional motifs across the YcaO superfamily. We therefore retrieved all nonredundant YcaO sequences, which were subsequently compared in a pairwise manner in the form of a sequence similarity network (SSN)26 and in all-by-all manner in the form of a maximum-likelihood tree (SI, Figures S12 and S13). Using the Rapid ORF Description and Evaluation Online (RODEO) bioinformatics tool,27 the sequences were binned based on genomic context into the following three groups: (i) TfuA-associated and methanogenic YcaOs that putatively catalyze thioamidation; (ii) E1- and ocin-ThiF-associated YcaOs that putatively catalyze cyclodehydration; and (iii) bottromycin-associated YcaOs that putatively catalyze macrolactamidation (SI, data set 1). The distribution of these three groups on the SSN indicates that thioamide-forming YcaOs are more similar to each other than to cyclodehydratases. In contrast, the cyclodehydratases are considerably more sequence divergent. Notably, despite the greater sequence diversity among putative cyclodehydratases, the Pro-rich C-termini (defined as two or more Pro residues in the last 10 residues) are present in ∼90% of proteins across all clades, consistent with a potential role in catalysis (SI, Figure S12). Given their broad distribution on the phylogenetic tree, cyclodehydratase activity within the YcaO superfamily likely emerged early from an ancestral YcaO and then was broadly distributed. Emanating from the TfuA-associated (putative thioamide-forming) YcaOs is a distinct clade that includes the YcaO domain fused to a tetratripeptide repeat domain (TPR, PF00515), as well as the clade encompassing the EcYcaO, suggesting that these lineages evolved from a single ancestor that catalyzed thioamide formation (SI, Figure S12). TfuA- and E1-associated YcaOs are present in two domains of life and in a number of phyla, including Actinobacteria, Proteobacteria, Firmicutes, Cyanobacteria, Bacteroidetes, and Euryarchaeota (SI, Figure S13). Notably, there are two clades of YcaO proteins that include archaeal representatives. The first clade represents the methanogenic, thioamide-forming YcaOs while the other clade is E1-associated and thus predicted to be involved in azoline formation. Their distinct locations suggest that archaea that produce ribosomal peptides bearing azoline heterocycles might have acquired the genes from bacteria and that bacteria that produce thioamide-containing polypeptides likely acquired the genes from archaea (SI, Figure S13).
Discussion
The combined structural, biochemical, and bioinformatic studies of MjYcaO provide a molecular framework for understanding the mechanism for backbone activation in peptide thioamidation and provide a context for parsing the functionalities of YcaOs involved in azoline and lactamidine formation. This extrapolation of mechanistic details is further supported by the demonstration that MjYcaO can also carry out cyclodehydration on an appropriate alternative substrate, albeit at reduced efficiency. These data illustrate how suitable alterations within an otherwise conserved active site can elaborate a seemingly divergent array of chemical modifications on peptide substrates.
Prior studies on RiPP biosynthetic systems suggest that leader peptide binding serves not only to enforce substrate proximity but also to allosterically activate the cognate biosynthetic enzymes. For example, the biosynthesis of the lanthipeptide lacticin 481 is supported by the LctM synthetase even when the leader and core sequences of the substrate are provided to the enzyme in trans, and fusion of the leader sequence to the enzyme provides a catalyst capable of processing the core peptide in the absence of the leader sequence.2828 Similarly, supply of the leader peptide in trans, or fusion of a leader sequence to the N-terminus of the YcaO, accelerates azoline installation using only the core sequence to a rate comparable to wild-type enzyme with full-length substrate for cyclodehydratases from the cyanobactin12,29 and LAP classes.18 While there are no available structural data for any RiPP YcaO enzyme in both the leader peptide-free and peptide-bound states, a comparison of the structures of MjYcaO with that of LynD provides insight into the basis for leader sequence-mediated rate enhancement. The structure of LynD in complex with a leader peptide shows inter-subunit interactions with loop residues near the active site. Specifically, hydrophobic packing and electrostatic interactions between the conserved LEEL motif in the leader and a helix that includes residues Leu395–Leu402 of LynD stabilize a proposed “active form” of the cyclodehydratase.12 The equivalent segment in MjYcaO corresponds to a region between Val52–Ala63 that undergoes a conformational movement upon binding to the peptide substrate. The stabilization of the Leu395–Leu402 region in LynD by the leader peptide would also position several polar residues along the trajectory of the substrate peptide-binding site in MjYcaO, and candidate residues in LynD that are poised to interact with the substrate include Thr401, His404, and Ser406 (SI, Figure S14). These observations support the molecular model for how the peptide processing in the cyclodehydratase YcaOs is enhanced upon leader peptide binding. In contrast, MjYcaO is able to carry out thioamidation and cyclodehydration in a leader peptide-independent manner because the corresponding loop region is much shorter and more amenable to conformational movement.
