

Abstract
Background
Tramadol is often prescribed to treat pain and associated physical disability in osteoarthritis (OA). Due to the pharmacologic mechanism of tramadol, it may lead to fewer associated adverse effects (i.e. gastrointestinal bleeding or renal problems) compared to non‐steroidal anti‐inflammatory drugs (NSAIDs). This is an update of a Cochrane Review originally published in 2006.
Objectives
To determine the benefits and harms of oral tramadol or tramadol combined with acetaminophen or NSAIDs in people with osteoarthritis.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE and Embase databases, as well as the US National Institutes of Health and World Health Organization trial registries up to February 2018. We searched the LILACS database up to August 2015.
Selection criteria
We included randomized controlled trials (RCTs) that evaluated the effect of tramadol, or tramadol in combination with acetaminophen (paracetamol) or NSAIDs versus placebo or any comparator in people with osteoarthritis.
Data collection and analysis
We used standard methodologic procedures expected by Cochrane.
Main results
We included 22 RCTs (11 more than the previous review) of which 21 RCTs were included in meta‐analyses for 3871 participants randomized to tramadol alone or tramadol in combination with another analgesic and 2625 participants randomized to placebo or active control. Seventeen studies evaluated tramadol alone and five evaluated tramadol plus acetaminophen. Thirteen studies used placebo controls and eleven studies used active controls (two trials had both placebo and active arms). The dose of tramadol ranged from 37.5 mg to 400 mg daily; all doses were pooled. Most trials were multicenter with a mean duration of two months. Participants were predominantly women with hip or knee osteoarthritis, with a mean age of 63 years and moderate to severe pain. There was a high risk of selection bias as only four trials reported both adequate sequence generation and allocation concealment. There was a low risk for performance bias as most studies blinded participants. There was a high risk of attrition bias as 10/22 trials showed incomplete outcome data. Most of the trials were funded by the pharmaceutical industry.
Moderate quality evidence (downgraded due to risk of bias) indicated that tramadol alone and in combination with acetaminophen had no important benefit on pain reduction compared to placebo control (tramadol alone: 4% absolute improvement, 95% confidence interval (CI) 3% to 5%; 8 studies, 3972 participants; tramadol in combination with acetaminophen: 4% absolute improvement, 95% CI 2% to 6%; 2 studies, 614 participants).
Fifteen out of 100 people in the tramadol group improved by 20% (which corresponded to a clinically important difference in pain) compared to 10/100 in the placebo group (5% absolute improvement). Twelve out of 100 people improved by 20% in the tramadol in combination with acetaminophen group compared to 7/100 in the placebo group (5% absolute improvement).
Moderate quality evidence (downgraded due to risk of bias) indicated that tramadol alone and in combination with acetaminophen led to no important benefit in physical function compared to placebo (tramadol alone: 4% absolute improvement, 95% CI 2% to 6%; 5 studies, 2550 participants; tramadol in combination with acetaminophen: 4% absolute improvement, 95% CI 2% to 7%; 2 studies, 614 participants).
Twenty‐one out of 100 people in the tramadol group improved by 20% (which corresponded to a clinically important difference in physical function) compared to 16/100 in the placebo group (5% absolute improvement). Fifteen out of 100 people improved by 20% in the tramadol in combination with acetaminophen group compared to 10/100 in the placebo group (5% absolute improvement).
Moderate quality evidence (downgraded due to risk of bias) indicated that, compared to placebo, there was a greater risk of developing adverse events with tramadol alone (risk ratio (RR) 1.34, 95% CI 1.24 to 1.46; 4 studies, 2039 participants) and tramadol in combination with acetaminophen compared to placebo (RR 1.91, 95% CI 1.32 to 2.76; 1 study, 308 participants). This corresponded to a 17% increase (95% CI 12% to 23%) with tramadol alone and 22% increase (95% CI 8% to 41%) with tramadol in combination with acetaminophen.
The three most frequent adverse events were nausea, dizziness and tiredness. Moderate quality evidence (downgraded due to risk of bias) indicated that there was a greater risk of withdrawing from the study because of adverse events with tramadol alone compared to placebo (RR 2.64, 95% CI 2.17 to 3.20; 9 studies, 4533 participants), which corresponded to a 12% increase (95% CI 9% to 16%).
Low quality evidence (downgraded due to risk of bias and inconsistency) indicated that there was a greater risk of withdrawing from the study because of adverse events with tramadol in combination with acetaminophen compared to placebo (RR 2.78, 95% CI 1.50 to 5.16; 2 studies, 614 participants), which corresponded to a 8% absolute improvement (95% CI 2% to 19%).
Low quality evidence (downgraded due to risk of bias and imprecision) indicated that there was a greater risk of developing serious adverse events with tramadol alone compared to placebo (110/2459 participants with tramadol compared to 22/1153 participants with placebo; RR 1.78, 95% CI 1.11 to 2.84; 7 studies, 3612 participants), which corresponded to a 1% increase (95% CI 0% to 4%). There were no serious adverse events reported in one small study (15 participants) of tramadol with acetaminophen compared to placebo.
Authors' conclusions
Moderate quality evidence indicates that compared to placebo, tramadol alone or in combination with acetaminophen probably has no important benefit on mean pain or function in people with osteoarthritis, although slightly more people in the tramadol group report an important improvement (defined as 20% or more). Moderate quality evidence shows that adverse events probably cause substantially more participants to stop taking tramadol. The increase in serious adverse events with tramadol is less certain, due to the small number of events.
Plain language summary
Tramadol for osteoarthritis
This summary of a Cochrane Review presents what we know from research about the benefits and harms of tramadol (a pain reliever) for treating osteoarthritis (OA). We examined the published research up to 1 February 2018 and found 22 studies involving 3871 people taking tramadol and 2625 people in a comparator group. Compared with placebo (dummy treatment), moderate quality evidence showed that taking tramadol for up to three months had no important benefit on mean pain or function, although slightly more people in the tramadol group reported an important improvement (defined as 20% or more). Also, people may have had more side effects that them to stop taking it, such as nausea, vomiting, dizziness, constipation, tiredness and headache. We were less certain of the risk of serious effects due to the small number of events. Most of the trials were funded by the pharmaceutical industry.
What is osteoarthritis and what is tramadol?
OA is a disease of the joints, such as the knee or hip. When the joint loses cartilage, the bone grows to try and repair the damage. Instead of making things better, the bone grows abnormally and makes things worse. For example, the bone can make the joint painful and unstable. This can affect physical function or ability to use the knee.
Tramadol is an opioid used to treat OA. Unlike other pain relievers such as non‐steroidal anti‐inflammatory drugs (NSAIDs), it does not cause bleeding in the stomach and intestines, or kidney problems. It also does not affect the cartilage at the end of the bones. However, tramadol may not decrease swelling.
What are the results of this review?
People in the 22 included trials took various daily doses of tramadol or a placebo, an NSAID or a different pain reliever. Most of them were women, with an average age of 63 years, and with moderate to severe pain. The length of the studies ranged from one week to three months. The results below are for tramadol alone compared to placebo. There were similar results for tramadol in combination with acetaminophen.
Pain (0 to 100 visual analog scale (VAS); lower scores mean less pain)
People who took tramadol alone rated their pain to be four points lower than placebo (4% absolute improvement). People who took tramadol alone rated their pain to be 50.3; people who took a placebo rated their pain to be 54.3.
Ten percent of people who took placebo had a clinically important improvement (at least 20%) in pain and 15% who took tramadol group had a clinically important improvement (5% more people).
Physical function (Western Ontario and McMaster Universities Arthritis Index (WOMAC) 0 to 1700 scale; lower scores mean better physical function)
People who took tramadol alone rated their physical function to be 68 points lower than placebo (4% absolute improvement). People who took tramadol alone rated their physical function to be 991; people who took placebo rated their physical function to be 1059.
Twenty‐one percent of people who took tramadol had a clinically important improvement in physical function and 16% of people who took placebo had a clinically important improvement (5% more people).
Total side effects
Sixty‐six out of 100 people may have had side effects when taking tramadol alone compared to 49 out of 100 people when taking a placebo (17% more people).
Withdrawals from study due to side effects
Nineteen out of 100 people withdrew from the study because of side effects when taking tramadol alone compared to seven out of 100 people when taking a placebo (12% more people).
Serious side effects Three out of 100 people had serious side effects when taking tramadol alone compared to two out of 100 people when taking a placebo (1% more people).
Summary of findings
Background
Description of the condition
Osteoarthritis (OA) is a disease characterized by joint pain, stiffness, distortion of joint architecture and functional limitations (Zhang 2008). It accounts for a substantial number of healthcare visits and costs across the world (Murray 2012).
OA, also known as degenerative arthritis, is one of the most frequent disorders and is the most common cause of disability in older adults (Lesnoff‐Caravaglia 2007). It frequently affects the hands, feet, and large weight‐bearing joints such as the hips and the knees (Buckwalter 2004). The prevalence of OA increases with age, since the ability of the articular cartilage to heal decreases, especially in people aged 50 years and older (Cross 2014; Lawrence 2008).
The enzymatic and mechanical breakdown of the matrix of the joint cartilage, and the cartilage's decreased capacity for regeneration are key features of the pathophysiology of OA. In OA, an excessive amount of proteases such as nitric oxide and other inflammatory cytokines are produced by chondrocytes (Lammert 2014). These mediators cause cellular injury, inhibit cartilage synthesis and render the chondrocytes susceptible to apoptosis. These inflammatory phenomena, in addition to promoting cartilage damage, stimulate A delta and C fibers in the synovium and surrounding tissues. This neural stimulation leads to peripheral and central sensitization, and chronic pain (Kean 2004).
Pain is the most common symptom of OA, and as pain levels rise, people experience a reduced range of motion and increasing disability (Bjordal 2004; Dieppe 2005). The pain and function limitations substantially reduce the quality of life of people with OA (Kean 2004). People with OA have a lower quality of life than people with gastrointestinal, cardiovascular or chronic respiratory illnesses (Reginster 2002).
Description of the intervention
The treatment goals for OA are to reduce pain and to maintain or improve (or both) functional status and quality of life (ACR 2000; Pendleton 2000; Tannenbaum 2000). Several clinical practice guidelines recommend non‐pharmacologic and pharmacologic therapies for the management of OA (Hochberg 2012; Jevsevar 2013; Richmond 2010; Zhang 2008).
Non‐pharmacologic therapies include weight reduction in obese people, physical therapy (for muscle strengthening), exercise and occupational therapy (e.g. training in the use of devices to assist ambulation) (Hochberg 2012; Jevsevar 2013; NCCCC 2008; Richmond 2010; Zhang 2008).
A wide variety of pharmacologic therapies are recommended by the American College of Rheumatology (ACR), the American Academy of Orthopaedic Surgeons (AAOS) and the Osteoarthritis Research Society International (OARSI) (Hochberg 2012; Richmond 2010; Zhang 2008), and are used to treat OA including analgesics such as tramadol (Hochberg 2012; Zhang 2008), non‐steroidal anti‐inflammatory drugs (NSAIDs) (Hochberg 2012; Jevsevar 2013; Richmond 2010), and acetaminophen (paracetamol; NCCCC 2008; Zhang 2008). Acetaminophen, although not associated with an increased risk of gastrointestinal events, is less effective than NSAIDs in reducing pain (NCCCC 2008; Towheed 2006).
NSAIDs are the cornerstone of pharmacologic therapy for the management of OA, relieving symptoms such as pain (Towheed 2006). However, their use is associated with gastrointestinal (Towheed 2006), cardiac (Zhang 2008), and renal problems (NCCCC 2008), especially in elderly people (Pelletier 2016). There has been ongoing debate that cyclo‐oxygenase‐2 (COX‐2)‐selective NSAIDs have increased cardiac adverse events compared to traditional NSAIDs. However, one non‐inferiority trial published in the New England Journal of Medicine demonstrated that the COX‐2‐selective NSAID, celecoxib, was not inferior to naproxen or ibuprofen in terms of cardiac adverse events (Nissen 2016).
Tramadol has been increasingly used for pain relief in people with OA due to having potentially fewer adverse events than with NSAIDs (Cepeda 2006). The ACR and the OARSI recommend tramadol in managing OA pain (Hochberg 2012; Zhang 2008), because in contrast to NSAIDs, it does not produce gastrointestinal bleeding, renal (Zhang 2004) or cardiovascular problems (Pelletier 2016). Tramadol is an opioid that acts on the neurotransmission of norepinephrine and serotonin and modifies the transmission of pain impulses (Pelletier 2016). This dual action makes tramadol an attractive option, although dependency is a concern, as with all opiates.
Although the analgesic effect of tramadol for acute and neuropathic pain has been established, there are few systematic reviews that evaluate the benefits of tramadol for OA. One systematic review included 11 trials comparing tramadol to placebo and other active controls (Cepeda 2006). Another systematic review included 18 trials comparing opioids including tramadol to placebo (Avouac 2007). These systematic reviews demonstrated a significant decrease in pain as well as benefits on physical function.
How the intervention might work
The mechanism for benefits of tramadol in OA is unclear: tramadol lacks peripheral action (i.e. it has no local effects on the joints) and its benefits may decline with chronic use (i.e. development of tolerance), as part of its action is opioid‐related. Nonetheless, the central action of tramadol could be of great benefit as this action could decrease the central neuronal sensitization produced by the persistent nociceptive peripheral input (Jett 1997). In addition, tolerance may not be a problem in this people with OA, since systematic reviews have shown that 44% of participants prescribed opioids for chronic non‐cancer pain continued to take opioids for up to 24 months (Kalso 2004).
Why it is important to do this review
Clinical studies are the best way to determine whether people with OA benefit from using tramadol. This review examined the clinical benefit and harms of tramadol for people with OA.
Objectives
To determine the benefits and harms of oral tramadol or tramadol combined with acetaminophen or NSAIDs in people with osteoarthritis.
Methods
Criteria for considering studies for this review
Types of studies
We included randomized controlled trials (RCTs). Published studies, as well as unpublished studies were eligible.
Types of participants
We included studies in adults (i.e.18 years and older) with OA affecting any joints. We included studies that evaluated participants who met one of the following: 1. the ACR clinical criteria for OA; and 2. radiographic evidence of OA. We excluded studies that evaluated other types of arthritis (e.g. rheumatoid arthritis, non‐osteoarthritic joint pain or back pain) or that did not provide data specific to participants with OA. We also excluded studies of tramadol for postoperative pain.
Types of interventions
We included studies that compared tramadol (with or without acetaminophen or NSAIDs) with either a placebo or an active treatment.
Types of outcome measures
We included studies that reported the effect of tramadol on pain intensity, physical function and adverse events of tramadol. If a trial reported many time points, we recorded the last time point.
