

Abstract
Eosinophilic granulomatosis with polyangiitis (EGPA) is a rare form of vasculitis characterised by atopic manifestations, inflammation of small-sized to medium-sized arteries and veins, hypereosinophilia and tissue infiltration with eosinophils. Cardiac complications occur most commonly in the absence of antineutrophil cytoplasmic antibodies. Cardiac complications include coronary arteritis (rare), pericarditis, myocarditis, endocardial fibrosis (Loeffler’s endocarditis) and intracavitary thrombosis of the left or right ventricle. This is the first reported case of large non-infectious valvular vegetations associated with EGPA.
Keywords: cardiovascular medicine, valvar diseases, immunology, pathology, vasculitis
Background
Eosinophilic granulomatosis with polyangiitis (EPGA), formerly known as the Churg–Strauss syndrome or allergic granulomatosis and angiitis, is an autoimmune disorder characterised by atopic manifestations, hypereosinophilia and inflammation of small-sized to medium-sized blood vessels.1–6 Four of the following six criteria are required for the diagnosis of EGPA: (1) asthma, (2) eosinophilia (>1500 cells/μL), (3) presence of mononeuropathy or polyneuropathy, (4) unfixed pulmonary infiltrates, (5) presence of paranasal sinus abnormalities and (6) histological evidence of extravascular eosinophils (sensitivity: 85%, specificity: 99.7%).1 Cardiovascular manifestations of EGPA are attributable to tissue infiltration by eosinophils and vasculitis.7–16 They may include pericardial, myocardial and endocardial abnormalities including valve involvement.8–16 Coronary arteritis occurs infrequently in patients with EGPA.8–16 In this report, we describe the case of a patient with EGPA who developed non-infectious vegetations involving the aortic valve, mitral valve and chordae tendineae requiring aortic and mitral valve replacement.
Case presentation
A middle-aged Caucasian man presented for evaluation of increasing shortness of breath. He had a prolonged history of asthma, allergic rhinitis and chronic sinusitis. Dyspnoea was accompanied by orthopnoea and lower extremity swelling. He also complained of pain in multiple joints, numbness and tingling of the upper and lower extremities, sharp lancinating retrosternal chest pain and drenching night sweats. Although his shortness of breath recently increased in severity, he had complained of persistent dyspnoea on exertion since the age of 35 years. At that age he was noted to have marked eosinophilia. Evaluation for causes of eosinophilia including assessment for specific allergies, malignancy, parasitic infestation and specific autoimmune diseases was negative. Subsequent evaluation of dyspnoea included chest X-rays and CTs of the chest which showed expanding transient and migratory pulmonary parenchymal opacities and bilateral small pleural effusions (figure 1). Pulmonary function tests showed severe obstructive and restrictive defects and a moderate decrease in diffusing capacity for carbon monoxide. One year prior to his current presentation, he had undergone coronary angiography for evaluation of atypical chest pain associated with elevation of serum troponin T levels. His coronary arteries were angiographically normal. He had been treated continuously with prednisone at various doses since the age of 37 years and developed avascular necrosis of the right femoral head requiring hip replacement surgery.
Figure 1.
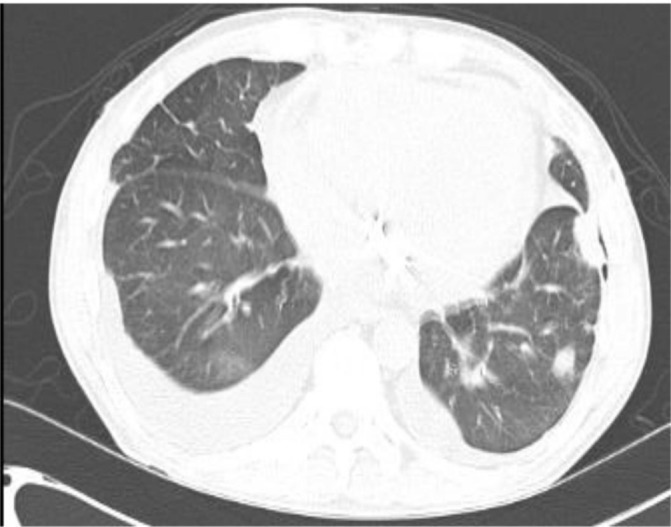
CT of the chest showing multiple parenchymal lung opacities and bilateral pleural effusions (right→left).