Studies with the BalhD cyclodehydratase demonstrated that extending the C-terminus by addition of a single Gly abolishes cyclodehydratase activity.2 Shortening the YcaO by replacing the C-terminal Pro with a stop codon or shifting this motif via removal of two upstream residues also abolishes activity. These data all support a role for the C-terminus of the cyclodehydratase YcaOs as a general base in catalysis. The abundance of Pro residues in cyclodehydratase YcaO C-termini positions the peptide backbone in an extended conformation, which points the C-terminal carboxylate toward the active site. The constellation of active site residues identified in our studies, which include Arg86, Arg177, and Thr/Ser154, are conserved among YcaO family members, consistent with activation of the backbone carbonyl as a universal feature, regardless of the chemical outcome. Deprotonation of the β-nucleophilic side chain, presumably by the C-terminal carboxylate, is necessary for efficient cyclodehydration.
Among YcaOs involved in cyanobactin biosynthesis, PatD (patellamide pathway) installs both thiazolines and oxazolines, a ∼90% similar YcaO, TruD (trunkamide pathway), chemoselectively catalyzes the formation of thiazolines, leaving Ser and Thr untouched.3030 A structure-based superposition of the MjYcaO–McrA structure onto that of TruD positions that C-terminal PTPMPF sequence in the latter directly adjacent to the β-nucleophilic side chain for deprotonation. The alternating Pro residues serve to keep the C-terminus of TruD in an extended confirmation that cannot adopt any secondary structure. In contrast, PatD contains a PTNIPF motif that lacks the alternating Pro motif, suggesting that difference in chemoselectivity between the enzymes may be due, at least in part, to suboptimal orientation of the C-terminal carboxylate (Figure 5c). The recent structure of the heterometic McbBCD complex involved in the biosynthesis of a LAP shows a similar extended orientation of the C-terminal carboxylate.31 Prior kinetic studies on the cyclodehydratases involved in LAP biosynthesis demonstrated that thiazoline installation occurs faster than oxazoline installation by a factor of roughly 30-fold, for the Balh system,32,32,31 while the difference is nearly 1000-fold for the microcin B17 synthetase.33 Our biochemical and structural studies suggest that the differences between these two systems for the rate of oxazoline formation may be due to requirement of regiospecificity (the Balh study utilized an artificial substrate), and/or chemoselectivity, namely, due to suboptimal orientation of the McbD C-terminal carboxylate (MVPFP) for its function in deprotonating the β-nucleophilic side chain for intramolecular attack.
The demonstration of converting MjYcaO, an enzyme naturally involved in McrA thioamidation, into a competent cyclodehydratase enforces a conserved mechanism and evolutionary relationship between the two YcaO catalytic subtypes. To achieve a suitable substrate for cyclodehydration, we substituted the (+1) position relative to the thioamidation site with Cys. However, we observed natural variants of McrA that have the Cys residue at their (+1) position (n = 10, SI, Figure S4) despite the high conservation of McrA. Even though we were not able to investigate the modification of this variant because of the lack of information on the cognate YcaO, this observation and the biosynthetic plasticity of MjYcaO provide a possible evolutionary avenue from cyclodehydration to thioamidation. Specifically, a plausible evolutionary pathway may originate from an ancestral YcaO enzyme that installs azolines, but the producing organism exists in a sulfide-rich environment. Ring opening would produce a thioamide with restoration of the nucleophile at the (+1) position. Our previous work demonstrated that the installation of thioamides onto appropriate substrates by a YcaO cyclodehydratase, followed by subsequent chemical thiolysis,18 is consistent with this speculative evolutionary pathway. Notably, our analysis of McrA partial sequence variation shows that the (0) position of the 11-mer region is sometimes substituted by Asp (n = 11) or Ser (n = 17) (SI, Figure S4), which suggest the presence of thio-Asp or thio-Ser in yet uncharacterized McrA proteins. Another known McrA modification within the 11-mer region is didehydroaspartate at Asp(+5).34 In our current data set, there are 99 naturally occurring cases where Gly is substituted at this position, suggesting the absence of this modification. The work described here may advance efforts directed at engineering new activities using a YcaO-based approach, given that nature has employed minor active site changes in a common scaffold to achieve a variety of chemical outcomes.