Major outcomes
The major outcomes of interest were based on the recommendations for outcomes in OA trials (Altman 1996; Bellamy 1997).
Pain.
Physical function.
Number of participants experiencing any adverse event.
Number of participants who withdrew due to adverse events.
Number of participants experiencing any serious adverse events.
For trials which assessed results using more than one pain scale, we used the hierarchy of pain‐related outcomes described by Jüni 2006 by extracting data on the pain scale that was highest on this list:
global pain;
pain on walking;
WOMAC Osteoarthritis Index pain subscore;
composite pain scores other than WOMAC;
pain on activities other than walking;
rest pain or pain during the night;
WOMAC Global Algofunctional score;
Lequesne Osteoarthritis Index global score;
other algofunctional scale;
participant's global assessment;
physician's global assessment.
For trials which assessed results from more than one physical function scale, we used the same approach as noted above, using the following hierarchy:
global disability score;
walking disability;
WOMAC Disability subscore;
composite disability scores other than WOMAC;
disability other than walking;
WOMAC Global Scale;
Lequesne Osteoarthritis Index global score;
other algofunctional scale;
participant's global assessment;
physician's global assessment.
If a study reported pain or function outcomes at several time points, we extracted the measure at the end of the treatment period.
Minor outcomes
Symptoms of opioid dependence, such as craving or physical withdrawal symptoms.
Search methods for identification of studies
Electronic searches
Electronic databases
We searched the following databases:
the Cochrane Central Register of Controlled Trials (CENTRAL) in the Cochrane Library; 2018, Issue 1;
Ovid MEDLINE 1966 to 1 February 2018;
Ovid Embase 1980 to 1 February 2018;
LILACS 1982 to 13 August 2015.
Trial registries
US National Institutes of Health trial registry up to February 2018 (ClinicalTrials.gov).
World Health Organization trial registry up to February 2018 (apps.who.int/trialsearch).
Search terms
For the identification of the studies in MEDLINE, we used the MeSH/EMTREE terms: Appendix 1.
There were no language restrictions. We translated non‐English language articles and assessed them. Where applicable, we communicated with the authors to obtain information not presented or that was unclear in the manuscripts.
For each of the other databases, we based search strategies on the search strategy developed for MEDLINE, but revised appropriately. We searched bibliographies from all retrieved articles for additional studies.
Data collection and analysis
Selection of studies
Two review authors (KTA and either CC or JV) independently screened titles and abstracts for inclusion of all potentially relevant studies identified by the search. We retrieved the full text of all articles in which the record or abstract referred to a trial of tramadol and OA.
Two review authors (KTA and either CC or JV) independently screened the full text and identified studies for inclusion, and identified and recorded reasons for exclusion of the ineligible studies. We resolved disagreements through discussion, and, if required, by a third review author (VW, PT). We identified and excluded duplicates and collated multiple reports of the same study so that each study, rather than each report, was the unit of interest in the review. We recorded the selection process in sufficient detail to complete a PRISMA flow diagram (prisma‐statement.org/PRISMAStatement/Default.aspx).
Data extraction and management
Two review authors (KTA and either CC or JV) independently extracted information from each study using a standardized, piloted extraction form accompanied by a codebook. We resolved disagreements by discussion, and, if necessary, consulted a third review author (VW).
We extracted the following study characteristics.
Methods: study design, total duration of study, study setting and withdrawals.
Participants: number, mean age, age range, sex, disease duration, types of joints affected, inclusion criteria and exclusion criteria.
Interventions: generic and trade name of the intervention, type of control used, dosage, frequency, route of administration and duration of treatment.
Outcomes: major and minor outcomes specified and collected, and time points reported.
Characteristics of the design of the trial as outlined in the Assessment of risk of bias in included studies section.
Notes: type and source of financial support for trial, and notable declarations of interest of trial authors.
When necessary, we approximated means and measures of dispersion from figures in the reports. For cross‐over trials, we extracted data from the first period only to include in the meta‐analyses. Whenever possible, we used results from an intention‐to‐treat (ITT) analysis. If effect sizes could not be calculated, we contacted the authors for additional data. One review author (TEH) transferred data into Review Manager 5 (Review Manager 2014). We double‐checked that data were entered correctly by comparing the data presented in the systematic review with the study reports.
Assessment of risk of bias in included studies
The articles that fulfilled the inclusion criteria underwent quality appraisal. Two review authors (two of the following authors: JB, JV, NA, CC, TEH, LM) independently assessed risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We resolved disagreements through discussion, and by a third review author if a consensus could not be reached (KTA, JP, VW, AWSR).
We assessed the following risk of bias domains for RCTs:
random sequence generation;
allocation concealment;
blinding of participants and personnel;
blinding of outcome assessment;
incomplete outcome data;
selective outcome reporting;
other bias.
We graded each potential source of bias as high, low or unclear risk, and provided a quote from the study report together with a justification for our judgment in the 'Risk of bias' table (see Characteristics of included studies table). Randomization was adequate if it resulted in an unpredictable allocation schedule. Authors had to note that they used a random component (e.g. random assignments generated by a computer).
Allocation concealment was adequate if participants and investigators responsible for participant selection were unable to suspect before allocation which treatment was used. Authors had to indicate that they employed central randomization by a third party or used sequentially numbered, opaque, sealed envelopes.
Blinding of participants was adequate if experimental and control preparations were explicitly described as indistinguishable or if a study used a double‐dummy technique. Blinding of study personnel was adequate if authors explicitly stated that investigators were blinded. Blinding of outcome assessors was adequate if blinding of participants was considered adequate for self‐reported outcome measures. Blinding of outcome assessors was considered unclear if authors only explicitly stated that participants were blinded for outcome measures that were not self‐reported, as personnel interacting with participants may not have been blinded and may have influenced outcome assessment.
Outcome data were considered complete if reasons for any losses to follow‐up, treatment withdrawals or trial group changes were explained by the authors and the reasons for those occurrences were unlikely to be connected with their subsequent outcome. In addition, the occurrence of missing data had to be balanced between intervention and control groups.
Outcome reporting was considered complete if the outcomes listed in the protocol matched those reported in the article. Risk of bias for outcome reporting was considered unclear if the protocol was not available.
An article was deemed to have demonstrated a low risk of bias for 'other biases' if no other internal biases were identified. Analyses were considered adequate if all randomized participants were included in the analysis according to the ITT principle.
For the cross‐over RCTs, we answered the following questions to assess the risk of bias (see other bias section in the Assessment of risk of bias in included studies).
Was use of a cross‐over design appropriate?
Was it clear that the order of receiving treatments was randomized?
Could it be assumed that the trial was not biased from carry‐over effects?
Were unbiased data available?
Measures of treatment effect
Major outcomes
1. Pain
We extracted the mean and standard deviation (SD) of pain in each study group after treatment, and calculated the mean differences (MD). In cases where the studies reported the difference in pain with no measure of dispersion, we estimated the standard error (SE) of the difference from the P value and the number of participants in each group, as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). To pool the data, we chose generic inverse variance fixed‐effect models a priori. We also used random‐effects models to verify if the two models gave consistent results. We then transformed the MD between groups and the SE into a standardized mean difference (SMD) and the SE of that SMD. We calculated the corresponding number needed to treat for an additional beneficial outcome (NNTB) and the mean percentage of participants who acquired a minimally clinically important difference (MCID) using the Wells calculator (available at the CMSG Editorial office musculoskeletal.cochrane.org/). The Wells calculator obtains the NNTB by taking the reciprocal of the net proportion of the population benefiting from an intervention (1/P(b)). The net proportion benefiting is dependent on the proportions benefiting in control and treatment groups and the proportion worsening in control and treatment groups which are in turn derived from the normal distribution of the difference between the minimally important difference and the effect size of a trial (SMD) (Norman 2001).
We also calculated the risk ratio (RR) by dividing the percentage of participants who improved in the treatment group by the mean percentage of participants who improved in the control group (corresponding risk/assumed risk) and 95% confidence intervals (CI). We added information on the NNTB, the mean percentage of participants who acquire an MCID and the RR in the footnotes of the 'Summary of findings' tables. The MCID was 20% of a pain scale (Pham 2003; Tubach 2005). It is important to note that when results are not statistically significant but CIs are wide, results may be explained by uncertainty. Thus, it does not preclude the intervention from having an effect if more research was conducted.
2. Physical function
We extracted the mean and SD in each study arm after treatment and calculated the MDs and 95% CI. If studies reported the MD in physical function between treatments without a measure of dispersion, we estimated the SE of the difference from the P value and the number of participants in each group, as described in the Chapter 7 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). To pool the data, we chose generic inverse variance fixed‐effect models a priori. We also used random‐effects models to verify if the two models gave consistent results. We then transformed the MD between groups and the SE into a SMD and the SE of that SMD. We calculated the corresponding NNTB and the mean percentage of participants who acquired an MCID using the Wells calculator (available at the CMSG Editorial office musculoskeletal.cochrane.org/). We also calculated the RR by dividing the percentage of participants who improved in the treatment group by the mean percentage of participants who improved in the control group (corresponding risk/assumed risk). We added information on the NNTB, the mean percentage of participants who acquired an MCID and the RR in the footnotes of the 'Summary of findings' tables. The MCID was 20% for physical function (Pham 2003; Tubach 2005).
3. Harms of tramadol
To evaluate the harms of tramadol, we extracted the proportion of participants who developed any adverse events, withdrawals due to adverse events and serious adverse events. We relied upon the definitions used by authors of the trials. We calculated the RR and corresponding number needed to treat for an additional harmful effect (NNTH), with 95% CIs.
Minor outcomes
1. Withdrawal symptoms and propensity for abuse
To evaluate the data pertaining to withdrawal symptoms, we extracted the proportion of participants who developed these symptoms and the proportion of participants who developed a propensity for abuse. We calculated the RR and corresponding NNTH, with 95% CIs.
Unit of analysis issues
Where multiple trial arms were reported in a single trial, we included only the relevant arms.
Dealing with missing data
We contacted investigators or study sponsors in order to verify key study characteristics and obtain missing numerical outcome data where possible (e.g. when a study was identified as an abstract only or when data were not available for all participants).
For dichotomous outcomes, we calculated the withdrawal rate using the number of participants randomized in the group as the denominator.
For continuous outcomes, we calculated the MD or SMD based on the number of participants analyzed at that time point. If the number of participants analyzed was not presented for each time point, we used the number of randomized participants in each group at baseline.
Where possible, we computed missing SDs from other statistics such as SEs, CIs or P values, according to the methods recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Chapter 7; Higgins 2011).
Assessment of heterogeneity
To evaluate statistical heterogeneity, we performed a visual inspection of the forest plot to assess differences in results between the studies when there was a sufficient number of trials. We also used the I² and Chi² statistical tests (Higgins 2003). We assessed clinical heterogeneity in terms of participants, interventions, outcomes, study characteristics and setting of the included trials. As recommended in theCochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), the interpretation of an I² value of 0% to 40% might not be important; 30% to 60% may represent moderate heterogeneity; 50% to 90% may represent substantial heterogeneity and 75% to 100% represents considerable heterogeneity. The Chi² test was interpreted where a P ≤ 0.05 indicated evidence of statistical heterogeneity.
Assessment of reporting biases
We created and examined funnel plots to explore possible small‐study biases when there was a sufficient number of trials. To assess outcome reporting bias, we verified trial protocols against published reports.
Data synthesis
We summarized continuous outcomes using SMDs and expressed dichotomous outcomes as RRs. We used standard inverse‐variance fixed‐effect meta‐analyses to combine the trials (DerSimonian 1986). We also used random‐effects models to verify if the two models gave consistent results.SMD were also translated into MDs on commonly used scales.
GRADE and 'Summary of findings' tables
We created 'Summary of findings' tables for the major outcomes. We added the absolute and relative percent change in the 'Summary of findings' tables.
Two review authors (of: JB, JV, NA, CC, TEH) independently assessed the quality of the evidence. We used the five GRADE considerations (unclear risk of bias, consistency of effect, imprecision, indirectness and publication bias) to assess the quality of a body of evidence as it related to the studies which contributed data to the meta‐analyses for the prespecified outcomes. We reported the quality of evidence as high, moderate, low or very low. Low quality means our confidence in the effect estimate was limited, whereas the moderate quality means we were moderately confident in the effect estimate. We considered the following criteria for upgrading the quality of evidence, if appropriate: large effect, dose‐response gradient and plausible confounding effect. We used methods and recommendations described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011; Schünemann 2011). We used GRADEpro software to prepare the 'Summary of findings' tables (GRADEpro GDT). We justified all decisions to downgrade or upgrade the quality of studies using footnotes and made comments to aid the reader's understanding of the review where necessary. We resolved disagreements by consensus, and by a third review author if a consensus could not be reached (KTA, JP, VW, AWSR).
Subgroup analysis and investigation of heterogeneity
We analyzed placebo‐controlled studies and active‐controlled studies separately. We analyzed studies that evaluated tramadol alone or tramadol plus acetaminophen or NSAIDs together, as the results of these trials were similar.
Interpreting results and reaching conclusions
We followed the guidelines in the Cochrane Handbook for Systematic Reviews of Interventions for interpreting results and distinguished a lack of evidence of effect from a lack of effect (Schünemann 2011).
Results
Description of studies
Results of the search
In the updated search conducted in February 2018, we identified 347 records through database searching and 37 records through other sources. After eliminating duplicates, we screened 372 records. We excluded 332 records after reviewing their titles and abstracts, leaving 40 full‐text articles which were assessed for eligibility. Eighteen articles were then excluded after reviewing the full text of these articles: three articles were not relevant to chronic management of knee or hip OA, or OA was not evaluated separately in these articles; two articles did not evaluate tramadol and 13 were not RCTs. We included 22 articles in this review and we excluded 11 of these from the meta‐analyses due to missing data. Three of these articles were secondary publications of another trial (Gana 2006), with no new outcomes of interest (Florete 2008; Vorsanger 2007), and eight which were reports of protocols for which the full data could not be accessed (see Appendix 2). Thus, we included 11 articles in this review, which represented 11 RCTs in this updated search. When combined with the RCTs from the search of the earlier version of this systematic review (11 RCTs), it amounted to 22 RCTs (see PRISMA flow diagram in Figure 1). However, one of these could not be included in the meta‐analyses because it was a cross‐over trial in which results were presented for all treatment periods combined for all outcomes of interest (Thorne 2008). These data were presented separately in the results.
1.