Vital signs on admission were as follows: blood pressure: 85/66 mm Hg, heart rate: 78 beats/min, respiratory rate: 18 breaths/min and temperature: 37.0 °C. The oxygen saturation on room air was 96%. The heart examination showed a non-radiating grade 2, harsh midsystolic murmur at the right and left second intercostal spaces and a non-radiating grade 2, high-pitched holosystolic murmur at the lower left sternal border and apex. There was jugular venous distention and hepatojugular reflux. Peripheral pulses were normal. The patient appeared to be subjectively dyspnoeic and was using accessory respiratory muscles. Wheezes were audible over the left posterior lung base. There were no crackles. There was mild bilateral pedal, ankle and lower leg oedema.
Investigations
His leucocyte count was 24 300 cells/mm3, 32% of which were eosinophils. The remainder of the differential was as follows: granulocytes (58%), lymphocytes (5%), monocytes (4%) and basophils (1%). The respective blood haemoglobin and haematocrit values were 109 g/L and 32.0%. Red blood cell indices were normal. The platelet count was 182×109/L. The erythrocyte sedimentation rate was 68 mm/hour. Six sets of blood cultures were obtained following admission; all were negative for pathogenic bacteria and fungi. The plasma N-Terminal pro-hormone B-type Natriuretic Peptide was >35 000 pg/mL. A comprehensive metabolic panel showed the following abnormalities: sodium: 128 mg/dL, chloride: 97 mg/dL, creatinine: 1.18 mg/dL, calcium: 7.9 mg/dL, total protein: 6.5 g/dL, albumin: 2.8 g/dL and aspartate aminotransferase: 49 units/L; serum creatine kinase (CK) was 304 units/L, CK-MB (CK-muscle/brain) was 25.6 ng/mL, CK index was normal at 8.44 and serum troponin T was 2.36 ng/mL. His fluorescent antinuclear antibody (ANA) titre was 1:160 (speckled pattern); however, the ANA profile was negative. Serum rheumatoid factor was normal. Serum complement levels (C3, C4) were normal. Antinuclear cytoplasmic antibodies (p-ANCA performed using an immunofluorecence assay) were absent. Serological evaluation for HIV, hepatitis A, B and C, and Q fever were negative. Bartonella henselae, Aspergillus galactomannan and QuantiFERON Gold titres were negative as was the Legionella antigen. Bone marrow examination showed normal precursor cells and mature eosinophils. Genetic testing showed the absence of the FIP1L1-PDGF fusion gene, the BCR-ABL fusion gene, the CHIC 2 gene and FGFR1. His 12-lead ECG showed a QS pattern in leads V1 and V2 and non-specific ST-T abnormalities. His chest X-ray showed evidence of pulmonary vascular congestion and bilateral small pleural effusions. CT of the chest showed small bilateral pleural effusions and no evidence of pulmonary embolism. Heart size was normal. A limited transthoracic echocardiogram showed normal left ventricular (LV) chamber size and wall thickness, an LV ejection fraction of 35% and moderate global hypokinesis. There was a large ill-defined mass on the aortic valve and moderate mitral regurgitation. Subsequently, a transoesophageal echocardiogram was performed which showed a 19×16 mm irregular echo-dense mass that appeared fixed and attached to two and possibly all three aortic valve leaflets (figure 2). The peak gradient over the aortic valve was 25 mm Hg. There was no aortic regurgitation. In addition, there was a 11×9 mm irregular, mobile echo-dense mass that appeared to be attached to the anterior leaflet of the mitral valve (figure 2). There was moderate to moderately severe mitral regurgitation. An electromyogram and nerve conduction velocity of the upper and lower extremities showed evidence of moderate axonal sensory neuropathy involving the median, ulnar and peroneal nerves.
Figure 2.