Acknowledgments
We thank Keith Brister and the staff at LS-CAT at the Advanced Photon Source (Argonne National Laboratory, Chicago) for facilitating data collection. We thank Graham Hudson from the Mitchell group for acquiring the high-resolution mass spectral data. This work was supported in part by funding from the National Institutes of Health (GM097142 to D.A.M., and GM079038 to S.K.N.). The Bruker UltrafleXtreme MALDI TOF/TOF mass spectrometer was purchased in part with a grant from the National Institutes of Health (S10 RR027109 A).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.9b00124.
Detailed methods for protein expression, purification, biochemical, and crystallographic studies, and additional data and figures (PDF)
Data set 1: spreadsheet containing all RODEO-derived information on the YcaO superfamily, including NCBI accession identifiers, neighboring gene co-occurrence, and hidden Markov model analysis (XLSX)
Author Contributions
# S.-H.D. and A.L. contributed equally. S.-H.D, A.L., and N.M. designed and performed the experiments. All authors analyzed data and assisted in the writing and editorial process. D.A.M. and S.K.N. conceived of and oversaw the project.
The authors declare no competing financial interest.
Supplementary Material
References
- Burkhart B. J.; Schwalen C. J.; Mann G.; Naismith J. H.; Mitchell D. A. YcaO-dependent posttranslational amide activation: Biosynthesis, Structure, and Function. Chem. Rev. 2017, 117, 5389–5456. 10.1021/acs.chemrev.6b00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar K. L.; Chekan J. R.; Cox C. L.; Burkhart B. J.; Nair S. K.; Mitchell D. A. Discovery of a new ATP-binding motif involved in peptidic azoline biosynthesis. Nat. Chem. Biol. 2014, 10, 823–829. 10.1038/nchembio.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnison P. G.; Bibb M. J.; Bierbaum G.; Bowers A. A.; Bugni T. S.; Bulaj G.; Camarero J. A.; Campopiano D. J.; Challis G. L.; Clardy J.; Cotter P. D.; Craik D. J.; Dawson M.; Dittmann E.; Donadio S.; Dorrestein P. C.; Entian K.-D.; Fischbach M. A.; Garavelli J. S.; Goeransson U.; Gruber C. W.; Haft D. H.; Hemscheidt T. K.; Hertweck C.; Hill C.; Horswill A. R.; Jaspars M.; Kelly W. L.; Klinman J. P.; Kuipers O. P.; Link A. J.; Liu W.; Marahiel M. A.; Mitchell D. A.; Moll G. N.; Moore B. S.; Mueller R.; Nair S. K.; Nes I. F.; Norris G. E.; Olivera B. M.; Onaka H.; Patchett M. L.; Piel J.; Reaney M. J. T.; Rebuffat S.; Ross R. P.; Sahl H.-G.; Schmidt E. W.; Selsted M. E.; Severinov K.; Shen B.; Sivonen K.; Smith L.; Stein T.; Suessmuth R. D.; Tagg J. R.; Tang G.-L.; Truman A. W.; Vederas J. C.; Walsh C. T.; Walton J. D.; Wenzel S. C.; Willey J. M.; van der Donk W. A. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. 10.1039/C2NP20085F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. W.; Mitchell D. A.; Markley A. L.; Hensler M. E.; Gonzalez D.; Wohlrab A.; Dorrestein P. C.; Nizet V.; Dixon J. E. Discovery of a widely distributed toxin biosynthetic gene cluster. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 5879–5884. 10.1073/pnas.0801338105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland Brown L. C.; Acker M. G.; Clardy J.; Walsh C. T.; Fischbach M. A. Thirteen posttranslational modifications convert a 14-residue peptide into the antibiotic thiocillin. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 2549–2553. 10.1073/pnas.0900008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt E. W.; Nelson J. T.; Rasko D. A.; Sudek S.; Eisen J. A.; Haygood M. G.; Ravel J. Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 7315–7320. 10.1073/pnas.0501424102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. M.; Milne J. C.; Madison L. L.; Kolter R.; Walsh C. T. From peptide precursors to oxazole and thiazole-containing peptide antibiotics: microcin B17 synthase. Science 1996, 274, 1188–1193. 10.1126/science.274.5290.1188. [DOI] [PubMed] [Google Scholar]