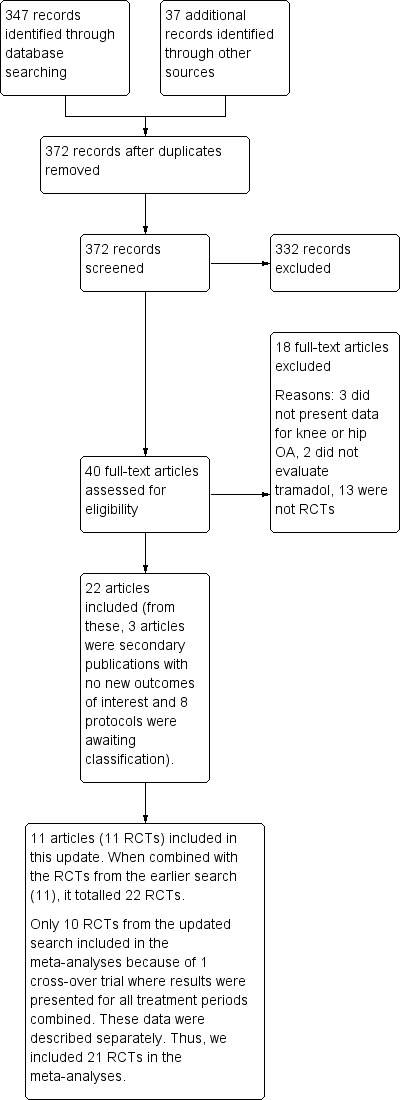
Study flow diagram. OA: osteoarthritis; RCT: randomized controlled trial.
Included studies
We included 22 RCTs in the present update (11 more than the previous review) of which 21 RCTs were included in meta‐analyses for 3871 participants who were randomized to tramadol alone or tramadol in combination with another analgesic and 2625 participants who were randomized to placebo or active‐control (see Characteristics of included studies table for further details). All studies were funded by the pharmaceutical industry with the exception of two (Bianchi 2003; Fujii 2014). All RCTs were parallel in design, with the exceptions of four studies, which used a cross‐over design (Bird 1995; Pavelka 1998; Peeva 2010; Thorne 2008). One study provided no data on the benefits of tramadol, as the aim of the study was to evaluate the tramadol‐sparing effect of naproxen (Schnitzer 1999). One cross‐over study provided data on benefits but this evidence could not be pooled with other studies as results were presented for all treatment periods combined (Peeva 2010). These two studies were included since they provided some data for the evaluation of harms. Only two trials were registered in the ClinicalTrials.gov register (Park 2012; Peeva 2010), although 10 trials included in the meta‐analyses were published after 2005.
Thirteen RCTs used placebo controls (Babul 2004; Burch 2007; DeLemos 2011; Emkey 2004; Fishman 2007; Fleischmann 2001; Gana 2006; Kean 2009; Malonne 2004; Peeva 2010; Schnitzer 1999; Silverfield 2002; Thorne 2008). In 11 studies, the active control was either acetaminophen 1500 mg/day (Bianchi 2003); NSAIDs (i.e. diclofenac 86.9 mg/day (Pavelka 1998), diclofenac 75 mg/day to 100 mg/day (Beaulieu 2008), celecoxib 200 mg/day (DeLemos 2011), loxoprofen 180 mg/day (Fujii 2014), meloxicam 7.5 mg or 15 mg or aceclofenac 100 mg (Park 2012), acetaminophen, indomethacin, brufen, diclofenac, feldene or mefenamate (Wilder‐Smith 2001) (since this group was not randomized, it was excluded from the analyses) and naproxen 500 mg/day (Peeva 2010)); or other opioids (i.e. transdermal fentanyl 25 μg/day to 50 μg/day (Fujii 2014), dihydrocodeine 120 mg/day (Wilder‐Smith 2001), dextropropoxyphene 300 mg/day (Jensen 1994), transdermal buprenorphine 5 μg/day to 20 μg/day (Karlsson 2009), and pentazocine 150 mg/day (Bird 1995)). Two studies used both placebo and active controls (DeLemos 2011; Peeva 2010). Seventeen studies evaluated tramadol alone (Babul 2004; Beaulieu 2008; Bianchi 2003; Bird 1995; Burch 2007; DeLemos 2011; Fishman 2007; Fleischmann 2001; Gana 2006; Jensen 1994; Karlsson 2009; Kean 2009; Malonne 2004; Pavelka 1998; Schnitzer 1999; Thorne 2008; Wilder‐Smith 2001), and five evaluated tramadol plus acetaminophen (Emkey 2004; Fujii 2014; Park 2012; Peeva 2010; Silverfield 2002).
The doses of tramadol used in the studies were variable, ranging from 37.5 mg to 400 mg daily. We assessed whether there were differences in the results depending on dose. Since the results were similar, we decided to pool the doses. All studies evaluated people with symptomatic OA of the hip or knee (or both) with 13 studies including participants with knee or hip OA and nine studies with knee OA. All studies included people aged 18 years or older, and 12 studies included people aged 35 years or older. Participants were predominantly women, with a mean age of 63 years. Most studies included participants with moderate to severe pain, with several studies defining this as a visual analog scale (VAS) score of at least 40 mm on a 100 mm scale. The mean number of participants in the tramadol group was 176 (range 10 to 815) and in the control groups was 119 (range 10 to 405). The mean length of the studies was 54 days (range 3 days to 91 days). Nine studies were 12 weeks long and four studies were eight weeks long; the remainder of the studies varied in duration.
Assessed primary and secondary outcomes
Major outcomes: pain and physical function
A total of 19 RCTs reported major outcomes, such as pain or physical function (or both) outcome of interest (i.e. RCTs with outcomes rated the highest on the hierarchy of outcomes described in the methods) (Babul 2004; Beaulieu 2008; Bianchi 2003; Bird 1995; Burch 2007; DeLemos 2011; Emkey 2004; Fishman 2007; Fleischmann 2001; Fujii 2014; Gana 2006; Jensen 1994; Karlsson 2009; Kean 2009; Malonne 2004; Park 2012; Pavelka 1998; Silverfield 2002; Wilder‐Smith 2001). All these RCTs reported the effect of tramadol on pain. Eleven RCTs reported the effect of tramadol on physical function (Babul 2004; Beaulieu 2008; DeLemos 2011; Emkey 2004; Fleischmann 2001; Gana 2006; Jensen 1994; Kean 2009; Park 2012; Pavelka 1998; Silverfield 2002).
Six RCTs evaluated pain using the WOMAC Pain subscale (Beaulieu 2008; DeLemos 2011; Fishman 2007; Kean 2009; Pavelka 1998; Silverfield 2002). Eight RCTs evaluated pain intensity using a VAS (Babul 2004; Bianchi 2003; Burch 2007; Emkey 2004; Fujii 2014; Gana 2006; Jensen 1994; Malonne 2004). One trial evaluated pain intensity during movement using a 4‐point Likert scale (Wilder‐Smith 2001), and the other trials evaluated pain intensity using a 4‐point Likert scale (Bird 1995; Fleischmann 2001), a numerical rating scale (Park 2012), and the Box scale 11 (Karlsson 2009).
Ten RCTs evaluated physical function using the WOMAC Physical Function subscale (Babul 2004; Beaulieu 2008; DeLemos 2011; Emkey 2004; Fleischmann 2001; Gana 2006; Kean 2009; Park 2012; Silverfield 2002; Thorne 2008). One trial used the WOMAC Total score (Pavelka 1998). One trial used an overall assessment of the therapy scored on a 5‐point Likert scale that was then transformed into a dichotomous scale (Jensen 1994). We used this overall assessment as a proxy for physical function.
Major outcomes: harms
All RCTs assessed harms outcomes, such as any adverse events, withdrawal due to adverse events, and serious adverse events. These included 10 trials assessing the number of participants with any adverse events, 16 trials assessing the number of participants who withdrew because of adverse events and 14 trials assessing the number of participants with serious adverse events.
Minor outcomes: withdrawal symptoms and propensity for abuse
Three trials reported withdrawal symptoms or propensity for abuse, or both (Beaulieu 2008; DeLemos 2011; Gana 2006). Another trial reported that there was no evidence of abuse but did not mention if it was assessed (Emkey 2004).
Excluded studies
In the full‐text screening, we excluded 18 full‐text articles in this update. Three articles did not present data for knee or hip OA, two articles did not evaluate tramadol and 13 did not report RCTs. A list of some of the excluded studies from both abstract/title and full‐text screening phases is shown in Characteristics of excluded studies table.
Risk of bias in included studies
Figure 2 shows the assessment of risk of bias for each included study and Figure 3 shows a summary of the risk of bias across the different types of bias.
2.
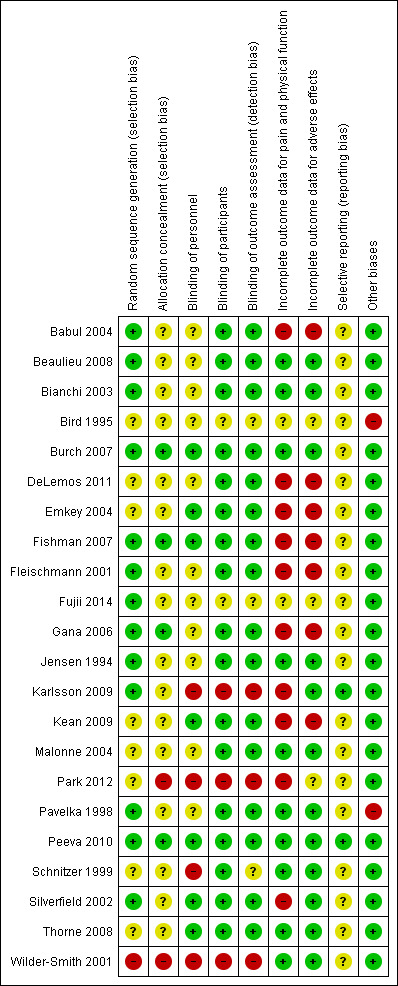
Risk of bias summary: review authors' judgments about each risk of bias item for each included study.
3.
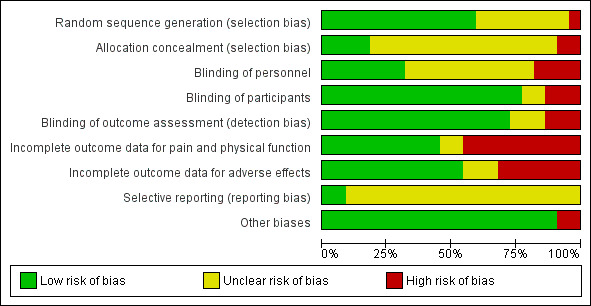
Risk of bias graph: review authors' judgments about each risk of bias item presented as percentages across all included studies.
Allocation
Random sequence generation
Thirteen trials reported adequate sequence generation, eight trials reported unclear sequence generation (Bird 1995; DeLemos 2011; Emkey 2004; Kean 2009; Malonne 2004; Park 2012; Schnitzer 1999; Thorne 2008), and one trial reported inadequate sequence generation (Wilder‐Smith 2001).
Four trials reported adequate allocation concealment (Burch 2007; Fishman 2007; Gana 2006; Peeva 2010), while most others had unclear allocation concealment, except for two trials that had inadequate allocation concealment (Park 2012; Wilder‐Smith 2001).
Blinding
All but five trials were at low risk of bias for blinding of participants and all but six had a low risk of bias for blinding of outcome measures. However, most trials had unclear or high risk of bias for blinding of personnel while seven trials showed a low risk of bias. All but four trials were described as double‐blind (Fujii 2014; Karlsson 2009; Park 2012; Wilder‐Smith 2001). Twelve trials reported the use of indistinguishable interventions to blind participants (Babul 2004; Bianchi 2003; Burch 2007; DeLemos 2011; Emkey 2004; Fleischmann 2001; Gana 2006; Jensen 1994; Malonne 2004; Pavelka 1998; Schnitzer 1999; Silverfield 2002), whereas four trials used double‐dummy techniques (Beaulieu 2008; Fishman 2007; Kean 2009; Peeva 2010). Though described as double‐blind, the authors of two trials did not describe blinding procedures (Bird 1995; Thorne 2008).
Incomplete outcome data
Ten trials were at a low risk of bias for incomplete outcome data for pain and physical function (Beaulieu 2008; Bianchi 2003; Burch 2007; Jensen 1994; Malonne 2004; Pavelka 1998; Peeva 2010; Schnitzer 1999; Thorne 2008; Wilder‐Smith 2001), two were unclear (Bird 1995; Fujii 2014), and the reminder showed a high risk of bias. Twelve trials were at a low risk of bias for incomplete outcome data for adverse events, three were unclear (Bird 1995; Fujii 2014; Park 2012), and the reminder showed a high risk of bias. Eleven trials described their benefits analysis to be according to the ITT principle (Babul 2004; DeLemos 2011; Emkey 2004; Fishman 2007; Fleischmann 2001; Gana 2006; Karlsson 2009; Kean 2009; Park 2012; Silverfield 2002; Thorne 2008), but some of these trials provided numbers for final analyses that were different from the numbers of randomized participants for pain and physical function outcomes. Among studies which did not employ the ITT principle and for which benefits data were usable for the purposes of this review update, exclusion of participants from the analysis of benefits outcomes ranged from 10% to 45% in the experimental groups and from 6% to 60% in the control groups.
Selective reporting
All outcomes mentioned in trials' methods were reported in most studies. However, we could only find the protocol of two trials (Peeva 2010; Park 2012). Most studies reported adverse events that were the most common.