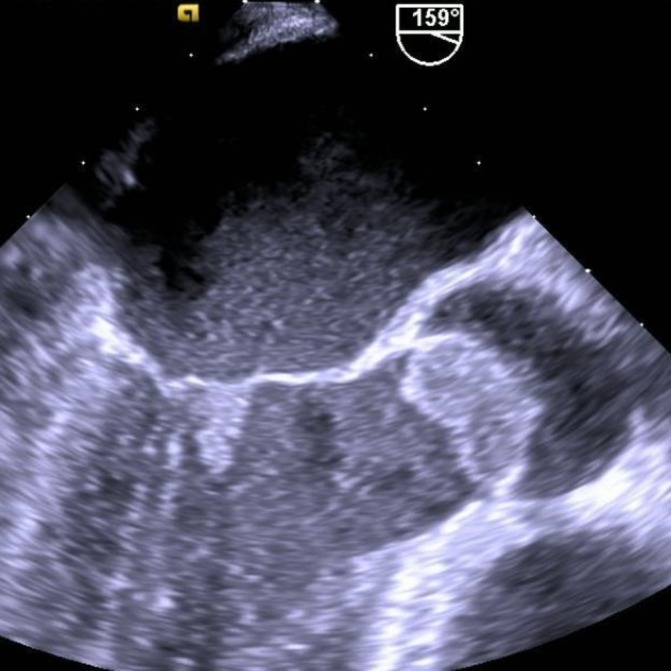
Transoesophageal echocardiographic images showing a large 19×16 mm vegetation attached to two and possibly all three aortic valve leaflets and a smaller 11×9 mm vegetation attached to the ventricular surface of the anterior mitral leaflet.
Differential diagnosis
The differential diagnosis for hypereosinophilic syndromes with cardiac involvement includes the idiopathic hypereosinophilic syndrome, EGPA, early giant cell myocarditis, hypersensitivity reactions (often induced by medication, parasitic infestation and malignancy).15 An appropriate travel history and a thorough evaluation for parasitic (particularly helminthic) infestation may help to narrow the diagnostic options. The presence of coronary arteritis which may result in myocardial infarction or sudden cardiac arrest and pericarditis favour the diagnosis of EGPA over the idiopathic hypereosinophilic syndrome15 In this case, infective endocarditis was excluded by negative blood cultures and absence of pathogens in the vegetations. Wegener’s granulomatosis is usually ANCA positive and not characterised by eosinophilia and was thought to be an unlikely cause of the vegetations.
Treatment
Based on the size of the aortic valve mass, the presence of a mitral valve mass and moderate or greater mitral regurgitation, the decision was made to proceed with aortic and mitral valve replacement. The surgical pathology report described the presence of a large friable aortic valve vegetation (2.2×1.5×1.2 cm) attached to the ventricular aspect of the right and non-coronary cusps which extended to the left coronary cusp, almost completely obstructing the LV outflow tract. A smaller vegetation was attached to the ventricular aspect of the anterior leaflet of the mitral valve. In addition, a vegetation was noted to be intertwined between the chordae tendineae extending to the anterior papillary muscle. The aortic valve was replaced with a 20 mm ATS mechanical aortic valve prosthesis, and the mitral valve was replaced with a 27 mm ATS mechanical mitral valve prosthesis. The surgical report described inflammation of the mitral valve, aortic valve and aortic root. The pericardium and epicardium also appeared to be inflamed. A wedge lung biopsy was obtained during surgery to rule out infectious or non-infectious causes of pulmonary infiltrates. Histological examination of the native valves showed myxoid degeneration with histiocytic, giant cell and eosinophilic infiltration (figure 3). No bacteria or fungi were noted and cultures of the valves were negative. Histological evaluation of the lung biopsy showed emphysematous changes and subpleural fibrosis.
Figure 3.
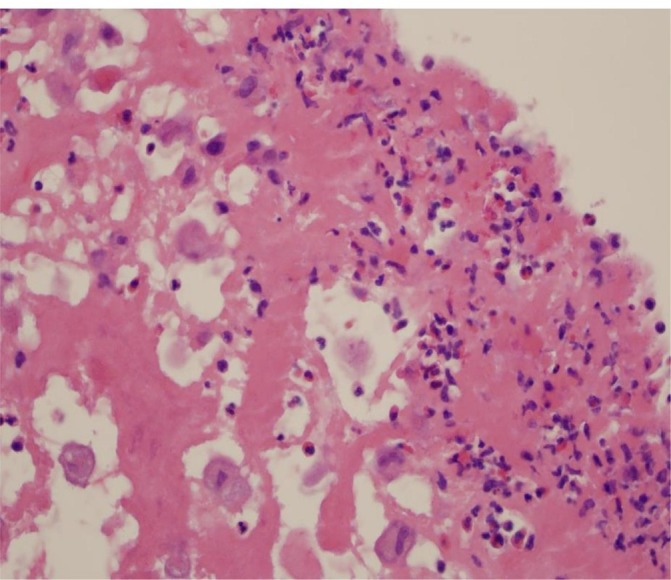
Histological section of the aortic valve vegetation showing areas of necrosis, histiocytes, multilobulated giant cells and eosinophils (H&E stain).
