

Abstract
The CACNA1H gene encodes the pore-forming α1 subunit of the T-type voltage-dependent calcium channel CaV3.2, expressed abundantly in the adrenal cortex. Mutations in CACNA1H are associated with various forms of primary aldosteronism (PA), including familial hyperaldosteronism type 4 (FH4). We describe a patient with refractory hypokalaemia and elevated aldosterone secretion independent of renin activity. Despite the absence of overt hypertension in this patient, the laboratory evaluation was consistent with a diagnosis of PA. Whole-exome sequencing revealed a de novo missense variant, R890H, in the voltage sensing domain of CACNA1H. Expression of the variant channel in cells resulted in decreased whole-cell current, consistent with a loss-of-function. We hypothesise this variant is the genetic cause of pathological aldosterone secretion in this patient, and thereby expand the current understanding of the genetic basis of FH4.
Keywords: fluid electrolyte and acid–base disturbances, genetics, adrenal disorders
Background
Primary aldosteronism (PA) is defined as excess production of aldosterone that is independent of circulating renin and angiotensin II. It is the most common cause of secondary arterial hypertension, with a prevalence estimated at 6% among patients diagnosed with hypertension in the primary care setting.1 2 PA often presents initially as hypertension and hypokalaemia, as was first described by Litynski in 19533 and subsequently characterised further by Conn in 19554 5; however, there is significant clinical variability in features of the disease. Emerging epidemiological data suggest that overt PA most commonly presents as normokalaemic hypertension,6 7 but the spectrum of disease includes renin-independent aldosterone secretion in individuals with normal blood pressure.8 9
As there is variation in the phenotype of PA, there is also variation in the underlying cause. The most common causes of PA are bilateral adrenal hyperplasia, accounting for roughly 60% of cases, and aldosterone-producing adenomas (APAs), causing another 30%.5 10 Less common causes include malignancy and familial hyperaldosteronism (FH). In the last 10 years, the use of next-generation sequencing has substantially expanded our understanding of the genetic basis of both sporadic and familial forms of PA.11 12 There are now five recognised subtypes of FH, each with distinct clinical features and genetic bases.13
Table 1 shows the genetic and clinical features of the previously described cases of FH type 4 (FH4) due to germline CACNA1H variants. The CACNA1H gene encodes the pore-forming α1 subunit of the T-type voltage-dependent calcium channel CaV3.2, expressed abundantly in the adrenal cortex. We report the case of a young woman with a long history of hypokalaemia found to have autonomous secretion of aldosterone consistent with a diagnosis of PA in the absence of adrenal adenoma or hyperplasia. Next generation sequencing discovered a novel germline missense variant to CACNA1H.
Table 1.
Genetic and clinical characteristics of published FH4 cases due to germline CACNA1H 21 22
| Age | Gender | Genetic variant | Blood pressure at diagnosis (mm Hg) | Serum K+ (mmol/L) |
Adrenal imaging | Additional clinical features |
| 3 years | M | p.Met1549Val | 140–160*/90–105 | 3.8 | No mass or hyperplasia | Developmentally delayed (spoke at age>2 years) |
| 7 years | F | p.Met1549Val | 150*/90 | 3.1 | No mass or hyperplasia | Mother also with PA. |
| 8 years | M | p.Met1549Val | 130–140*/80–90 | 3.6 | No mass or hyperplasia | Maternal uncle with hypertension at age 24 |
| 9 years | M | p.Met1549Val | 192*/144 | 3.7 | No mass or hyperplasia | Father with early onset hypertension |
| 2 months | M | p.Met1549Val | 170 s*/100 s | 4.1 | No mass or hyperplasia | … |
| 2 months | M | p.Met1549Ile | 110/70 | 1.7 | No mass or hyperplasia | Mild mental retardation and learning disabilities |
| 36 years | M | p.Ser196Leu | 155/95 | 2.7 | R adrenal nodule (21 mm); L adrenal nodule (11 mm) | Family history of hypertension and PA |
| 32 years | F | p.Ser196Leu | 153/97 | 2.9 | No mass or hyperplasia | Family history of hypertension and PA |
| 44 years | M | p.Pro2083Leu | 154/104 | 3.9 | No mass or hyperplasia | Family history of hypertension and PA |
| 39 years | M | p.Pro2083Leu | 160/98 | 2.8 | L adrenal thickening without individualised adenoma | Family history of hypertension and PA |
| 50 years | F | p.Val1951Glu | 142/87 | 2.5 | 8 mm left adrenal nodule | … |
Normal range of potassium: 3.3–4.6 mmol/L.