Other potential sources of bias
Some of the cross‐over trials did not appear to use adequate methods (Bird 1995; Pavelka 1998).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7
Summary of findings for the main comparison. Tramadol alone compared with placebo for osteoarthritis.
| Tramadol alone compared with placebo for osteoarthritis | ||||||
|
Patient or population: osteoarthritis of the hip or knee, or both Settings: outpatient clinics Intervention: tramadol alone Comparison: placebo | ||||||
| Outcomesa | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk** | Corresponding risk** | |||||
| Placebo | Tramadol alone | |||||
|
Pain assessed with: 0–100‐mm VAS pain intensity where 0 = no pain Follow‐up: range 1 week to 3 months |
The mean pain was 54.3 points | The mean pain in the intervention group was 4 points lower (3 lower to 5 lower)b | — | 3972 (8 RCTs) | ⊕⊕⊕⊝ Moderatee | Mean pain: 4% absolute improvement (95% CI 3% to 5% improvement),b 7% relative improvement (6% to 9% improvement), SMD –0.25 (95% CI –0.32 to –0.18)f NNTB 13 (95% CI 10 to 18)g A cross‐over study (Thorne 2008), which was not included in the meta‐analyses, showed improvement in pain in the intervention group compared to the placebo group (mean ± SD: 189.0 ± 105.0 versus 230.0 ± 115.4; P = 0.00). |
| 10 out of 100 improved by 20%c | 5 more out of 100 (3 more to 6 more) in the intervention group improved by 20%c | RR 1.50 (95% CI 1.30 to 1.60)d | ||||
|
Physical function assessed with: WOMAC Physical Function (scale 0 to 1700) Follow‐up: range 1 week to 3 months |
The mean physical function was 1059 | The mean physical function in the intervention group was 68 points lower (41 lower to 99 lower)b | — | 2550 (5 RCTs) | ⊕⊕⊕⊝ Moderatee | Mean function: 4% absolute improvement (95% CI 2% to 6% improvement),b 6% relative improvement (95% CI 4% to 9% improvement),f SMD –0.20 (95% CI –0.29 to –0.12), NNTB 13 (95% CI 9 to 21)g A cross‐over study (Thorne 2008), which was not included in the meta‐analyses, showed improvement in physical function in the intervention group compared to the placebo group (mean ± SD: 632.4 ± 361.3 vs 727.4 ± 383.4; P = 0.02). |
| 16 out of 100 improved by 20%c | 5 more out of 100 (3 more to 8 more) in the intervention group improved by 20%c | RR 1.31 (95% CI 1.19 to 1.50)d | ||||
|
Number of participants experiencing any adverse events Follow‐up: range 1 week to 3 months |
492 per 1000 | 659 per 1000 (610 to 718) | RR 1.34 (95% CI 1.24 to 1.46) | 2039 (4 RCTs) | ⊕⊕⊕⊝ Moderatee | 17% absolute worsening (95% CI 12% more to 23% more), 34% relative worsening (95% CI 24% more to 46% more), NNTH 6 (95% CI 5 to 9) A cross‐over study (Thorne 2008), which was not included in the meta‐analyses, showed that there was little or no difference in the total number of adverse events between the intervention group (79.8%) and placebo group (65.9%) (P = 0.08). |
|
Number of participants who withdrew due to adverse events Follow‐up: range 1 week to 3 months |
73 per 1000 | 194 per 1000 (159 to 235) | RR 2.64 (95% CI 2.17 to 3.20) | 4533 (9 RCTs) | ⊕⊕⊕⊝ Moderatee | 12% absolute worsening (95% CI 9% more to 16% more), 164% relative worsening (95% CI 117% more to 220% more), NNTH 9 (95% CI 7 to 12) In a cross‐over study (Thorne 2008), which was not included in the meta‐analyses, 15 participants withdrew after randomization due to adverse events, 12 of which were in the intervention group at the time of withdrawal. |
|
Number of participants experiencing any serious adverse events Follow‐up: range 1 week to 3 months |
19 per 1000 | 34 per 1000 (21 to 54) | RR 1.78 (95% CI 1.11 to 2.84) | 3612 (7 RCTs) | ⊕⊕⊝⊝ Lowe,h | 1% absolute worsening (95% CI 0% more to 4% more), 78% relative worsening (95% CI 11% more to 184% more), NNTH 68 (95% CI 29 to 477) In a cross‐over study (Thorne 2008), which was not included in the meta‐analyses, 1 serious adverse event occurred in a participant in the intervention group. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
**The assumed and the corresponded risk was calculated from the SMD and SE. CI: confidence interval; MCID: minimal clinically important difference; NNTB: number needed to treat for an additional beneficial outcome; NNTH: number needed to treat for an additional harmful outcome;RCT: randomized controlled trial;RR: risk ratio;SD: standard deviation; SE: standard error; SMD: standardized mean difference; WOMAC: Western Ontario and McMaster Universities Arthritis Index. | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aContinuous outcomes summarized using SMDs and SEs, and binary outcomes expressed as RRs. We used standard inverse‐variance fixed‐effect meta‐analysis to combine the trials in Review Manager 5 (Review Manager 2014). bAbsolute improvement on a common scale (e.g. 100 mm, 1700‐point scale) calculated by multiplying the SMD by the SD of the scale (in the control group at baseline) as suggested by the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). cAssumed and corresponding risks calculated from the SMD and SE, with improvement based on an MCID of 20% of the given scale using the Wells calculator (from the Cochrane Musculoskeletal Group Editorial office; musculoskeletal.cochrane.org/). dRR and its 95% CI calculated using the assumed risk of the control group and corresponding risk of the treatment group. The corresponding risk was divided by the assumed risk. eDowngraded one level due to unclear risk of bias (all trials had high or unclear risk of at least one type of bias). fRelative improvement percentage defined as relative to the control group risk at baseline. gNNTB corresponded to the number of participants that needed to be treated to see one participant improve. Improvement defined as reaching an MCID of 20% on the given scale. NNTB calculated using the Wells calculator (from the CMSG Editorial office; musculoskeletal.cochrane.org/ne.org/). hDowngraded one level due to imprecision.
Summary of findings 2. Tramadol in combination with acetaminophen compared with placebo for osteoarthritis.
| Tramadol in combination with acetaminophen compared with placebo for osteoarthritis | ||||||
|
Patient or population: osteoarthritis Settings: outpatient clinics Intervention: tramadol in combination with acetaminophen Comparison: placebo | ||||||
| Outcomesa | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk** | Corresponding risk** | |||||
| Placebo | Tramadol in combination with acetaminophen | |||||
|
Pain assessed with: self‐report VAS pain intensity (scale 0 to 100 where 0 = no pain) Follow‐up: range 10 days to 13 weeks |
The mean pain was 48.3 points | The mean pain in the intervention group was 4 points lower (2 lower to 6 lower)b | — | 614 (2 RCTs) | ⊕⊕⊕⊝ Moderatee |
Mean pain: 4% absolute improvement (95% CI 2% to 6% improvement),b 8% relative improvement (95% CI 4% to 12% improvement),f SMD –0.28 (95% CI –0.45 to –0.12), NNTB 14 (95% CI 9 to 33)g |
| 7 out of 100 improved by 20%c | 5 more out of 100 (2 more to 9 more) in the intervention group improved by 20%c | RR 1.71 (95% CI 1.29 to 2.29)d | ||||
|
Physical function assessed with: self‐report questionnaire WOMAC Physical Function (scale 0 to 10, where 0 = no limitation) Follow‐up: range 10 days to 13 weeks |
The mean physical function was 5.9 points |
The mean physical function in the intervention group was 0.4 points lower (0.2 lower to 0.7 lower)b | ‐ | 614 (2 RCTs) | ⊕⊕⊕⊝ Moderatee |
Mean function: 4% absolute improvement (95% CI 2% to 7% improvement),b 7% relative improvement (95% CI 3% to 12% improvement),f SMD –0.27 (95% CI –0.43 to –0.11), NNTB 12 (95% CI 8 to 30)g |
| 10 out of 100 improved by 20%c | 5 more out of 100 (2 more to 9 more) in the intervention group improved by 20%c | RR 1.50 (95% CI 1.20 to 1.90)d | ||||
|
Number of participants experiencing any adverse event Follow‐up: range 10 days to 13 weeks |
234 per 1000 | 447 per 1000 (309 to 646) | RR 1.91 (95% CI 1.32 to 2.76)d | 308 (1 RCT) | ⊕⊕⊕⊝ Moderatee |
22% absolute worsening (95% CI 8% more to 41% more), 91% relative worsening (95% CI 32% more to 176% more), NNTH 5 (95% CI 3 to 14) |
|
Number of participants who withdrew due to adverse events Follow‐up: range 10 days to 13 weeks |
45 per 1000 | 126 per 1000 (68 to 235) |
RR 2.78 (95% CI 1.50 to 5.16) |
614 (2 RCTs) | ⊕⊕⊝⊝ Lowe,h | 8% absolute worsening (95% CI 2% more to 19% more), 178% relative worsening (95% CI 50% more to 416% more), NNTH 13 (95% CI 6 to 44) |
|
Number of participants experiencing any serious adverse events Follow‐up: range 10 days to 13 weeks |
0 per 1000 | 0 per 1000 | Not estimable | 15 (1 RCT) | ⊕⊕⊝⊝ Lowi | No events reported: not estimable, NNTH not estimable |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
**The assumed and the corresponded risk was calculated from the SMD and SE. CI: confidence interval; MCID: minimal clinically important difference; NNTB: number needed to treat for an additional beneficial outcome; NNTH: number needed to treat for an additional harmful outcome;RR: risk ratio;SD: standard deviation; SE: standard error; SMD: standardized mean difference; WOMAC: Western Ontario and McMaster Universities Arthritis Index. | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aContinuous outcomes summarized using SMD and SE, and binary outcomes expressed as RR. We used standard inverse‐variance fixed‐effect meta‐analysis to combine the trials in Review Manager 5 (Review Manager 2014). bAbsolute improvement on a common scale (e.g. 100‐mm, 1700‐point scale) calculated by multiplying the SMD by the SD of the scale (in the control group at baseline) as suggested by the Cochrane Handbook for Systematic Reviews of Interventions (Section 12.6.4; Higgins 2011). cAssumed and corresponding risks calculated from SMD and SE, with improvement based on an MCID of 20% of the given scale using the Wells calculator (from the Cochrane Musculoskeletal Group Editorial office; musculoskeletal.cochrane.org/). dRR and its 95% CI calculated using the assumed risk of the control group and corresponding risk of the treatment group. The corresponding risk was divided by the assumed risk. eDowngraded one level due to unclear risk of bias (all trials had high or unclear risk of at least one type of bias). fThe relative improvement percentage was defined as relative to the control group risk at baseline. gNNTB corresponded to the number of participants that needed to be treated to see one participant improve. Improvement defined as reaching an MCID of 20% on the given scale. NNTB calculated using the Wells calculator (from the Cochrane Musculoskeletal Group Editorial office; musculoskeletal.cochrane.org/ne.org/). hDowngraded one level for imprecision (wide CI). iDowngraded two levels for serious for imprecision (no events).
Summary of findings 3. Tramadol alone compared with acetaminophen for osteoarthritis.
| Tramadol alone compared with acetaminophen for osteoarthritis | ||||||
|
Patient or population: osteoarthritis Settings: outpatient clinics Intervention: tramadol alone Comparison: acetaminophen | ||||||
| Outcomesa | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk** | Corresponding risk** | |||||
| acetaminophen | Tramadol alone | |||||
|
Pain assessed with: self‐report VAS pain intensity (scale 0 to 100 where 0 = no pain) Follow‐up: 1 week |
The mean pain was 38 points | The mean pain in the intervention group was 2 higher (12 lower to 16 higher)b | — | 20 (1 RCT) | ⊕⊝⊝⊝ Very lowe,f |
Mean pain: 2% absolute worsening (95% CI 16% worsening to 12% improvement),b 5% relative worsening (95% CI 42% worsening to 32% improvement),g SMD 0.13 (–0.80 to 1.06), NNTB not applicableh |
| 9 out of 100 improved by 20%c | 2 less out of 100 (8 less to 20 more)in the intervention group improved by 20%c | RR 0.78 (95% CI 0.11 to 3.22)d | ||||
| Physical function | — | — | — | — | — | Not reported |
| Number of participants experiencing any adverse event | — | — | — | — | — | Not reported |
|
Number of participants who withdrew due to adverse events Follow‐up: 1 week |
0 per 1000 | 0 per 1000 | RR 5.00 (95% CI 0.27 to 92.62) | 20 (1 RCT) | ⊕⊝⊝⊝ Very lowe,f |
NNTH not estimable |
| Number of participants experiencing any serious adverse events | — | — | — | — | — | Not reported |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
**The assumed and the corresponded risk was calculated from the SMD and SE. CI: confidence interval; MCID: minimal clinically important difference; NNTB: number needed to treat for an additional beneficial outcome; NNTH: number needed to treat for an additional harmful outcome;RR: risk ratio;SD: standard deviation; SE: standard error; SMD: standardized mean difference. | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aContinuous outcomes summarized using SMD and SE, and binary outcomes expressed as RR. We used standard inverse‐variance fixed‐effect meta‐analysis to combine the trials in Review Manager 5 (Review Manager 2014). bAbsolute improvement on a common scale (e.g. 100‐mm, 1700‐point scale) calculated by multiplying the SMD by the SD of the scale (in the control group at baseline) as suggested by the Cochrane Handbook for Systematic Reviews of Interventions (Section 12.6.4; Higgins 2011). cAssumed and corresponding risks calculated from the SMD and SE, with improvement based on an MCID of 20% of the given scale using the Wells calculator (from the Cochrane Musculoskeletal Group Editorial office; musculoskeletal.cochrane.org/). dRR and its 95% CI calculated using the assumed risk of the control group and corresponding risk of the treatment group. The corresponding risk was divided by the assumed risk. eDowngraded one level for unclear risk of bias (all trials had high or unclear risk of at least one type of bias). fDowngraded two levels for serious imprecision (few events and wide CI). gRelative improvement percentage defined as relative to the control group risk at baseline. hNNTB corresponded to the number of participants that needed to be treated to see one participant improve. Improvement was defined as reaching an MCID of 20% on the given scale. NNTB calculated using the Wells calculator (from the Cochrane Musculoskeletal Group Editorial office; musculoskeletal.cochrane.org/ne.org/). It was only calculated for statistically significant results.