Outcome and follow-up
The patient was discharged after 26 days of hospitalisation. He was treated with 70 mg of prednisone daily with a rapid taper during the hospitalisation. His discharge medication included antibiotics and prednisone 5 mg daily. He had been treated with 5 mg of prednisone daily chronically prior to admission for reactive airway disease. Transthoracic echocardiograms obtained 6 weeks, 3 months and 6 months after surgery showed an increase in LV volume and a progressive decline in LV ejection fraction to 25% despite appropriate medical therapy for LV systolic dysfunction. The patient remained clinically stable until 10 months after valve replacement surgery when he developed progressive shortness of breath, pulmonary vascular congestion on chest X-ray, marked elevation of the plasma NT-proBNP level, hypotension and non-blanching erythematous macules on the back, anterior chest, shoulders, forearms, palms and soles (figure 4). A punch biopsy of one of these lesions showed extravasation of red blood cells in the superficial dermis, thrombus formation in the deep dermis surrounded by infiltration with numerous lymphocytes and eosinophils as well as multiple histiocytes with phagocytised material (figure 5). These findings were consistent with those described with EGPA. The patient developed progressive cardiogenic shock despite vasopressor therapy. Transthoracic and transoesophageal echocardiography, fluoroscopy and cardiac MRI showed normal appearance and function of the prosthetic valves. Cardiac MRI showed moderate LV dilatation with severe global LV hypokinesis (LV ejection fraction was 15%), mild right ventricular dilation with mildly reduced right ventricular systolic function and biatrial enlargement. Delayed gadolinium enhancement was not observed on cardiac MRI. There was no evidence of myocardial infarction, myocarditis or scar. There was no evidence of thrombus in the ventricular cavities or in the atrial appendages. There was a small echo-free pericardial effusion. The patient declined further interventions and succumbed shortly thereafter. The family declined an autopsy.
Figure 4.
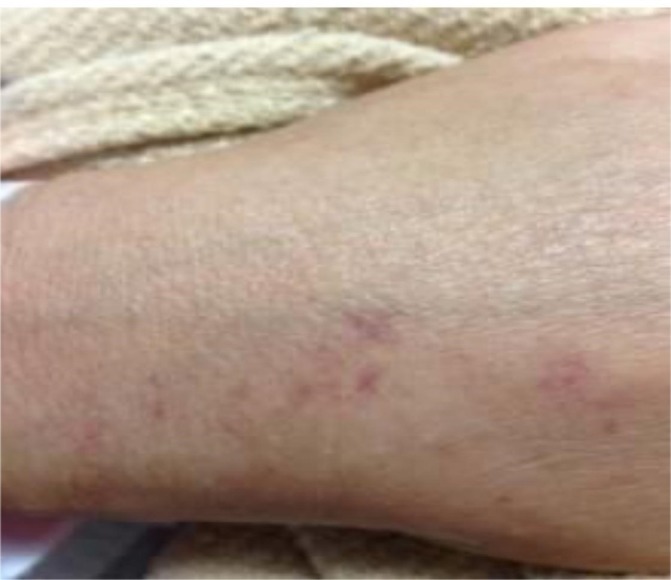
Vasculitic rash appearing shortly before death.
Figure 5.