*Indicates systolic blood pressure >95th percentile for age.
FH4, familial hyperaldosteronism type 4; PA, primary aldosteronism.
Case presentation
A 31-year-old woman with hypermobile Ehlers-Danlos syndrome and postural orthostatic tachycardia syndrome (POTS) presented for evaluation of chronic hypokalaemia associated with profound weakness. The patient was a ballet dancer in childhood, but hyperextensible joints soon lead to multiple sprained ankles and stress fractures. As a college student, she was diagnosed with POTS with associated symptomatic hypotension, for which she was treated with propranolol and midodrine as needed. The patient also reported extreme weakness and was found to be hypokalaemic. The lowest serum potassium level captured was 2.6 mmol/L (normal range 3.5–5.1 mmol/L). For many years, the patient was receiving intravenous infusions every 2–4 weeks consisting of 2 L of lactated Ringer’s (LR) solution supplemented with a ranged dose of 40–60 mEq potassium and 4–5 g magnesium. Attempts to treat with oral potassium repletion resulted in intolerable gastrointestinal side effects. When measured during clinic visits, the patient’s blood pressure ranged from 96 to 133/59–95 mm Hg. In addition to propranolol and midodrine as needed, other medications at the time of evaluation included flecanide, colchicine, lorazepam as needed, buprenorphine, diltiazem, calcium and vitamin D supplementation. She was of Ashkenazi Jewish descent and family history was negative for PA, early onset hypertension or hypokalaemia. The patient had no personal history of epilepsy and had met normal developmental milestones.
Prior evaluation had revealed elevated serum aldosterone (11 and 38 ng/dL; normal range for upright, morning level <29 ng/dL) with suppressed plasma renin activity (0.26 and 0.17 pg/dL, respectively; normal range 0.25–5.82 pg/dL), and hypokalaemia (2.9 and 3.3 mmol/L, respectively; normal range 3.5–5.1 mmol/L). A 24 hours urine collection with a volume of 5.66 L was notable for a creatinine index of 23.53 mg/kg, indicating adequate collection, and total potassium excretion of 117.2 mEq/day (reference range 25–125 mEq/day). A confirmatory saline suppression test for the diagnosis of PA was aborted due to the development of severe cardiac symptoms during the infusion. MRI abdomen, protocolled specifically for adrenal imaging, demonstrated normal appearing adrenal glands without adenoma. Low-dose spironolactone was initiated at 12.5 mg /day, which modestly improved the serum potassium levels, yet the patient still required monthly potassium infusions for symptomatic relief due to ongoing weakness.
To further evaluate the high aldosterone state, a laboratory assessment was made prehydration and posthydration with 2 L LR solution, supplemented with 50 mEq K and 5 g Mg, and infused over 10 hours. Results demonstrated persistently elevated serum aldosterone despite volume repletion (table 2). Of note, spironolactone was continued during the testing.
Table 2.