Summary of findings 4. Tramadol alone compared with NSAIDs for osteoarthritis.
| Tramadol alone compared with NSAIDs for osteoarthritis | ||||||
|
Patient or population: osteoarthritis Settings: outpatient clinics Intervention: tramadol alone Comparison: NSAIDs | ||||||
| Outcomesa | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk** | Corresponding risk** | |||||
| NSAIDs | Tramadol alone | |||||
|
Pain assessed with: self‐report VAS pain intensity (scale 0 to 500 where 0 = no pain) Follow‐up: range 4 weeks to 12 weeks |
The mean pain was 300.8 points | The mean pain in the intervention group was 22 points higher (7 higher to 37 higher)b | — | 952 (3 RCTs) | ⊕⊕⊕⊝ Moderatee |
Mean pain: 4% absolute worsening (95% CI 1% to 7% worsening),b 7% relative worsening (95% CI 2% to 12% worsening),f SMD 0.21 (95% CI 0.07 to 0.36), NNTB 12 (95% CI 7 to 35)g |
| 17 out of 100 improved by 20%c | 5 less out of 100 (8 less to 2 more) in the intervention group improved by 20%c | RR 0.71 (95% CI 0.53 to 0.88)d | ||||
|
Physical function assessed with: self‐report questionnaire WOMAC Physical Function (scale 0 to 1700, where 0 = no limitation) Follow‐up: range 4 weeks to 12 weeks |
The mean physical function was 1019 points | The mean physical function in the intervention group was 82 higher (32 higher to 131 higher)b |
— | 952 (3 RCTs) | ⊕⊕⊝⊝ Lowe,h |
Mean physical function: 5% absolute worsening (95% CI 2% to 8% worsening),b 8% relative worsening (95% CI 3% to 13% worsening),f SMD 0.23 (95% CI 0.09 to 0.37), NNTB 11 (95% CI 7 to 27)g |
| 17 out of 100 improved by 20%c | 5 less out of 100 (7 less to 2 less) in the intervention group improved by 20%c | RR 0.71 (95% CI 0.59 to 0.88)d | ||||
|
Number of participants experiencing any adverse event Follow‐up: 8 weeks |
591 per 1000 | 774 per 1000 (609 to 987) | RR 1.31 (95% CI 1.03 to 1.67) | 128 (3 RCTs) | ⊕⊕⊕⊝ Moderatee |
18% absolute worsening (95% CI 2% more to 40% more), 31% relative worsening (95% CI 3% more to 67% more), NNTH 6 (95% CI 3 to 57) |
|
Number of participants who withdrew due to adverse events Follow‐up: range 8 weeks to 12 weeks |
112 per 1000 | 210 per 1000 (142 to 309) | RR 1.88 (95% CI 1.27 to 2.76) | 929 (2 RCTs) | ⊕⊕⊝⊝ Lowe,h | 10% absolute worsening (95% CI 3% more to 20% more), 88% relative worsening (95% CI 27% more to 176% more), NNTH 10 (95% CI 5 to 33) |
|
Number of participants experiencing any serious adverse events Follow‐up: range 4 weeks to 8 weeks |
21 per 1000 | 4 per 1000 (0 to 90) | RR 0.21 (95% CI 0.01 to 4.34) | 188 (2 RCTs) | ⊕⊝⊝⊝ Very lowe,i | NNTH not applicable |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
**The assumed and the corresponded risk was calculated from the SMD and SE. CI: confidence interval; MCID: minimal clinically important difference; NNTB: number needed to treat for an additional beneficial outcome; NNTH: number needed to treat for an additional harmful outcome;RR: risk ratio;SD: standard deviation; SE: standard error; SMD: standardized mean difference WOMAC: Western Ontario and McMaster Universities Arthritis Index. | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aContinuous outcomes summarized using SMD and SE, and binary outcomes expressed as RR. We used standard inverse‐variance fixed‐effect meta‐analysis to combine the trials in Review Manager 5 (Review Manager 2014). bAbsolute effect on a common scale (e.g. 100‐mm, 1700‐point scale) calculated by multiplying the SMD by the SD of the scale (in the control group at baseline) as suggested by the Cochrane Handbook for Systematic Reviews of Interventions (Section 12.6.4; Higgins 2011). cAssumed and corresponding risks calculated from the SMD and SE, with improvement based on an MCID of 20% of the given scale using the Wells calculator (from the Cochrane Musculoskeletal Group Editorial office; musculoskeletal.cochrane.org/). dRR and its 95% CI calculated using the assumed risk of the control group and corresponding risk of the treatment group. The corresponding risk was divided by the assumed risk. eDowngraded one level for unclear risk of bias (all trials had high or unclear risk of at least one type of bias). fRelative improvement percentage defined as relative to the control group risk at baseline. gNNTB corresponded to the number of participants that needed to be treated to see one participant improve. Improvement defined as reaching an MCID of 20% on the given scale. NNTB calculated using the Wells calculator (from the Cochrane Musculoskeletal Group Editorial office; musculoskeletal.cochrane.org/ne.org/). hDowngraded one level for inconsistency. iDowngraded two levels for serious imprecision (few events and wide CI).
Summary of findings 5. Tramadol alone compared with other opioids for osteoarthritis.
| Tramadol alone compared with other opioids for osteoarthritis | ||||||
|
Patient or population: osteoarthritis Settings: outpatient clinics Intervention: tramadol alone Comparison: other opioids | ||||||
| Outcomesa | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk** | Corresponding risk** | |||||
| Other opioids | Tramadol alone | |||||
|
Pain assessed with: self‐report VAS pain intensity (scale 0 to 100 mm where 0 = no pain) Follow‐up: range 2 weeks to 12 weeks |
The mean pain was 36 points | The mean pain in the intervention group was 3 points lower (9 lower to 3 higher)b | — | 411 (4 RCTs) | ⊕⊕⊕⊝ Moderatee |
Mean pain: 3% absolute improvement (95% CI 3% worsening to 9% improvement),b 8% relative improvement (95% CI 8% worsening to 25% improvement),f SMD –0.11 (95% CI –0.33 to 0.12) NNTB not applicableg |
| 23 out of 100 improved by 20%c | 3 more out of 100 (4 less to 11 more) in the intervention group improved by 20%c | RR 1.13 (95% CI 0.83 to 1.48)d | ||||
|
Physical function assessed with participants rating their overall assessment of the therapy at end of study as good or better Follow‐up: 2 weeks |
505 per 1000 | 667 per 1000 (525 to 848) | RR 1.32 (95% CI 1.04 to 1.68) | 190 (1 RCT) | ⊕⊕⊕⊝ Moderatee |
16% absolute improvement (95% CI 2% more to 34% more),b 32% relative improvement (95% CI 4% more to 68% more),f NNTB: 7 (95% CI 3 to 50)g |
|
Number of participants experiencing any adverse event Follow‐up: range 2 weeks to 12 weeks |
541 per 1000 | 536 per 1000 (471 to 612) | RR 0.99 (95% CI 0.87 to 1.13) | 438 (3 RCTs) | ⊕⊝⊝⊝ Very lowe,h,i |
1% absolute worsening (95% CI 7% fewer to 7% more), 1% relative worsening (95% CI 13% fewer to 13% more), NNTH not applicable |
|
Number of participants who withdrew due to adverse events Follow‐up: range 2 weeks to 12 weeks |
138 per 1000 | 311 per 1000 (209 to 464) |
RR 2.26 (95% CI 1.52 to 3.37) |
438 (3 RCTs) | ⊕⊕⊝⊝ Lowe,h |
17% absolute worsening (95% CI 7% more to 33% more), 126% relative worsening (95% CI 52% more to 237% more), NNTH 6 (95% CI 3 to 14) |
|
Number of participants experiencing any serious adverse events Follow‐up: range 2 weeks to 12 weeks |
0 per 1000 | 0 per 1000 | RR 7.42 (95% CI 0.39 to 141.00) | 495 (4 RCTs) | ⊕⊝⊝⊝ Very lowe,j | 0% absolute worsening (95% CI 0% fewer to 0% fewer), 642% relative worsening (95% CI 13% fewer to 41% more), NNTH not applicable |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
**The assumed and the corresponded risk was calculated from the SMD and SE. CI: confidence interval; MCID: minimal clinically important difference; NNTB: number needed to treat for an additional beneficial outcome; NNTH: number needed to treat for an additional harmful outcome;RR: risk ratio;SD: standard deviation; SE: standard error; SMD: standardized mean difference. | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aContinuous outcomes summarized using SMD and SE, and binary outcomes expressed as RR. We used standard inverse‐variance fixed‐effect meta‐analysis to combine the trials in Review Manager 5 (Review Manager 2014). bAbsolute effect on a common scale (e.g. 100‐mm, 1700‐point scale) calculated by multiplying the SMD by the SD of the scale (in the control group at baseline) as suggested by the Cochrane Handbook for Systematic Reviews of Interventions (Section 12.6.4; Higgins 2011). cAssumed and corresponding risks calculated from the SMD and SE, with improvement based on an MCID of 20% of the given scale using the Wells calculator (from the Cochrane Musculoskeletal Group Editorial office; mumusculoskeletal.cochrane.org/). dRR and its 95% CI calculated using the assumed risk of the control group and corresponding risk of the treatment group. The corresponding risk was divided by the assumed risk. eDowngraded one level for unclear risk of bias (all trials had high or unclear risk of at least one type of bias). fRelative improvement percentage defined as relative to the control group risk at baseline. gNNTB corresponded to the number of participants that needed to be treated to see one participant improve. Improvement was defined as reaching an MCID of 20% on the given scale. NNTB calculated using the Wells calculator (from the Cochrane Musculoskeletal Group Editorial office; musculoskeletal.cochrane.org/ne.org/). It was only calculated for statistically significant results. hDowngraded one level for inconsistency. iDowngraded one level for imprecision (wide CI). jDowngraded two levels for serious imprecision (few events and wide CI).
Summary of findings 6. Tramadol in combination with acetaminophen compared with NSAIDs for osteoarthritis.
| Tramadol in combination with acetaminophen compared with NSAIDs for osteoarthritis | ||||||
|
Patient or population: osteoarthritis Settings: outpatient clinics Intervention: tramadol in combination with acetaminophen Comparison: NSAIDs | ||||||
| Outcomesa | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk** | Corresponding risk** | |||||
| NSAIDs | Tramadol in combination with acetaminophen | |||||
|
Pain assessed with: self‐report VAS pain intensity (scale 0 to 10 mm where 0 = no pain) Follow‐up: range 8 weeks to 12 weeks |
The mean pain was 5.2 points | The mean pain in the intervention group was 0.3 points higher (0.4 lower to 0.9 higher)b | — | 226 (2 RCTs) | ⊕⊕⊕⊝ Moderatee |
Mean pain: 3% absolute worsening (95% CI 9% worsening to 4% improvement),b 6% relative worsening (95% CI 17% worsening to 8% improvement),f SMD 0.12 (–0.16 to 0.39), NNTB not applicableg |
| 47 out of 100 improved by 20%c | 5 less out of 100 (15 less to 6 more) in the intervention group improved by 20%c | RR 0.89 (95% CI 0.68 to 1.13)d | ||||
|
Physical function assessed with: WOMAC Physical Function on a 96‐point scale Follow‐up: 8 weeks |
The mean physical function was 21.40 | The mean physical function in the intervention group was 2 points higher (2 lower to 6 higher) | — | 91 (1 RCT) | ⊕⊕⊝⊝ Lowe,h |
Mean physical function: 2% absolute worsening (95% CI 7% worsening to 2% improvement),b 9% relative worsening (95% CI 28% worsening to 9% improvement),f SMD 0.20 (–0.21 to 0.61) NNTB not applicableg |
| 22 out of 100 improved by 20%c | 5 less out of 100 (13 less to 7 more) in the intervention group improved by 20%c | RR 0.77 (95% CI 0.41 to 1.32)d | ||||
|
Number of participants experiencing any adverse event Follow‐up: 8 weeks |
600 per 1000 | 702 per 1000 (522 to 942) | RR 1.17 (95% CI 0.87 to 1.57) | 97 (1 RCT) | ⊕⊕⊝⊝ Lowe,h |
10% absolute worsening (95% CI 8% improvement to 34% worsening), 17% relative worsening (95% CI 13% improvement to 57% worsening), NNTH not applicable |
| Number of participants who withdrew due to adverse events | — | — | — | — | — | Not reported |
|
Number of participants experiencing any serious adverse events Follow‐up: 3 days |
0 per 1000 | 0 per 1000 (0 to 0) | Not estimable | 15 (1 RCT) | ⊕⊕⊝⊝ Lowi |
NNTH not estimable |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
**The assumed and the corresponded risk was calculated from the SMD and SE. CI: confidence interval; MCID: minimal clinically important difference; NNTB: number needed to treat for an additional beneficial outcome; NNTH: number needed to treat for an additional harmful outcome;RR: risk ratio;SD: standard deviation; SE: standard error; SMD: standardized mean difference; WOMAC: Western Ontario and McMaster Universities Arthritis Index. | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aContinuous outcomes summarized using SMD and SE, and binary outcomes expressed as RR. We used standard inverse‐variance fixed‐effect meta‐analysis to combine the trials in Review Manager 5 (Review Manager 2014). bAbsolute effect on a common scale (e.g. 100‐mm, 1700‐point scale) calculated by multiplying the SMD by the SD of the scale (in the control group at baseline) as suggested by the Cochrane Handbook for Systematic Reviews of Interventions (Section 12.6.4; Higgins 2011). cAssumed and corresponding risks calculated from the SMD and SE, with improvement based on an MCID of 20% of the given scale using the Wells calculator (from the Cochrane Musculoskeletal Group Editorial office; musculoskeletal.cochrane.org/). dRR and its 95% CI calculated using the assumed risk of the control group and corresponding risk of the treatment group. The corresponding risk was divided by the assumed risk. eDowngraded one level for unclear risk of bias (all trials had high or unclear risk of at least one type of bias). fRelative improvement percentage defined as relative to the control group risk at baseline. gNNTB corresponded to the number of participants that needed to be treated to see one participant improve. Improvement defined as reaching an MCID of 20% on the given scale. NNTB calculated using the Wells calculator (from the Cochrane Musculoskeletal Group Editorial office; musculoskeletal.cochrane.org/ne.org/). It was only calculated for statistically significant results. hDowngraded one level for imprecision (wide CI). iDowngraded two levels for serious imprecision (no events).
Summary of findings 7. Tramadol in combination with acetaminophen compared with other opioids for osteoarthritis.
| Tramadol in combination with acetaminophen compared with other opioids for osteoarthritis | ||||||
|
Patient or population: osteoarthritis Settings: outpatient clinics Intervention: tramadol in combination with acetaminophen Comparison: other opioids | ||||||
| Outcomesa | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk** | Corresponding risk** | |||||
| Other opioids | Tramadol in combination with acetaminophen | |||||
|
Pain assessed with: self‐report VAS pain intensity (scale 0 to 10 mm where 0 = no pain) Follow‐up: 12 weeks |
The mean pain was 6.4 points | The mean pain of the intervention group was 0.3 points higher (1.8 higher to 1.3 lower)b | — | 130 (1 RCT) | ⊕⊕⊝⊝ Lowe,f |
Mean pain: 3% absolute worsening (95% CI 18% worsening to 13% improvement),b 5% relative worsening (95% CI 28% worsening to 20% improvement),f SMD 0.06 (95% CI –0.31, 0.43), NNTB not applicableh |
| 32 out of 100 improved by 20%c | 2 less out of 100 (16 less to 13 more) in the intervention group improved by 20%c | RR 0.96 (95% CI 0.67 to 1.27)d | ||||
| Physical function | — | — | — | — | — | Not reported |
| Number of participants experiencing any adverse event | — | — | — | — | — | Not reported |
| Number of participants who withdrew due to adverse events | — | — | — | — | — | Not reported |
| Number of participants experiencing any serious adverse events | — | — | — | — | — | Not reported |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
**The assumed and the corresponded risk was calculated from the SMD and SE. CI: confidence interval; MCID: minimal clinically important difference; NNTB: number needed to treat for an additional beneficial outcome; RR: risk ratio;SD: standard deviation; SE: standard error; SMD: standardized mean difference. | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aContinuous outcomes summarized using SMD and SE, and binary outcomes expressed as RR. We used standard inverse‐variance fixed‐effect meta‐analysis to combine the trials in Review Manager 5 (Review Manager 2014). bAbsolute effect on a common scale (e.g. 100‐mm, 1700‐point scale) calculated by multiplying the SMD by the SD of the scale (in the control group at baseline) as suggested by the Cochrane Handbook for Systematic Reviews of Interventions (Section 12.6.4; Higgins 2011). cAssumed and corresponding risks calculated from the SMD and SE, with improvement based on an MCID of 20% of the given scale using the Wells calculator (from the Cochrane Musculoskeletal Group Editorial office; musculoskeletal.cochrane.org/). dRR and its 95% CI calculated using the assumed risk of the control group and corresponding risk of the treatment group. Corresponding risk was divided by the assumed risk. eDowngraded one level for unclear risk of bias (all trials had high or unclear risk of at least one type of bias). fDowngraded one level for imprecision. gRelative improvement percentage is defined as relative to the control group risk at baseline. hNNTB corresponded to the number of participants that needed to be treated to see one participant improve. Improvement defined as reaching an MCID of 20% on the given scale. NNTB calculated using the Wells calculator (from the Cochrane Musculoskeletal Group Editorial office; musculoskeletal.cochrane.org/ne.org/). It was only calculated for statistically significant results.