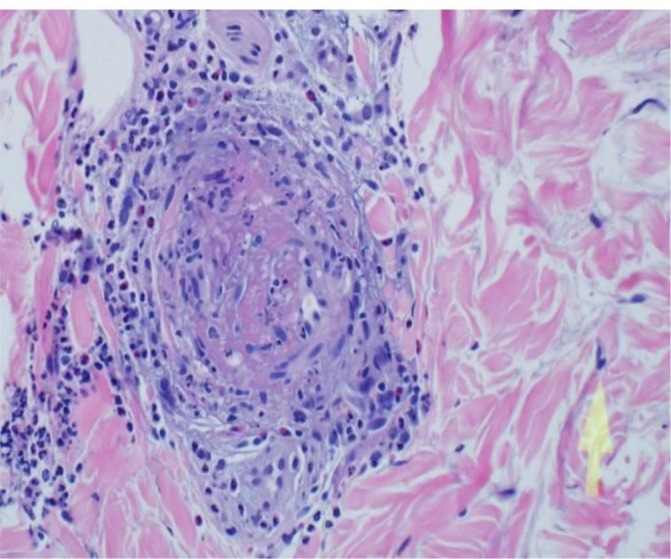
Histological section from a skin biopsy of the vasculitic rash showing thrombosis and eosinophilic infiltration (H&E stain).
Discussion
This patient manifested all the American College of Rheumatology criteria for EGPA including asthma, eosinophilia >1500 cells/µL, polyneuropathy, unfixed pulmonary infiltrates not explained by other disease states, chronic sinusitis and the presence of tissue eosinophils.1 Cardiac involvement occurs in 50%–62% of patients with EGPA and is a common cause of morbidity and mortality.8–18 Previously described cardiac complications of EGPA include vasculitis of the epicardial coronary arteries, vasculitis of the small intramyocardial vasculature (arteries, capillaries, venules), granulomatous myocardial infiltration, eosinophilic myocarditis, myocardial fibrosis, right ventricular and LV endocardial fibrosis, right ventricular and/or LV thrombus formation, restrictive or dilated cardiomyopathy, mitral regurgitation due to papillary muscle dysfunction, acute pericarditis, pericardial effusion with or without tamponade, constrictive pericarditis, supraventricular and ventricular arrhythmias, atrioventricular and intraventricular block, and sudden cardiac death. Higher peripheral blood eosinophil counts and the absence of ANCA positivity have been associated with higher risk of cardiac involvement in EGPA.1–19
Endocardial involvement in patients with EGPA is similar to that of other hypereosinophilic syndromes (idiopathic hypereosinophilic syndrome and its variants, tropical endomyocardial fibrosis).17–23 The pathological lesion most commonly associated with hypereosinophilic syndromes is referred to as Loeffler’s endocarditis.17–23 Loeffler’s endocarditis occurs less commonly with EGPA than with other hypereosinophilic syndromes.16–22 It evolves in three stages. The first stage is characterised by endomyocardial infiltration with eosinophils and lymphocytes.17–23 Degranulation of eosinophils results in the release of cationic protein, eosinophil major protein, eosinophil myeloperoxidase and Charcot–Leyden crystal protein. These substances are cytotoxic and are thought to produce endomyocardial necrosis. The second stage is characterised by thrombus formation in areas of endomyocardial damage (the left and right ventricular apex are most commonly affected).14–23 The third stage is characterised by endocardial fibrosis which may be local or diffuse and may result in restrictive cardiomyopathy. Endocardial fibrosis is recognised by late gadolinium enhancement on cardiac MRI.16 17 Valvular function in Loeffler’s endocarditis may be affected by fibrotic tethering of chordae tendineae or fibroinflammatory involvement of valvular endothelium.17–23 Mitral and tricuspid regurgitation are the most commonly reported forms of valve dysfunction in patients with EGPA, but are infrequently observed.8–17 Large valvular vegetations have been described in the idiopathic hypereosinophilic syndrome and with vasculitides such as Wegener’s granulomatosis.15 24–26 To our knowledge this case is the first description of non-infectious valvular vegetations in a patient with EGPA. In the absence of myocardial tissue from biopsy or autopsy, we are uncertain as to the cause of this patient’s decline in LV systolic function after surgery. Eosinophilic infiltration of myocardium is a possibility, but cardiac MRI showed no evidence of myocarditis or endocardial fibrosis. Severing of the chordae tendineae to facilitate mitral valve replacement is also a possible explanation.