Results of lab testing prehydration and posthydration
| Prehydration | Posthydration | Reference range | |
| Aldosterone (ng/dL) | 17 | 45 | <29 (upright) |
| Plasma renin activity (pg/dL) | 0.23 | 0.27 | 0.25–5.82 |
| Potassium, serum (mmol/L) | 3.8 | 4.1 | 3.5–5.1 |
| Potassium, 24 hours urine (mmol/day) | 38.5 | 61.4 | 25–125 |
| Sodium, 24 hours urine (mmol/day) | 94 | 99 | 40–220 |
Investigations
Given the unusual nature of her presentation, including a laboratory work-up suggesting PA without overt hypertension, trio whole exome sequencing was performed with DNA from the patient and both her parents. Results of whole exome sequencing (Personalis, Inc.) revealed a de novo missense germline variant, NM_0 21 098 c.2917G->A (p.R890H), in the voltage-sensing domain of the CaV3.2 protein encoded by the CACNA1H gene. The variant was present in the patient and not detected in either parent. This variant lies within a transmembrane domain of the protein that is highly conserved across vertebrates, and the pathogenicity score is 1.0, suggesting it is very likely to be deleterious. We cannot provide a specific allele frequency; however, this variant is absent from the g.NOMAD database, which contains over 280 000 previously reported sequences of the CACNA1H gene.14
Expression of the mutated CaV3.2 channel in cultured cells revealed a marked decrease in whole-cell current compared with the wild-type (WT) channel (figure 1). Activation and inactivation kinetics were identical between the mutant and WT channels.
Figure 1.
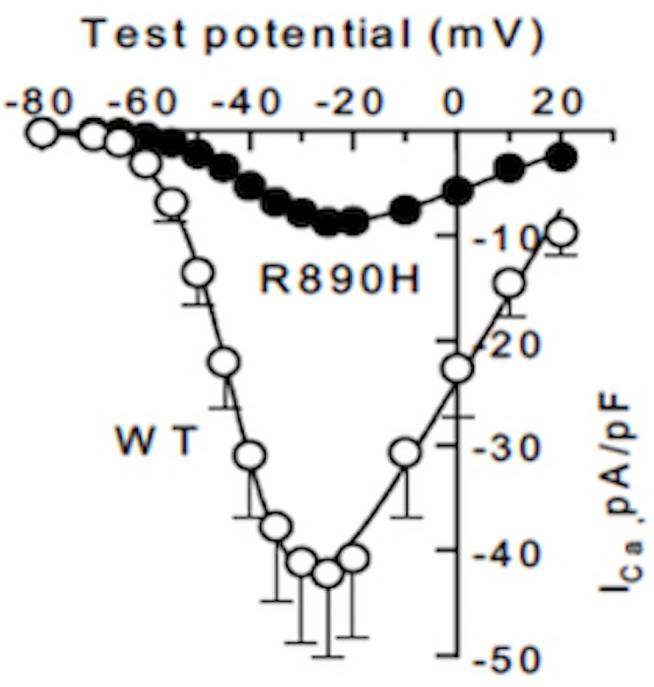
Current–voltage relationship (WT n=10 replicates, R890 H, n=8 replicates.). WT, wild type.
Outcome and follow-up
The patient was started on amiloride, which helped to maintain a higher serum potassium level. As a result, the frequency of potassium infusions and the concentration of potassium administered during the infusion was able to be reduced. However, at least monthly potassium infusions are still required and over time have resulted in sclerotic veins that make intravenous access increasingly difficult. A trial of adrenocorticotropic hormone (ACTH) gel has been ongoing currently, given in vitro evidence that ACTH induces CaV3.2 current and messenger RNA expression.15 ACTH has resulted in slightly longer intervals between required infusions.
Discussion
In this case, the evaluation of hypokalaemia led to the eventual diagnosis of PA, and the use of genetic testing provided an increased understanding of the possible molecular mechanisms driving excessive aldosterone production. The differential diagnosis of hypokalaemia includes causes of renal potassium loss, gastrointestinal potassium loss and transcellular potassium shifts. The history did not reveal an apparent cause of gastrointestinal potassium loss, and the patient was not taking medications known to cause significant cellular potassium shifts; therefore, the evaluation was focused on aetiologies of renal potassium loss. Nearly all potassium that is filtered at the glomerulus is reabsorbed in the proximal tubule and the loop of Henle, while potassium that is excreted is secreted in the connecting and collecting tubules.16 Aldosterone regulates renal potassium excretion by directly stimulating potassium secretion in the cells of the collecting duct. The physiological response to hypokalaemia is to minimise potassium excretion,16 but in the case of our patient, the results of the 24 hours urine collection demonstrated inappropriate renal potassium wasting (<30 mEq/day of urinary potassium excretion is expected in the setting of hypokalaemia).