Major outcomes
Benefits
Pain
Placebo‐controlled studies
Eight trials including 2647 participants in the tramadol alone groups and 1325 participants in the placebo control groups contributed to the analysis of knee or hip pain. Two trials including 350 participants in the combined tramadol/acetaminophen groups and 264 in the placebo control groups contributed to the analysis of knee or hip pain.
We found moderate quality evidence that tramadol alone had a small and probably not clinically important pain reduction compared to placebo control interventions (SMD –0.25, 95% CI –0.32 to –0.18; Table 1). Fifteen out of 100 people in the tramadol group improved by 20% (which corresponded to a clinically important difference in pain) compared to 10/100 in the placebo group (see Table 1). On a scale of 0 to 100, people in the tramadol group had a 4‐point reduction in pain (95% CI 3 to 5; 8 studies, 3972 participants). This translated into an NNTB of 13 (95% CI 10 to 18). There was no substantial clinical heterogeneity of the participants (i.e. there were no important differences in sample, intervention or control group characteristics). An I2 statistic of 0% indicated a low degree of statistical heterogeneity. A visual inspection of the funnel plot suggested symmetry (see Figure 4). Applying the GRADE criteria to value the overall quality of the evidence, we downgraded once due to unclear risk of bias in the included trials (see Table 1).
4.
Funnel plot of comparison: 1 Tramadol versus placebo, outcome: 1.1 Pain.
One cross‐over study, which was not included in the meta‐analyses, showed that WOMAC Osteoarthritis Index subscale scores for pain were statistically significantly better with tramadol compared to placebo (189.0 (SD 105.0) with tramadol versus 230.0 (SD 115.4) with placebo; P = 0.0001), but these differences were probably not clinically important (Thorne 2008). The study also measured pain intensity using an ordinal scale (0 = none, 1 = mild, 2 = moderate, 3 = severe, 4 = excruciating) and a VAS scale (from 0 mm to 100 mm). Tramadol had statistically significantly lower VAS pain intensity scores compared with placebo (37.4 (SD 23.9) with tramadol versus 45.1 (SD 24.3) with placebo; P = 0.0009), but these differences were probably not clinically important.
We found moderate quality evidence that tramadol in combination with acetaminophen had a small but probably not clinically important pain reduction compared to placebo control interventions (SMD –0.28, 95% CI –0.45 to –0.12; Table 2). Twelve out of 100 people improved in a clinically important way in the tramadol in combination with acetaminophen group compared to 7/100 in the placebo group (see Table 2). On a scale of 0 to 100, people in the tramadol in combination with acetaminophen group had a 4‐point reduction in pain (95% CI 2 to 6; 2 studies, 614 participants). This translated into an NNTB of 14 (95% CI 9 to 33). There was no substantial clinical heterogeneity of the participants (i.e. there were no important differences in sample, intervention or control group characteristics). An I2 statistic of 0% indicated a low degree of heterogeneity. Applying the GRADE criteria to the overall quality of the evidence, we downgraded once due to unclear risk of bias in the included trials (see Table 2).
Active‐controlled studies
Eleven trials including 1017 participants in experimental groups and 668 participants in active control groups (which included acetaminophen, NSAIDs and opioids as active controls) contributed to the analyses of knee or hip pain.
Due to very low quality evidence, we were uncertain whether tramadol alone reduced pain compared to acetaminophen (SMD 0.13, 95% CI –0.80 to 1.06; Table 3; Bianchi 2003). Seven out of 100 people improved in a clinically important way with tramadol compared to 9/100 people in the acetaminophen group (see Table 3). Compared to the acetaminophen group, people in the tramadol group had a 2‐point increase in pain (95% CI –12 to 16 on a scale of 0 to 100; 1 study, 20 participants). Applying the GRADE criteria to the overall quality of the evidence, we downgraded three times due to unclear risk of bias and the serious imprecision in the included trials (see Table 3).
We found moderate quality evidence that tramadol alone was less effective in pain reduction compared to NSAIDs (SMD 0.21, 95% CI 0.07 to 0.36; Table 4; Beaulieu 2008; DeLemos 2011; Pavelka 1998). Twelve out of 100 people improved in a clinically important way in the tramadol group compared to 17/100 in the NSAID group (see Table 4). Compared to NSAIDs, people in the tramadol group had a 4‐point worsening in pain on a scale of 0 to 100 (95% CI 1 to 7; 3 studies, 952 participants). This translated into an NNTB of 12 (95% CI 7 to 35). There was no substantial clinical heterogeneity of the participants (i.e. there were no important differences in sample, intervention or control group characteristics). An I2 statistic of 23% indicated a low degree of statistical heterogeneity. Applying the GRADE criteria to the overall quality of the evidence, we downgraded once due to unclear risk of bias in the included trials (see Table 4).
We found moderate quality evidence that tramadol alone did not lead to pain reduction compared to other opioids (SMD –0.11, 95% CI –0.33 to 0.12; 4 studies, 411 participants; Table 5; Bird 1995; Jensen 1994; Karlsson 2009; Wilder‐Smith 2001). Twenty‐six out of 100 people improved in a clinically important way in the tramadol group compared to 23/100 people in the other opioid group (see Table 5). There was no substantial clinical heterogeneity of the participants (i.e. there were no important differences in sample, intervention or control group characteristics). An I2 statistic of 0% indicated a low degree of statistical heterogeneity. Applying the GRADE criteria to the overall quality of the evidence, we downgraded once due to unclear risk of bias in the included trials (see Table 5).
We found moderate quality evidence that tramadol in combination with acetaminophen did not lead to pain reduction compared to NSAIDs (SMD 0.12 95% CI –0.16 to 0.39; Table 6; Fujii 2014; Park 2012). Forty‐two out of 100 people improved in a clinically important way in the tramadol in combination with acetaminophen group compared to 47/100 people in the NSAIDs group (2 studies, 226 participants; Table 6). There was no substantial clinical heterogeneity of the participants (i.e. there were no important differences in sample, intervention or control group characteristics). An I2 statistic of 35% indicated a low degree of statistical heterogeneity. Applying the GRADE criteria to the overall quality of the evidence, we downgraded once due to unclear risk of bias in the included trials (see Table 6).
We found low quality evidence that tramadol in combination with acetaminophen did not lead to pain reduction compared to other opioids (SMD 0.06, 95% CI –0.31 to 0.43; 2 studies, 226 participants; Table 7; Fujii 2014). Thirty out of 100 people improved in a clinically important way in the tramadol in combination with acetaminophen group compared to 32/100 people in the other opioids group (1 study, 130 participants; see Table 7). Applying the GRADE criteria to the overall quality of the evidence, we downgraded twice due to unclear risk of bias in the included trials and the imprecision between trials (see Table 7).
Physical function
Placebo‐controlled trials
Five studies including 1789 participants in the tramadol alone groups and 761 participants in the placebo control groups contributed to the analysis of physical function (Babul 2004; DeLemos 2011; Fleischmann 2001; Gana 2006; Kean 2009). Two studies including 350 participants in the tramadol in combination with acetaminophen groups and 264 participants in the placebo control groups contributed to the analysis of physical function (Emkey 2004; Silverfield 2002).
We found moderate quality evidence that tramadol alone had a small but probably not clinically important benefit in physical function compared to placebo (SMD –0.20, 95% CI –0.29 to –0.12; Table 1). Twenty‐one percent of people in the tramadol group improved by 20%, which corresponded to a clinically important difference in physical function, and 16% of people improved in a clinically important way in the placebo group (see Table 1). On a scale of 0 to 100, people in the tramadol group had a 4‐point improvement in physical function (95% CI 2 to 6; 5 studies, 2550 participants). This translated into an NNTB of 13 (95% CI 9 to 21). There was no substantial clinical heterogeneity of the participants (i.e. there were no important differences in sample, intervention or control group characteristics). An I2 statistic of 54% indicated a moderate degree of statistical heterogeneity. One trial was responsible for the statistical heterogeneity (DeLemos 2011). The trial had three intervention groups with different doses of tramadol (100 mg, 200 mg and 300 mg) and had both a placebo and active control group. The trial indicated no statistically significant or clinically important difference in physical function (SMD –0.02, 95% CI –0.18 to 0.14). Applying the GRADE criteria to the overall quality of the evidence, we downgraded once due to unclear risk of bias in the included trials (see Table 1).
One study, which was not included in the meta‐analyses, found that WOMAC Osteoarthritis Index subscale scores for physical function were statistically significantly better with tramadol alone compared to placebo, but these differences were probably not clinically important (632.4 (SD 361.3) with tramadol alone versus 727.4 (SD 383.4) with placebo; P = 0.0205; Thorne 2008).
We found moderate quality evidence that tramadol in combination with acetaminophen had a small but probably not clinically important benefit in physical function compared to placebo (SMD –0.27, 95% CI –0.43 to –0.11; Table 2). Fifteen percent of people improved in a clinically important way in the tramadol in combination with acetaminophen group compared to 10% in the placebo group (see Table 2 for the main comparison). On a scale of 0 to 100, people in the tramadol in combination with acetaminophen group had a 4‐point improvement in physical function (95% CI 2 to 7; 2 studies, 614 participants). This translated into an NNTB of 12 (95% CI 8 to 30). There was no substantial clinical heterogeneity of the participants (i.e. there were no important differences in sample, intervention or control group characteristics). An I2 statistic of 0% indicated low degree of heterogeneity. Applying the GRADE criteria to the overall quality of the evidence, we downgraded once due to unclear risk of bias in the included trials (see Table 2).
Active‐controlled trials
Five studies including 769 participants in experimental groups and 410 participants in active control groups (which included NSAIDs and other opioids) contributed to the analysis of physical function.
We found low quality evidence that tramadol alone had a small but probably not clinically important worsening in physical function compared to NSAIDs (SMD 0.23, 95% CI 0.09 to 0.37; Table 4; Beaulieu 2008; DeLemos 2011; Pavelka 1998). Twelve out of 100 people improved in a clinically important way with tramadol alone compared to 17/100 people in the NSAIDs group (see Table 4). On a scale of 0 to 100, people in the tramadol group had a 5‐point worsening in physical function compared to the NSAIDs group (95% CI 2 to 8; 3 studies, 952 participants). This translated into an NNTB of 11 (95% CI 7 to 27). There was no substantial clinical heterogeneity of the participants (i.e. there were no important differences in sample, intervention or control group characteristics). However, an I2 statistic of 70% indicated a high degree of statistical heterogeneity. One trial was responsible for most of the statistical heterogeneity (Pavelka 1998). This trial has a small sample size (54 participants) and included participants as young as 18 years old, contrary to most studies. It employed a cross‐over design and only data obtained during the first four weeks of the trial were analyzed. The SMD, though not statistically significant, indicated an improvement in physical function compared to NSAIDs, contrary to the other trials comparing tramadol alone and NSAIDs (SMD –0.38, 95% CI –0.92 to 0.16). Applying the GRADE criteria to the overall quality of the evidence, we downgraded twice due to unclear risk of bias and inconsistency (see Table 4).
We found moderate quality evidence that tramadol alone had a small but probably not clinically important benefit in overall assessment at end of therapy compared to other opioids (RR 1.32, 95% CI 1.04 to 1.68; 1 study, 190 participants; Table 5; Jensen 1994). According to the only study in this analysis, 67/100 people defined their overall assessment at end of therapy as good or better in the tramadol group compared to 51/100 people in the opioids group. This translated into an NNTB of 7 (95% CI 3 to 50). Applying the GRADE criteria to the overall quality of the evidence, we downgraded once due to unclear risk of bias (see Table 5).
We found low quality evidence that tramadol in combination with acetaminophen did not improve physical function compared to NSAIDs (SMD 0.20, 95% CI –0.21 to 0.61; 1 study, 91 participants; Table 6; Park 2012). Seventeen out of 100 people improved in a clinically important way in the tramadol in combination with acetaminophen group compared to 22/100 people in the NSAIDs group (see Table 6). Applying the GRADE criteria to the overall quality of the evidence, we downgraded twice due to unclear risk of bias and imprecision (see Table 6).
Harms
Any adverse events
Placebo‐controlled trials
Four trials including 1366 participants in the tramadol alone groups and 673 participants in the placebo control groups reported the number of participants presenting with any adverse events (Babul 2004; Fishman 2007; Gana 2006; Malonne 2004). One trial including 197 participants in the combined tramadol/acetaminophen groups and 111 participants in the placebo control group reported the number of participants presenting with any adverse events (Silverfield 2002). There were 1424 people with adverse events in these five trials. Tramadol may cause adverse effects such as nausea, vomiting, dizziness, constipation, tiredness, headache, sweating and abdominal pain.
We found moderate quality evidence that there was a greater risk of developing any adverse events with tramadol alone compared to placebo (RR 1.34, 95% CI 1.24 to 1.46; 4 studies, 2039 participants; Table 1). Sixty‐six out of 100 people may have developed adverse effects when taking tramadol alone compared to 49/100 people when taking placebo, which corresponded to 17 more people out of 100 who develop adverse events. The NNTH to cause one additional participant to experience an adverse event was 6 (95% CI 5 to 9). An I2 statistic of 62% indicated a high degree of heterogeneity. One trial was responsible for most of the statistical heterogeneity (Malonne 2004). This could be explained by the duration of the trial, which was two weeks as compared to the 12‐week duration for the other trials, as well as the population, which was exclusively white as compared to the slightly more diverse population of the other studies. It had a higher RR than the other studies (RR 2.33, 95% CI 1.53 to 3.55). Applying the GRADE criteria to the overall quality of the evidence, we downgraded once due to unclear risk of bias (see Table 1).
In one study which was not included in the meta‐analyses, 94 (79.8%) participants reported adverse events during tramadol treatment while 88 (65.9%) participants reported adverse events during placebo treatment (Thorne 2008). The difference in the overall number of adverse events between treatment groups was not clinically significant (P = 0.0833).