Current recommendations for the treatment of EGPA include therapy with glucocorticoids such as prednisolone, followed by administrations of an immunosuppressants such as cyclophosphamide and azathioprine.27 A systematic review of treatment of EGPA by Bosch et al suggested that azathioprine and possibly methotrexate may be useful in maintaining remission.27 In 2017, the humanised monoclonal antibody mepolizumab was approved by the Food and Drug Administration for use in EGPA.28 Our patient was treated with high-dose glucocorticoids, but did not receive cyclophosphamide, azathioprine, leflunomide, rituximab, intravenous immunoglobulin or plasma exchange. Failure to initiate one or more of these therapies probably occurred due to the delay in confirming the diagnosis of EGPA, which occurred shortly before death, and because the patient declined further therapy once the diagnosis of EGPA was confirmed. It is possible that earlier diagnosis and more aggressive pharmacotherapy might have changed the course of this patient’s illness. Mepolizumab was not available prior to the patient’s demise.
In conclusion, non-infectious valvular vegetations should be added to the list of cardiovascular complications of EGPA.
Learning points.
Previously described cardiac complications of eosinophilic granulomatosis with polyangiitis (EGPA) include coronary arteritis (rare), pericarditis, myocarditis, endocardial fibrosis (Loeffler’s endocarditis) and thrombus formation in the left or right ventricle.
Cardiac complications occur in 50%–62% of patients with EGPA, mainly in those who are antineutrophil cytoplasmic antibody negative.
Large non-infectious valvular vegetations may occur in patients with EGPA.
Footnotes
Contributors: KK involved with writing up the clinic case and gathering the data for case reporting. Drafting and editing the case report to final form. SB and MAA involved in the conception, planning and analysis, and interpretation of the data. Involved in providing insight into format for write-up and data presentation.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent for publication: Not required.
References
- 1. Masi AT, Hunder GG, Lie JT, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum 1990;33:1094–100. [DOI] [PubMed] [Google Scholar]
- 2. Churg J, Strauss L. Allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Am J Pathol 1951;27:277–301. [PMC free article] [PubMed] [Google Scholar]
- 3. Chumbley LC, Harrison EG, DeRemee RA. Allergic granulomatosis and angiitis (Churg-Strauss syndrome). Report and analysis of 30 cases. Mayo Clin Proc 1977;52:477–84. [PubMed] [Google Scholar]
- 4. Guillevin L, Lhote F, Gayraud M, et al. Churg-Strauss syndrome. A prospective study in 342 patients. Medicine 1996;75:17–25. [DOI] [PubMed] [Google Scholar]
- 5. Guillevin L, Pagnour C, Mouthon L. Churg-Strauss syndrome. Semin Respir Crit Care Med 2004;25:535–45. 10.1055/s-2004-836145 [DOI] [PubMed] [Google Scholar]
- 6. Keogh K, Specks U. Churg-Strauss syndrome. Semin Respir Crit Care Med 2006;27:148–57. 10.1055/s-2006-939518 [DOI] [PubMed] [Google Scholar]
- 7. Khoury P, Grayson PC, Klion AD. Eosinophils in vasculitis: characteristics and roles in pathogenesis. Nat Rev Rheumatol 2014;10:474–83. 10.1038/nrrheum.2014.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pelà G, Tirabassi G, Pattoneri P, et al. Cardiac involvement in the Churg-Strauss syndrome. Am J Cardiol 2006;97:1519–24. 10.1016/j.amjcard.2005.11.088 [DOI] [PubMed] [Google Scholar]
- 9. Dennert RM, van Paassen P, Schalla S, et al. Cardiac involvement in Churg-Strauss syndrome. Arthritis Rheum 2010;62:NA–634. 10.1002/art.27263 [DOI] [PubMed] [Google Scholar]
- 10. Cereda AF, Pedrotti P, De Capitani L, et al. Comprehensive evaluation of cardiac involvement in eosinophilic granulomatosis with polyangiitis (EGPA) with cardiac magnetic resonance. Eur J Intern Med 2017;39:51–6. 10.1016/j.ejim.2016.09.014 [DOI] [PubMed] [Google Scholar]