Laboratory testing demonstrated an elevated serum aldosterone level despite suppressed plasma renin activity; however, because of the apparent absence of hypertension and the overlapping diagnosis of POTS, further testing was done to confirm the aetiology of the hyperaldosterone state. Aldosterone secretion is regulated by extracellular hyperkalaemia, ACTH, and the renin-angiotensin II axis, the latter of which is activated in states of decreased intravascular volume.17 Because of comorbid medical conditions, at the time of the evaluation for PA, the patient was on multiple medications that lower blood pressure and was not able to discontinue them for an extended period of time due to severe symptoms. Therefore, we cannot state with complete certainty that the patient did not meet the current clinical definition of overt hypertension. In addition, several studies suggest that POTS is associated with a reduction in total blood volume and perturbation of the renin–angiotensin–aldosterone axis.18 19 To confirm that the hyperaldosterone state was renin- and angiotensin II-independent, a modified saline suppression test was conducted. Laboratory testing prehydration and posthydration with intravenous fluids demonstrated persistently elevated aldosterone levels and suppressed plasma renin activity, suggesting aldosterone secretion was independent of intravascular volume status. Despite being unable to discontinue spironolactone during this evaluation, the effects of which are to artificially increase renin and, to a lesser extent, aldosterone secretion, the fact that the plasma renin activity remained suppressed confirmed a diagnosis of PA. Of note, the 2016 updates to the Endocrine Society Guidelines for the management of PA now state that in the setting of spontaneous hypokalaemia, plasma renin below detection levels, and a serum aldosterone concentration >20 ng/dL, there may be no need for further confirmatory testing to make the diagnosis of PA.20
With the diagnosis of PA confirmed, the use of gene sequencing led to the discovery of a novel de novo germline variant of CACNA1H this is the likely cause of increased aldosterone production. The CACNA1H gene encodes the pore-forming α1 subunit of the T-type voltage-dependent calcium channel CaV3.2, expressed abundantly in the adrenal cortex. In a recent study of 40 patients with preadolescent hypertension as a result of PA, five subjects shared an identical heterozygous CACNA1H M1549V mutation. This mutation resulted in increased aldosterone production via alterations in intracellular calcium influx, the signal for cholesterol transport into mitochondria and induction of steroidogenesis.13 As shown in table 1, other gain-of-function variants of CACNA1H have been described in FH4,21 implicating CACNA1H as a susceptibility gene for PA with different phenotypes. The patient described here had no evidence on imaging of an APA and no family history of PA or early onset hypertension. Together with the results of the modified saline suppression test, the novel R890H variant in the CACNA1H gene was felt to likely account for upregulated renin- and angiotensin II-independent aldosterone secretion consistent with PA. Although the variant protein appeared to reduce whole-cell current consistent with a loss-of-function when expressed in cultured cells, the precise physiological effect of the R890H CACNA1H gene variant will require further investigation. We postulate that the amino acid change within this transmembrane region causes a decrease in whole-cell current, as per the R890H electrophysiology characterisation described. We do not understand at this time how the amino acid change results in increased aldosterone production and cellular proliferation.
In a case of refractory hypokalaemia, exome trio sequencing, along with a high clinical suspicion for PA, led to the correct diagnosis. We describe a new genotype and phenotype of FH4, that of a young woman with hypokalaemia and the absence of overt hypertension resulting from a germline R890H variant to CACNA1H.
Patient’s perspective.
I was fourteen years old the first time a doctor called me ‘interesting’ to my face. Interesting. A fifty-ton brick. Perhaps he thought I wouldn’t understand his euphemism; on the contrary, I had been hearing doctors call me ‘interesting’ since I was five years old, though always in hushed tones to their colleagues. I dreamt of becoming ‘textbook’.