We found moderate quality evidence that there was a greater risk of developing any adverse events with tramadol in combination with acetaminophen compared to placebo (RR 1.91, 95% CI 1.32 to 2.76; 1 study, 308 participants; Table 2). Forty‐five out of 100 people may have developed adverse effects when taking tramadol in combination with acetaminophen compared to 23/100 people when taking a placebo, which corresponded to 22 more people out of 100 who developed adverse events. The NNTH to cause one additional participant to experience an adverse event was 5 (95% CI 3 to 14; Table 2). Applying the GRADE criteria to the overall quality of the evidence, we downgraded once due to unclear risk of bias (see Table 2).
Active‐controlled trials
Five trials reported the number of participants presenting with any adverse event including 326 participants in experimental groups (tramadol alone and tramadol in combination with acetaminophen) and 331 participants in active control groups (including NSAIDs and opioids). There were 403 people with adverse events in these five trials.
We found moderate quality evidence that there was a greater risk of developing any adverse events with tramadol alone compared to NSAIDs (RR 1.31, 95% CI 1.03 to 1.67; 1 study, 128 participants; see Table 4; Beaulieu 2008). Seventy‐seven out of 100 people may have developed adverse effects when taking tramadol alone compared to 59 out of 100 people when taking NSAIDs, which corresponded to 18 more people out of 100 who develop adverse events. The NNTH to cause one additional participant to experience an adverse event was 6 (95% CI 3 to 57). Applying the GRADE criteria to the overall quality of the evidence, we downgraded once due to unclear risk of bias (see Table 4).
Due to very low quality evidence, we were uncertain whether tramadol alone had an increased risk of adverse events compared to other opioids (RR 0.99, 95% CI 0.87 to 1.13; 3 studies, 438 participants; Table 5; Bird 1995; Jensen 1994; Karlsson 2009). An I2 statistic of 90% indicated a high degree of heterogeneity for the trials comparing tramadol alone to other opioids. One trial was responsible for some of the statistical heterogeneity (Jensen 1994). This trial indicated that tramadol increased the risk of overall adverse events compared to other opioids, contrary to the findings of the other trials. This could be explained by the high dose of tramadol given to the treatment group (tramadol 100 mg three times daily). Applying the GRADE criteria to the overall quality of the evidence, we downgraded three times due to unclear risk of bias, inconsistency and imprecision (see Table 5).
We found low quality evidence that in one trial comparing tramadol in combination with acetaminophen to NSAIDs, there was no increase in risk of developing any adverse events (RR 1.17 95% CI 0.87 to 1.57; 1 study, 91 participants; Table 6; Park 2012). Applying the GRADE criteria to the overall quality of the evidence, we downgraded twice due to unclear risk of bias and imprecision (see Table 6).
Withdrawals due to adverse events
Placebo‐controlled trials
Nine trials with 2979 participants in the tramadol alone groups and 1555 participants in the placebo control groups contributed to the meta‐analyses of participants withdrawn or dropped out because of adverse events (Babul 2004; Burch 2007; DeLemos 2011; Fishman 2007; Fleischmann 2001; Gana 2006; Kean 2009; Malonne 2004; Schnitzer 1999). Two trials with 350 participants in the tramadol in combination with acetaminophen groups and 264 participants in the placebo control groups contributed to the meta‐analyses of participants withdrawn or dropped out because of adverse events (Emkey 2004; Silverfield 2002). There were 796 people with adverse events that made them stop the treatment in the 11 trials.
We found moderate quality evidence that participants who received tramadol alone had a greater risk of withdrawing from the study because of adverse events compared to participants who received placebo (RR 2.64, 95% CI 2.17 to 3.20; 9 studies, 4533 participants; Table 1). Nineteen out of 100 people withdrew due to adverse events when taking tramadol alone and 7/100 people withdrew due to adverse events when taking a placebo, which corresponded to 12 more people out of 100 people who withdrew due to adverse events. The NNTH to cause one additional participant to withdraw due to adverse events was 9 (95% CI 7 to 12; Table 1). An I2 statistic of 55% indicated a moderate degree of heterogeneity. Two trials were responsible for the statistical heterogeneity (Kean 2009; Malonne 2004). This could be explained by the duration of the Malonne 2004 trial, which was two weeks as compared to the 12‐week duration of the other trials, and the population, which was exclusively white as compared to the slightly more diverse population of the other studies. While both trials indicated that participants who received tramadol alone had a greater risk of withdrawing from the study because of adverse events compared to participants who received placebo (Kean 2009: RR 5.04, 95% CI 2.94 to 8.62; Malonne 2004: RR 25.73, 95% CI 3.54 to 187.02), they had higher RRs and wider CIs than the other trials. Applying the GRADE criteria to the overall quality of the evidence, we downgraded twice due to unclear risk of bias and inconsistency (see Table 1).
In one study which was not included in the meta‐analyses, 15 participants withdrew after randomization due to adverse events, 12 of whom were receiving tramadol at the time of their withdrawal (Thorne 2008).
We found low quality evidence that participants receiving tramadol in combination with acetaminophen had a greater risk of withdrawing from the study due to adverse events compared to participants who received placebo (RR 2.78, 95% CI 1.50 to 5.16; 2 studies, 614 participants; Table 2). Thirteen out of 100 people withdrew due to adverse events when taking tramadol in combination with acetaminophen and 5/100 people withdrew due to adverse events when taking placebo, which corresponded to 8 more people out of 100 people who withdrew due to adverse events. The NNTH to cause one additional participant to withdraw due to adverse events was 13 (95% CI 6 to 44; Table 2). An I2 statistic of 0% indicated a low degree of heterogeneity. Applying the GRADE criteria to the overall quality of the evidence, we downgraded twice due to unclear risk of bias and imprecision (see Table 2).
Active‐controlled trials
Six trials with 891 participants in the tramadol alone groups and 496 participants in the active control groups (including acetaminophen, NSAIDs and opioids) contributed to the meta‐analyses of participants withdrawn or dropped out because of adverse events. There were 275 adverse events that made them stop the treatment in the 11 trials.
Due to very low quality evidence, we were uncertain whether tramadol alone had a greater risk of withdrawing from the study because of adverse events compared to acetaminophen (RR 5.00, 95% CI 0.27 to 92.62; 1 study, 20 participants; Table 3; Bianchi 2003). Applying the GRADE criteria to the overall quality of the evidence, we downgraded three times due to unclear risk of bias and serious imprecision (see Table 3).
We found low quality evidence that participants receiving tramadol alone had a greater risk of withdrawing from the study because of adverse events compared to NSAIDs (RR 1.88, 95% CI 1.27 to 2.76; 2 studies, 929 participants; Table 4; Beaulieu 2008; DeLemos 2011). Twenty‐one out of 100 people withdrew due to adverse events when taking tramadol alone and 11/100 people withdrew due to adverse events when taking NSAIDs, corresponding to 10 more people out of 100 people who withdrew due to adverse events. The NNTH to cause one additional participant to withdraw due to adverse events was 10 (95% CI 5 to 33; see Table 4 for the main comparison). An I2 statistic of 60% indicated a high degree of heterogeneity. Applying the GRADE criteria to the overall quality of the evidence, we downgraded twice due to unclear risk of bias and inconsistency (see Table 4).
We found low quality evidence that participants receiving tramadol alone had an increased risk of withdrawing due to adverse events compared to other opioids (RR 2.26, 95% CI 1.52 to 3.37; 3 studies, 438 participants; Table 5; Bird 1995; Jensen 1994; Karlsson 2009). Thirty‐one out of 100 people withdrew due to adverse events when taking tramadol alone and 14/100 people withdrew due to adverse events when taking other opioids, corresponding to 17 more people out of 100 people who withdrew due to adverse events. The NNTH to cause one additional withdrawal due to adverse events was 6 (95% CI 3 to 14). An I2 statistic of 70% indicated a high degree of heterogeneity among studies comparing tramadol alone to other opioids. One trial was responsible for most of the statistical heterogeneity (Bird 1995). This trial had a short duration of two weeks and a small sample size of 30 participants. It indicated that participants receiving tramadol had a decreased risk of withdrawing due to adverse events compared to other opioids (RR 0.67, 95% CI 0.22 to 2.01), contrary to the findings of the other trials. Applying the GRADE criteria to the overall quality of the evidence, we downgraded twice due to the unclear risk of bias and inconsistency (see Table 5).
Serious adverse events
Placebo‐controlled trials
Seven trials with 2460 participants in the tramadol alone groups and 1153 participants in the placebo control groups contributed to the analysis of participants experiencing any serious adverse event (Babul 2004; Burch 2007; DeLemos 2011; Fishman 2007; Fleischmann 2001; Gana 2006; Malonne 2004). One trial with eight participants in the tramadol in combination with acetaminophen groups and seven participants in the placebo control groups contributed to the analysis of participants experiencing any serious adverse event (Peeva 2010). There were 132 people with serious adverse events in these eight trials. Serious adverse events reported in the tramadol groups included unstable angina, chest pain, breast cancer, diverticulitis, grand mal convulsions, prostate cancer, popliteal bursitis, small intestinal obstruction, cholelithiasis, pancreatitis and abdominal pain. However, authors of the trials mentioned only a few serious adverse events that may have been related to tramadol use. These were syncope, subendocardial myocardial infraction, renal insufficiency combined with an elevation of liver enzymes with inflammation of the liver, gastritis and drug withdrawal syndrome.
We found low quality evidence that participants receiving tramadol alone had a greater risk of developing serious adverse events compared to participants who received placebo (RR 1.78, 95% CI 1.11 to 2.84; 7 studies, 3612 participants; Table 1). Three out of 100 people developed serious adverse events when taking tramadol alone and 2/100 people developed serious adverse events when taking a placebo, corresponding to 1 more person out of 100 people who developed serious adverse events. The NNTH to cause one additional participant to experience a serious adverse event was 68 (95% CI 29 to 477; Table 1). An I2 statistic of 0% indicated low heterogeneity between trials. Applying the GRADE criteria to the overall quality of the evidence, we downgraded twice due to unclear risk of bias and imprecision (see Table 1).
In one study which was not included in the meta‐analyses, one serious adverse event occurred during the double‐blind phase of the study (atrial flutter in one participant with a history of supraventricular tachycardia who received tramadol 150 mg) (Thorne 2008).
We found low quality evidence that no serious adverse events were reported in a study of tramadol in combination with acetaminophen compared to placebo (1 study, 15 participants; Table 2; Peeva 2010). Applying the GRADE criteria to the overall quality of the evidence, we downgraded twice due to serious imprecision (see Table 2).
Active‐controlled trials
Seven trials with 348 participants in the tramadol groups and 350 participants in the active control groups evaluated the number of participants experiencing serious adverse events but only two contributed to the meta‐analyses since five trials did not report any serious adverse events. There were five people with serious adverse events in these seven trials. Some of the serious adverse events were gastrointestinal bleeding and severe pancreatitis. Due to very low quality evidence, we were uncertain whether tramadol alone compared to NSAIDs had an increased risk of developing serious adverse events (RR 0.21, 95% CI 0.01 to 4.34; 2 studies, 188 participants; Table 4; Beaulieu 2008; Pavelka 1998). Applying the GRADE criteria to the overall quality of the evidence, we downgraded three times due to unclear risk of bias and serious imprecision (see Table 4).
Similarly, due to very low quality evidence, we were uncertain whether participants who received tramadol alone had an increased risk of developing serious adverse events compared to participants who received other opioids (RR 7.42, 95% CI 0.39 to 141.00; 4 studies, 495 participants; Table 5; Bird 1995; Jensen 1994; Karlsson 2009; Wilder‐Smith 2001). Applying the GRADE criteria to the overall quality of the evidence, we downgraded three times due to unclear risk of bias and serious imprecision (see Table 5).
We found low quality evidence that there was no risk of developing serious adverse events for both the participants who received tramadol in combination with acetaminophen and those who received NSAIDs (1 study, 15 participants; Table 6; Peeva 2010). Applying the GRADE criteria to the overall quality of the evidence, we downgraded twice due to serious imprecision (see Table 6).
Minor outcomes
Symptoms of opioid dependence: withdrawal symptoms and propensity for abuse
Four trials reported withdrawal symptoms or the propensity for abuse, or both (Beaulieu 2008; DeLemos 2011; Emkey 2004; Gana 2006). However, only one trial provided the data required to contribute to the systematic review (Beaulieu 2008). This trial assessed propensity for abuse using the Drug Liking Index, a 9‐point scale. There was no difference between treatment groups in these scores. For the number of participants who liked the drug effect, tramadol had similar harms compared to NSAIDs (RR 1.03, 95% CI 0.62 to 1.74). Another trial reported that there was no evidence of abuse, but did not mention how it was assessed (Emkey 2004).
The other trials assessed this variable in various ways. One trial assessed the psychic dependence using the 49‐item Short Form Addiction Research Center Inventory questionnaire and the 16‐item Physical Dependence Questionnaire (PDQ) (DeLemos 2011). The rates of 12 related symptoms were different among treatment groups. About 5% to 10% more participants in the tramadol group reported these symptoms compared to the placebo group. In one of the studies, physical dependence was assessed using a 16‐item PDQ, where there was one serious adverse event related to study treatment and three drug withdrawal syndromes (non‐serious adverse events) in the tramadol group (Gana 2006). Thus, 4/815 (0.5%) participants in the tramadol group reported this event. There was a different frequency of physical dependence symptoms between treatment groups one week after discontinuation.
Two trials excluded participants that had any history of substance abuse (Beaulieu 2008; Emkey 2004), and two other trials excluded participants who had had substance abuse in the past six months (DeLemos 2011; Gana 2006).
Discussion
Summary of main results
In our systematic review and meta‐analyses, we found tramadol alone or in combination with acetaminophen demonstrated no clinically important difference compared to placebo in terms of mean pain relief and improvement of physical function in people with OA. The benefits were small, and may not have been clinically important since the 4‐point improvement in pain was lower than the MCID that we defined as 20 points on a 100‐point scale, although there were slightly more people in the tramadol group who were clinically important responders for both pain and physical function. Adverse events were also higher for participants using tramadol compared to placebo, which may limit the usefulness of tramadol. The quality of this evidence was moderate for benefit outcomes and low to moderate for harm outcomes for placebo‐controlled trials, mostly because of unclear risk of bias, as well as imprecision, especially for the harm outcomes. The quality of the evidence was very low to moderate for active‐controlled trials, mostly because of unclear risk of bias, imprecision and inconsistency.