- San Millan J. L.; Kolter R.; Moreno F. Plasmid genes required for microcin B17 production. J. Bacteriol. 1985, 163 (3), 1016–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorgey P.; Lee J.; Kordel J.; Vivas E.; Warner P.; Jebaratnam D.; Kolter R. Posttranslational modifications in microcin B17 define an additional class of DNA gyrase inhibitor. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 4519–4523. 10.1073/pnas.91.10.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar K. L.; Melby J. O.; Mitchell D. A. YcaO domains use ATP to activate amide backbones during peptide cyclodehydrations. Nat. Chem. Biol. 2012, 8, 569–575. 10.1038/nchembio.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart B. J.; Hudson G. A.; Dunbar K. L.; Mitchell D. A. A prevalent peptide-binding domain guides ribosomal natural product biosynthesis. Nat. Chem. Biol. 2015, 11, 564–570. 10.1038/nchembio.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehnke J.; Mann G.; Bent A. F.; Ludewig H.; Shirran S.; Botting C.; Lebl T.; Houssen W.; Jaspars M.; Naismith J. H. Structural analysis of leader peptide binding enables leader-free cyanobactin processing. Nat. Chem. Biol. 2015, 11, 558–563. 10.1038/nchembio.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crone W. J.; Vior N. M.; Santos-Aberturas J.; Schmitz L. G.; Leeper F. J.; Truman A. W. Dissecting bottromycin biosynthesis using comparative untargeted metabolomics. Angew. Chem., Int. Ed. 2016, 55, 9639–9643. 10.1002/anie.201604304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak D. D.; Mahanta N.; Mitchell D. A.; Metcalf W. W. Post-translational thioamidation of methyl-coenzyme M reductase, a key enzyme in methanogenic and methanotrophic archaea. eLife 2017, 6, e29218. 10.7554/eLife.29218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz L.; Adam S.; Santos-Aberturas J.; Truman A. W.; Koehnke J. Macroamidine formation in bottromycins is catalyzed by a divergent YcaO enzyme. J. Am. Chem. Soc. 2017, 139, 18158–18161. 10.1021/jacs.7b09898. [DOI] [PubMed] [Google Scholar]
- Schwalen C. J.; Hudson G. A.; Kosol S.; Mahanta N.; Challis G. L.; Mitchell D. A. In vitro biosynthetic studies of bottromycin expand the enzymatic capabilities of the YcaO superfamily. J. Am. Chem. Soc. 2017, 139, 18154–18157. 10.1021/jacs.7b09899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travin D. Y.; Metelev M.; Serebryakova M.; Komarova E. S.; Osterman I. A.; Ghilarov D.; Severinov K. Biosynthesis of translation inhibitor klebsazolicin proceeds through heterocyclization and N-terminal amidine formation catalyzed by a single YcaO enzyme. J. Am. Chem. Soc. 2018, 140, 5625–5633. 10.1021/jacs.8b02277. [DOI] [PubMed] [Google Scholar]
- Dunbar K. L.; Mitchell D. A. Insights into the mechanism of peptide cyclodehydrations achieved through the chemoenzymatic generation of amide derivatives. J. Am. Chem. Soc. 2013, 135, 8692–8701. 10.1021/ja4029507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa M.; Kawasaki T.; Hayakawa Y. Cloning and heterologous expression of the thioviridamide biosynthesis gene cluster from Streptomyces olivoviridis. Appl. Environ. Microbiol. 2013, 79, 7110–7113. 10.1128/AEM.01978-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahnt J.; Buchenau B.; Mahlert F.; Kruger M.; Shima S.; Thauer R. K. Post-translational modifications in the active site region of methyl-coenzyme M reductase from methanogenic and methanotrophic archaea. FEBS J. 2007, 274, 4913–4921. 10.1111/j.1742-4658.2007.06016.x. [DOI] [PubMed] [Google Scholar]