While I would have to wait another decade for a formal diagnosis, I always knew on some primal level that my body just did not seem to ‘fit’. I felt like a walking museum of medical oddities: musculoskeletal, neurological, gastroenterological, cardiac, pulmonary, endocrine, renal, immune. I fought, truly fought, to pursue my studies and (hopefully) one day become a biomedical researcher, but from my preteen years onward, I would wake up every day quaking in pain, too frail to sit up. Though I managed to attend some high school and college, my highest academic degree remains my middle school diploma. As a child and young adult, I had been an avid dancer, pianist, actor, choreographer, composer, political activist, entrepreneur. One by one, my passions slipped through my fingertips as I turned my focus 100% to simply surviving each day. I sought out a stream of esteemed specialists for answers, only to find that far too many dismissed me on first glance, attributing my symptoms to various somatization disorders. I knew in my soul that multiple systems were not randomly failing, but that there must be a common link.
At 22, as my friends entered post-collegiate life, and I gave up my apartment and moved back home with my family, my medical team landed on a diagnosis. We were quite confident that Ehlers-Danlos syndrome (EDS) and a commonly associated form of autonomic dysfunction called postural orthostatic tachycardia syndrome (POTS) could explain everything. EDS and POTS seem like natural catch-all conditions, since connective tissue and the autonomic nervous system affect so many functions, and EDS tends to manifest a little differently in each patient. However, we soon learned that here, too, I was anomalous. Within the next two years, I developed a slew of symptoms that were not known to be associated with EDS, including chronic inflammatory conditions like pericarditis and peritonitis. My already-fragile health imploded, to put it mildly. Most troublingly, my serum potassium level, which had always trended low, began sinking until I was chronically in critical condition. I thought I knew what weakness felt like until my potassium level slipped to the mid-2 range. My arm would cramp reaching for a glass of water. Maybe I would have the energy to drink it, maybe not; I felt oddly disconnected from myself. I tried multiple formulations of oral potassium supplementation, but they all wreaked havoc on my GI system, and I grew weaker still from the resulting blood loss. We decreased the oral potassium to every other day (the most that I could tolerate) and relied primarily on IV repletion, increasing the potassium concentration in the regular IV infusions I had been receiving for several years due to POTS-related chronic dehydration and hypotension. Since some specialists I met characterized these infusions as ‘just salt water’, please allow me to state for the record: IV potassium hurts. Due to my soft tissue fragility, my veins could only handle IV infusions at a slow rate, so each infusion spanned a minimum of twelve hours, depending on how many tries it took to place my IV – sixteen attempts was my record. I would writhe with pain and nausea as the fiery potassium infused, and even with my slow rate, the relatively sudden shift in fluid balance would send me into shock. Over time, the infusions aggravated my delicate veins more and more, prompting recurrent venous rupture, IV infiltration, and horrific-looking hematomas. After multiple harrowing stays in cardiac critical care, I prepared to say goodbye to my loved ones.
All the while, in addition to trying to understand the short-term causes of my hypokalemia, we were still searching for answers to explain my overall decline in health. Were these new complications part of a novel form of Ehlers-Danlos syndrome, a secondary condition, or something else altogether? In an incredible stroke of luck, I serendipitously crossed paths with a geneticist Dr. ‘N’, who took me on as a regular patient and teamed up with my other doctors to coordinate my care. He noted that it was unusual for patients with such severe cases of EDS to have no family history of the condition, and speculated that my mutation was either a rare de novo or recessive variant, a suspicion we could confirm with trio exome testing. My parents, who have been endlessly loving and supportive every day of my life, were genuinely thrilled to be able to help, and I think we all found some sense of poetic justice in using their respective health to better understand my illness. When I met with Dr. N again after he had completed the sequencing and analysis, he grinned gleefully as he described narrowing down my candidate genes to a likely culprit. That gene, CACNA1H, represented hope. We didn’t have any solid answers yet, but we knew more of the right questions to ask. Receiving the genetic diagnosis and finally starting to learn why my body is its weird, anomalous self unleashed a world of possibility that I never would have dared to imagine on my own. That hope is one of two main reasons why I am able to keep going, almost a decade after I thought I was a goner.