- 11. Neumann T, Manger B, Schmid M, et al. Cardiac involvement in Churg-Strauss syndrome: impact of endomyocarditis. Medicine 2009;88:236–43. 10.1097/MD.0b013e3181af35a5 [DOI] [PubMed] [Google Scholar]
- 12. Misra DP, Shenoy SN. Cardiac involvement in primary systemic vasculitis and potential drug therapies to reduce cardiovascular risk. Rheumatol Int 2017;37:151–67. 10.1007/s00296-016-3435-1 [DOI] [PubMed] [Google Scholar]
- 13. Morgan JM, Raposo L, Gibson DG. Cardiac involvement in Churg-Strauss syndrome shown by echocardiography. Br Heart J 1989;62:462–6. 10.1136/hrt.62.6.462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Agard C, Rendu E, Leguern V, et al. Churg-Strauss syndrome revealed by granulomatous acute pericarditis: two case reports and a review of the literature. Semin Arthritis Rheum 2007;36:386–91. 10.1016/j.semarthrit.2006.12.002 [DOI] [PubMed] [Google Scholar]
- 15. Ogbogu PU, Rosing DR, Horne MK. Cardiovascular manifestations of hypereosinophilic syndromes. Immunol Allergy Clin North Am 2007;27:457–75. 10.1016/j.iac.2007.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Szczeklik W, Miszalski-Jamka T, Mastalerz L, et al. Multimodality assessment of cardiac involvement in Churg-Strauss syndrome patients in clinical remission. Circ J 2011;75:649–55. 10.1253/circj.CJ-10-0772 [DOI] [PubMed] [Google Scholar]
- 17. Alter P, Maisch B. Endomyocardial fibrosis in Churg-Strauss syndrome assessed by cardiac magnetic resonance imaging. Int J Cardiol 2006;108:112–3. 10.1016/j.ijcard.2005.02.019 [DOI] [PubMed] [Google Scholar]
- 18. Seo JS, Song JM, Kim DH, et al. A case of loeffler’s endocarditis associated with churg-strauss syndrome. J Cardiovasc Ultrasound 2010;18:21–4. 10.4250/jcu.2010.18.1.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Akbar G, Vikram E, Freudenberger R, et al. Eosinophilic heart disease: a case report and review of the literature. J Cardiol Cardiovasc Ther 2017;4:555631. [Google Scholar]
- 20. Parrillo JE, Borer JS, Henry WL, et al. The cardiovascular manifestations of the hypereosinophilic syndrome. Prospective study of 26 patients, with review of the literature. Am J Med 1979;67:572–82. [DOI] [PubMed] [Google Scholar]
- 21. Spry CJ, Take M, Tai PC. Eosinophilic disorders affecting the myocardium and endocardium: a review. Heart Vessels Suppl 1985;1:240–2. 10.1007/BF02072402 [DOI] [PubMed] [Google Scholar]
- 22. Sen T, Ponde CK, Udwadia ZF. Hypereosinophilic syndrome with isolated Loeffler’s endocarditis: complete resolution with corticosteroids. J Postgrad Med 2008;54:135–7. [DOI] [PubMed] [Google Scholar]
- 23. Jin X, Ma C, Liu S, et al. Cardiac involvements in hypereosinophilia-associated syndrome: Case reports and a little review of the literature. Echocardiography 2017;34:1242–6. 10.1111/echo.13573 [DOI] [PubMed] [Google Scholar]
- 24. Lacoste C, Mansencal N, Ben M’rad M, et al. Valvular involvement in ANCA-associated systemic vasculitis: a case report and literature review. BMC Musculoskelet Disord 2011;12:50 10.1186/1471-2474-12-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Espitia O, Droy L, Pattier S, et al. A case of aortic and mitral valve involvement in granulomatosis with polyangiitis. Cardiovasc Pathol 2014;23:363–5. 10.1016/j.carpath.2014.07.007 [DOI] [PubMed] [Google Scholar]
- 26. Stöllberger C, Finsterer J, Zlabinger GJ, et al. Antineutrophil cytoplasmic autoantibody-negative antiproteinase 3 syndrome presenting as vasculitis, endocarditis, polyneuropathy and Dupuytren’s contracture. J Heart Valve Dis 2003;12:530–4. [PubMed] [Google Scholar]
- 27. Bosch X, Guilabert A, Espinosa G, et al. Treatment of antineutrophil cytoplasmic antibody associated vasculitis: a systematic review. JAMA 2007;298:655–9. 10.1001/jama.298.6.655 [DOI] [PubMed] [Google Scholar]
- 28.FDA News Release. FDA approves first drug for eosinophilic granulomatosis with polyangiitis, a rare disease formally known as Churg-Strauss syndrome, 2017. [Google Scholar]