- Basu M. K.; Selengut J. D.; Haft D. H. ProPhylo: partial phylogenetic profiling to guide protein family construction and assignment of biological process. BMC Bioinf. 2011, 12, 434. 10.1186/1471-2105-12-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanta N.; Liu A.; Dong S.; Nair S. K.; Mitchell D. A. Enzymatic reconstitution of ribosomal peptide backbone thioamidation. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 3030–3035. 10.1073/pnas.1722324115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong S. H.; Tang W.; Lukk T.; Yu Y.; Nair S. K.; van der Donk W. A. The enterococcal cytolysin synthetase has an unanticipated lipid kinase fold. eLife 2015, 4, e07607. 10.7554/eLife.07607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y. O.; Levengood M. R.; Ihnken L. A.; Knowlton A. K.; van der Donk W. A. Lacticin 481 synthetase as a general serine/threonine kinase. ACS Chem. Biol. 2009, 4, 379–385. 10.1021/cb800309v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repka L. M.; Chekan J. R.; Nair S. K.; van der Donk W. A. Mechanistic understanding of lanthipeptide biosynthetic enzymes. Chem. Rev. 2017, 117, 5457–5520. 10.1021/acs.chemrev.6b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlt J. A.; Bouvier J. T.; Davidson D. B.; Imker H. J.; Sadkhin B.; Slater D. R.; Whalen K. L. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta, Proteins Proteomics 2015, 1854, 1019–1037. 10.1016/j.bbapap.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tietz J. I.; Schwalen C. J.; Patel P. S.; Maxson T.; Blair P. M.; Tai H. C.; Zakai U. I.; Mitchell D. A. A new genome-mining tool redefines the lasso peptide biosynthetic landscape. Nat. Chem. Biol. 2017, 13, 470–478. 10.1038/nchembio.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levengood M. R.; Patton G. C.; van der Donk W. A. The leader peptide is not required for post-translational modification by lacticin 481 synthetase. J. Am. Chem. Soc. 2007, 129, 10314–10315. 10.1021/ja072967+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardar D.; Pierce E.; McIntosh J. A.; Schmidt E. W. Recognition sequences and substrate evolution in cyanobactin biosynthesis. ACS Synth. Biol. 2015, 4, 167–176. 10.1021/sb500019b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh J. A.; Donia M. S.; Schmidt E. W. Insights into heterocyclization from two highly similar enzymes. J. Am. Chem. Soc. 2010, 132, 4089–4091. 10.1021/ja9107116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghilarov D.; Stevenson C. E. M.; Travin D. Y.; Piskunova J.; Serebryakova M.; Maxwell A.; Lawson D. M.; Severinov K. Architecture of Microcin B17 Synthetase: An Octameric Protein Complex Converting a Ribosomally Synthesized Peptide into a DNA Gyrase Poison. Mol. Cell 2019, 73, 749–762. 10.1016/j.molcel.2018.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melby J. O.; Dunbar K. L.; Trinh N. Q.; Mitchell D. A. Selectivity, directionality, and promiscuity in peptide processing from a Bacillus sp. Al Hakam cyclodehydratase. J. Am. Chem. Soc. 2012, 134, 5309–5316. 10.1021/ja211675n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshaw P. J.; Roy R. S.; Kelleher N. L.; Walsh C. T. Kinetics and regioselectivity of peptide-to-heterocycle conversions by microcin B17 synthetase. Chem. Biol. 1998, 5, 373–384. 10.1016/S1074-5521(98)90071-0. [DOI] [PubMed] [Google Scholar]
- Wagner T.; Kahnt J.; Ermler U.; Shima S. Didehydroaspartate modification in methyl-Coenzyme M reductase catalyzing methane formation. Angew. Chem., Int. Ed. 2016, 55, 10630–10633. 10.1002/anie.201603882. [DOI] [PubMed] [Google Scholar]
- Wallace A. C.; Laskowski R. A.; Thornton J. M. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng., Des. Sel. 1995, 8, 127–134. 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.