The other reason is, of course, ‘my people’, to whom I can only offer my most profound gratitude. Doctors, you need to know that community is as vital to your patients’ health as any medication. My family, my friends, and my medical team make me want to keep trying on the inevitable days when I cannot find the motivation within myself. My family has not only graced me with boundless love and every kind of care, but endeavored to provide all of the resources I need, a gift that I do not take lightly. They have steadfastly powered through the drudgery that comes with having a critically ill child. As for my friends, I never could have imagined it would be possible to maintain such beautiful friendships while being as sick as I am. Inspiration matters. I could not have braved these ordeals without them, nor without my broader medical team, comprised of the nurses, techs, medical assistants, pharmacists, and schedulers who execute each plan and whose (frequently underappreciated) hard work often represents the line between life and death. Finally, as for my doctors: they transcend words. Since I am not a conventional patient with a conventional condition, it makes sense that my doctors and I have had to forge a new kind of relationship: part doctor/patient, part teacher/student, part mentor/mentee, part friends, and part mutual cheerleaders. I know that doctors and patients are ‘supposed’ to maintain a polite, professional distance, but I tend to believe that my close relationships with my doctors represent the primary key to our success. My doctors don’t only bring their expertise to the table, but the full force of their curiosity and wisdom and our shared personal motivation. Whether they are sending me journal articles to consider or silly texts to make me laugh, I know they are as committed as I am. The truth is that being sick is a painful, dehumanizing business, and participating in my own care as a colleague is empowering in a way I could never begin to describe. I cannot know if I will be able to learn my way out of my illness, but I have to try; I now have a scrap of hope, and the work is always unquestionably interesting.
Learning points.
Mutations of CACNA1H show phenotypic variance and are associated with different forms of primary aldosteronism (PA), including aldosterone-producing adenoma, familial hyperaldosteronism, early onset PA, and, as described here, PA without hypertension.
Primary aldosteronism should be suspected in patients with refractory hypokalaemia, regardless of the presence of hypertension.
Gene sequencing, particularly whole exome sequencing, can serve as a useful diagnostic tool in cases of enigmatic conditions, including common diseases with rare presentations.
Footnotes
Contributors: MP and RLN initiated this project and were involved in the clinical management. MP is the guarantor. RLN and EP-R were responsible for the genetic testing, including laboratory testing of the mutated calcium channel. KW wrote the majority of the case report, with all authors contributing to the revision process.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Correction notice: This article has been corrected since it was published Online First, patient’s perspective has been added to this article.
Patient consent for publication: Obtained.
References
- 1. Mosso L, Carvajal C, González A, et al. Primary aldosteronism and hypertensive disease. Hypertension 2003;42:161–5. 10.1161/01.HYP.0000079505.25750.11 [DOI] [PubMed] [Google Scholar]
- 2. Monticone S, Burrello J, Tizzani D, et al. Prevalence and Clinical Manifestations of Primary Aldosteronism Encountered in Primary Care Practice. J Am Coll Cardiol 2017;69:1811–20. 10.1016/j.jacc.2017.01.052 [DOI] [PubMed] [Google Scholar]
- 3. Litynski M. [Hypertension caused by tumors of the adrenal cortex]. Pol Tyg Lek 1953;8:204–8. [PubMed] [Google Scholar]
- 4. Conn JW. The evolution of primary aldosteronism: 1954-1967. Harvey Lect 1966;62:257–91. [PubMed] [Google Scholar]
- 5. Bravo EL, Tarazi RC, Dustan HP, et al. The changing clinical spectrum of primary aldosteronism. Am J Med 1983;74:641–51. 10.1016/0002-9343(83)91022-7 [DOI] [PubMed] [Google Scholar]
- 6. Vaidya A, Mulatero P, Baudrand R, et al. The Expanding Spectrum of Primary Aldosteronism: Implications for Diagnosis, Pathogenesis, and Treatment. Endocr Rev 2018;39:1057–88. 10.1210/er.2018-00139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mulatero P, Stowasser M, Loh KC, et al. Increased diagnosis of primary aldosteronism, including surgically correctable forms, in centers from five continents. J Clin Endocrinol Metab 2004;89:1045–50. 10.1210/jc.2003-031337 [DOI] [PubMed] [Google Scholar]
- 8. Baudrand R, Guarda FJ, Fardella C, et al. Continuum of Renin-Independent Aldosteronism in Normotension. Hypertension 2017;69:950–6. 10.1161/HYPERTENSIONAHA.116.08952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Markou A, Pappa T, Kaltsas G, et al. Evidence of primary aldosteronism in a predominantly female cohort of normotensive individuals: a very high odds ratio for progression into arterial hypertension. J Clin Endocrinol Metab 2013;98:1409–16. 10.1210/jc.2012-3353 [DOI] [PubMed] [Google Scholar]
- 10. Mattsson C, Young WF. Primary aldosteronism: diagnostic and treatment strategies. Nat Clin Pract Nephrol 2006;2:198–208. quiz, 1 p following 230 10.1038/ncpneph0151 [DOI] [PubMed] [Google Scholar]
- 11. Prada ETA, Burrello J, Reincke M, et al. Old and New Concepts in the Molecular Pathogenesis of Primary Aldosteronism. Hypertension 2017;70:875–81. 10.1161/HYPERTENSIONAHA.117.10111 [DOI] [PubMed] [Google Scholar]
- 12. Zennaro MC, Boulkroun S, Fernandes-Rosa F. Genetic Causes of Functional Adrenocortical Adenomas. Endocr Rev 2017;38:516–37. 10.1210/er.2017-00189 [DOI] [PubMed] [Google Scholar]
- 13. Lenzini L, Prisco S, Caroccia B, et al. Saga of Familial Hyperaldosteronism: Yet a New Channel. Hypertension 2018;71:1010–4. 10.1161/HYPERTENSIONAHA.118.11150 [DOI] [PubMed] [Google Scholar]
- 14. Karczewski KJ, Francioli LC, Tiao G, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of- function intolerance across human protein-coding genes.
- 15. Liu H, Enyeart JA, Enyeart JJ. ACTH induces Cav3.2 current and mRNA by cAMP-dependent and cAMP-independent mechanisms. J Biol Chem 2010;285:20040–50. 10.1074/jbc.M110.104190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gilbert S, Weiner D, Bomback A, et al. Nation Kidney Foundation’s Primer On Kidney Diseases. Philadelphia, PA: Elsevier, Inc, 2018:97–106. [Google Scholar]
- 17. Kronenberg H, Melmed S, Polonsky K, et al. Williams Textbook of Endocrinology. 11th ed Philadelphia, PA: Saunders, 2008. [Google Scholar]
- 18. Mustafa HI, Garland EM, Biaggioni I, et al. Abnormalities of angiotensin regulation in postural tachycardia syndrome. Heart Rhythm 2011;8:422–8. 10.1016/j.hrthm.2010.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Raj SR, Biaggioni I, Yamhure PC, et al. Renin-aldosterone paradox and perturbed blood volume regulation underlying postural tachycardia syndrome. Circulation 2005;111:1574–82. 10.1161/01.CIR.0000160356.97313.5D [DOI] [PubMed] [Google Scholar]
- 20. Funder JW, Carey RM, Mantero F, et al. The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2016;101:1889–916. 10.1210/jc.2015-4061 [DOI] [PubMed] [Google Scholar]
- 21. Scholl UI, Stölting G, Nelson-Williams C, et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife 2015;4:e06315 10.7554/eLife.06315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Daniil G, Fernandes-Rosa FL, Chemin J, et al. CACNA1H Mutations Are Associated With Different Forms of Primary Aldosteronism. EBioMedicine 2016;13:225–36. 10.1016/j.ebiom.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]