Tramadol alone had similar benefits to other opioids in terms of pain relief and showed a small benefit in terms of physical function compared to other opioids, although probably not clinically important. The quality of this evidence was moderate, due to the unclear risk of bias in included trials. Tramadol alone showed lower benefits than NSAIDs for pain and physical function. However, these benefits were only small, and not clinically important. The quality of this evidence was moderate for pain and low for physical function, due to the risk of bias, as well as imprecision in the physical function analyses. Tramadol in combination with acetaminophen had similar benefits to NSAIDs and opioids for pain and physical function. The quality of this evidence was low to moderate, due to the unclear risk of bias and imprecision. Tramadol alone had similar harms to NSAIDs and opioids, except for withdrawals due to adverse events which were higher for tramadol compared to these controls, and overall adverse events which were higher for tramadol compared to NSAIDs. Tramadol in combination with acetaminophen also had similar harms to NSAIDs. Studies in which participants received tramadol alone had more adverse events than studies in which participants received tramadol in combination with acetaminophen, when compared to placebo. This could be explained by the higher dose of tramadol in the studies in which participants received tramadol alone, compared to a lower dose in studies of tramadol in combination with acetaminophen. The quality of the evidence was very low to moderate because of unclear risk of bias, imprecision of estimates and inconsistency between trials. Although a higher propensity for abuse when using tramadol was not established, it was rarely studied, which precluded any conclusions being made.
Overall completeness and applicability of evidence
This review had several limitations. Most trials were short in duration, with no trials that were longer than 13 weeks. The trials also varied in terms of duration (i.e. range of one week to three months) with a mean duration of two months. Trials allowed participants to take a wide range of dosages of tramadol (i.e. 37.5 mg/day to 400 mg/day) and rarely reported the mean dose of tramadol actually received by participants. Only four studies compared different doses of tramadol (DeLemos 2011; Fishman 2007; Gana 2006; Kean 2009), but doses were pooled in this review since they did not show different results. Most studies permitted additional use of analgesics, usually for pain other than due to OA. Some trials also permitted the use of other cointerventions such as physical therapy if its use was stable during the study. The inclusion of these cointerventions could have influenced the results.
Participants were predominantly women, with a mean age of 63 years and with moderate to severe pain, which represents most people with OA. Even though this review included trials for OA of the knee or hip (or both), it is important to note that a significant proportion of the trials were conducted with participants with knee OA only (13 studies including participants with knee or hip OA and nine studies with knee OA).
Most studies were conducted in high‐income countries. Studies were conducted in North America (13 studies) and Europe (seven studies, with one of these in both North America and Europe), Asia (two studies) and Africa (one study). Two studies were conducted in low‐ and middle‐income countries (i.e. Romania and South Africa) (Burch 2007; Wilder‐Smith 2001).
Thirteen studies included predominantly white participants while one study included predominantly African participants (study in South Africa) and eight studies did not mention it in their inclusion criteria or results. Since there are complex pain disparities related to ethnicity (Green 2003), and there are ethnic differences in the experience of chronic pain (Riley 2002), findings of included studies may not be applicable to people with OA of all races and ethnicities.
Three studies were conducted with participants who had previously failed other treatments or benefited from other treatments, which may add to clinical heterogeneity between studies. All studies listed multiple morbidities as exclusion criteria. Therefore, review findings may not be applicable to people with OA who have multiple morbidities. It is known that RCTs regularly exclude participants who have multiple morbidities, which is not representative of the general population that a physician would encounter in clinical practice. Findings that are applied in clinical practice should consider the complexity of effective treatment of these participants (Fortin 2006).
The included studies did not permit analysis of specified outcomes in this subset of the population. In the included studies, women of childbearing age were regularly excluded if they were pregnant, lactating or not using adequate contraception. The findings of this review may not be applicable to these women.
Based on the analyzed evidence, further research about the benefits and harms of tramadol for people with OA should include participants from different ethnicities and with multiple morbidities. Other desirable characteristics are: more head‐to‐head comparisons with active comparators, relevant outcomes recommended for OA, and study designs to accomplish low levels of risk of bias, and that are independently funded.
Quality of the evidence
Most of the trials were funded by the pharmaceutical industry and it was not possible to explore the role of this factor in explaining the estimated treatment effect due to the low number of trials that were not funded by industry. Many trials also had an unclear risk of bias for allocation concealment, blinding of personnel and selective outcome reporting, and a few of the trials were at high risk of bias for incomplete outcome data.
Some of the most important factors in determining the analgesic benefits and harms of tramadol are the dose being tested and the duration of the treatment. In this systematic review, the mean dose of tramadol varied between trials (range 37.5 mg to 400 mg daily), as well as the mean length of the trials of 57 days varied (3 to 91 days), which may explain some of the heterogeneity found in some analyses.
There was no substantive clinical heterogeneity (i.e. there were no important differences in sample, intervention or control group characteristics) but some comparisons showed moderate to high levels of heterogeneity between trials. However, given the acceptable lack of clinical heterogeneity, all trials were kept in the analyses.
Few studies assessed withdrawal symptoms or propensity for abuse and in various ways which precluded pooling this data. Since studies excluded participants with a history of substance abuse, it could lead to under‐reporting of the problem since participants who may abuse tramadol are likely to have a history of substance abuse. Studies should be conducted to address this potential adverse event in a more standardized manner. Studies addressing this issue should also be conducted for longer duration than 91 days (13 weeks), which was the longest duration in the trials assessed in this systematic review.
Potential biases in the review process
We performed a systematic literature search, which included both published (i.e. from various electronic databases) and unpublished trials (i.e. from protocol registries). This approach helped to ensure that all publications were identified and thus reduced potential publication bias. However, there are eight protocols of completed trials for which we could not obtain data, which remains a limitation (Studies awaiting classification). We contacted authors of each trial and protocol at least twice to obtain any missing information. In the cases of these protocols, we could not obtain more data. The funnel plot for trials contributing data for the pain relief effect of tramadol compared to placebo showed no publication bias, but there is still potentially a publication bias since we could not include any data from the protocols for which data was not published (see Appendix 2 with list of studies and authors/companies that were reached).
Two review authors independently performed selection of trials, risk of bias assessment and data extraction, and consulted a third review author if an agreement could not be reached. This process helped to ensure the accuracy of data and to reduce the risk of bias (Egger 2001; Gøtzsche 2007).
Agreements and disagreements with other studies or reviews
The previously published Cochrane systematic review on tramadol for OA included 11 trials that compared tramadol to placebo and other active controls (Cepeda 2006). We included data from 10 of these in our meta‐analyses and included data from 10 additional trials. Compared to this earlier published review, results showed slightly less pain relief (4 more points on a 100‐mm VAS for tramadol alone and tramadol in combination with acetaminophen versus 8.5 more points on a 100‐mm VAS for trials of tramadol alone and tramadol in combination with acetaminophen) and slightly higher physical function compared to placebo (4 out of 100 on a WOMAC Function scale for tramadol alone and tramadol in combination with acetaminophen versus 3 out of 100 points on a VAS for trials of tramadol alone and tramadol in combination with acetaminophen). We included 11 more trials compared to two other systematic reviews (Avouac 2007; Cepeda 2006), which may be the reason that we found smaller effects when estimating pain relief.
The current review also showed that tramadol was somewhat less effective in reducing pain and improving physical function compared to NSAIDs, which is a new finding compared to older systematic reviews of tramadol and other opioids, which could not reach conclusions on this comparison because of the small number of trials (Avouac 2007; Cepeda 2006).
The current review showed that the same types of adverse effects were reported for tramadol compared to placebo in the last published Cochrane Review of tramadol for OA (Cepeda 2006) and others (Avouac 2007).
As with the previous Cochrane Review, it was difficult to provide a robust estimate of adverse events when tramadol was compared to other drugs as there were few trials (Cepeda 2006). However, this review found an increased risk of withdrawals due to adverse events in the tramadol group compared to other NSAIDs and opioids. Similar to other systematic reviews of tramadol, there was not enough controlled trial evidence to make conclusions about withdrawal symptoms and propensity for abuse, so although observational evidence suggested that tramadol may have been well tolerated compared to other opioids, the same precautions against addiction and abuse should be taken as with other opioids. Long term RCTs of high quality are needed to investigate further potential harms for people with OA compared to placebo and other active treatments, such as acetaminophen, NSAIDs and other opioids.
Authors' conclusions
Implications for practice.
Based on moderate quality evidence, tramadol alone or in combination with acetaminophen probably has no important benefit on mean pain intensity or physical function over placebo in people with osteoarthritis. However, there were slightly more people in the tramadol group who achieved a clinically important response. Moderate quality evidence shows that adverse events probably cause substantially more participants to stop taking tramadol. The increase in serious adverse events with tramadol is less certain, due to the small number of events. Use of tramadol for osteoarthritis needs to consider the limited benefits with the likelihood of increasing the adverse effects.
Implications for research.
There is a need to study propensity for abuse in a more systematic method since it is rarely reported in trials.
What's new
| Date | Event | Description |
|---|---|---|
| 12 August 2019 | Amended | A typo in the abstract has been corrected |
History
Protocol first published: Issue 4, 2005 Review first published: Issue 3, 2006
| Date | Event | Description |
|---|---|---|
| 1 February 2018 | New citation required but conclusions have not changed | Updated search with 11 new studies. New author team. Conclusions similar to those of the previous review published in 2006. |
| 1 February 2018 | New search has been performed | Updated search – 11 new studies added. |
| 10 November 2008 | Amended | Converted to new review format. CMSG ID: C092‐R |
Acknowledgements
The Cochrane Musculoskeletal Review Group conducted the literature search. Jordi Pardo Pardo participated in the appraisal of the methodologic quality of studies. Chisa Cumberbatch participated in the appraisal of the methodologic quality of studies and performed some of the analysis. Nicole Auclair participated in the appraisal of the methodologic quality of studies.
Appendices
Appendix 1. MEDLINE search strategy
1. exp osteoarthritis/ 2. osteoarthr$.tw. 3. degenerative arthritis.tw. 4. tramadol.tw or tramadol.sh 5. ultracet.tw or ultracet.nm 6. or/1‐5 7. randomized controlled trial.pt. 8. controlled clinical trial.pt. 9. randomized controlled trials.sh. 10. random allocation.sh. 11. double blind method.sh. 12. single‐blind method.sh. 13. clinical trial.pt. 14. clinical trials.sh. 15. clinical trial.tw. 16. ((singl$ or doubl$ or trebl$ or tripl$) and (mask$ or blind$)).tw. 17. placebos.sh. 18. placebo$.tw. 19. random$.tw. 20. Research Design/ 21. comparative study.sh. 22. evaluation studies.sh. 23. follow‐up studies.sh. 24. prospective studies.sh. 25. control$.tw. 26. prospectiv$.tw. 27. volunteer$.tw. 28. or/7‐27 29. (animal not human).mp. 30. 28 not 29 31. and/6‐30
Appendix 2. Table of protocols awaiting classification
| Study name | Author | Registry | Identifier |
| Norspan® patches versus tramadol in subjects with chronic, moderate to severe osteoarthritis pain in the hip knee and/or lumbar spine | Dorthe Tvinnemose | Clinicaltrials.gov | NCT00426647 |
| A study comparing Norspan patch and oral tramadol | M Karlsson (principal investigator) | Clinicaltrials.gov | NCT01019265 |
| A comparative study of tramadol hydrochloride plus acetaminophen tablets maintenance versus non‐steroidal anti‐inflammatory drugs (NSAIDs) maintenance in participants with knee osteoarthritis | Janssen Korea (study director) | Clinicaltrials.gov | NCT00635349 |
| An efficacy, safety and effects on quality of life of tramadol/paracetamol [acetaminophen] as add‐on therapy in chronic osteoarthritis | Janssen Pharmaceutica (study director) | Clinicaltrials.gov | NCT01728246 |
| An efficacy and safety study of acetaminophen plus tramadol hydrochloride (JNS013) in participants with chronic pain | Janssen Pharmaceutica (study director) | Clinicaltrials.gov | NCT00736853 |
| A four‐arm study comparing the analgesic efficacy and safety of tramadol once a day 100, 200 and 300 mg versus placebo for the treatment of pain due to osteoarthritis of the knee | Not provided | Clinicaltrials.gov | NCT00832416 |
| A study to assess the ability of tramadol, naproxen and oxycodone to affect the pain thresholds of patients with osteoarthritis of the thumb | Pfizer CT.gov Call Center (study director) | Clinicaltrials.gov | NCT00743587 |
| A study of the tolerability of titrated dose tramadol/acetaminophen combination tablet in Korean patients with osteoarthritis | James WEI (principal investigator) | Clinicaltrials.gov | NCT01063842 |
Data and analyses
Comparison 1. Tramadol versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pain | 10 | Std. Mean Difference (Fixed, 95% CI) | ‐0.25 [‐0.32, ‐0.19] | |
| 1.1 Tramadol alone | 8 | Std. Mean Difference (Fixed, 95% CI) | ‐0.25 [‐0.32, ‐0.18] | |
| 1.2 Tramadol in combination with acetaminophen | 2 | Std. Mean Difference (Fixed, 95% CI) | ‐0.28 [‐0.45, ‐0.12] | |
| 2 Physical function | 7 | Std. Mean Difference (Fixed, 95% CI) | ‐0.22 [‐0.30, ‐0.14] | |
| 2.1 Tramadol alone | 5 | Std. Mean Difference (Fixed, 95% CI) | ‐0.20 [‐0.29, ‐0.12] | |
| 2.2 Tramadol in combination with acetaminophen | 2 | Std. Mean Difference (Fixed, 95% CI) | ‐0.27 [‐0.43, ‐0.11] | |
| 3 Number of participants experiencing any adverse events | 5 | 2347 | Risk Ratio (IV, Fixed, 95% CI) | 1.36 [1.26, 1.48] |
| 3.1 Tramadol alone | 4 | 2039 | Risk Ratio (IV, Fixed, 95% CI) | 1.34 [1.24, 1.46] |
| 3.2 Tramadol in combination with acetaminophen | 1 | 308 | Risk Ratio (IV, Fixed, 95% CI) | 1.91 [1.32, 2.76] |
| 4 Number of participants who withdrew due to adverse events | 11 | 5147 | Risk Ratio (IV, Fixed, 95% CI) | 2.65 [2.20, 3.19] |
| 4.1 Tramadol alone | 9 | 4533 | Risk Ratio (IV, Fixed, 95% CI) | 2.64 [2.17, 3.20] |
| 4.2 Tramadol in combination with acetaminophen | 2 | 614 | Risk Ratio (IV, Fixed, 95% CI) | 2.78 [1.50, 5.16] |
| 5 Number of participants experiencing any serious adverse events | 8 | 3627 | Risk Ratio (IV, Fixed, 95% CI) | 1.78 [1.11, 2.84] |
| 5.1 Tramadol alone | 7 | 3612 | Risk Ratio (IV, Fixed, 95% CI) | 1.78 [1.11, 2.84] |
| 5.2 Tramadol in combination with acetaminophen | 1 | 15 | Risk Ratio (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
1.1. Analysis.
Comparison 1 Tramadol versus placebo, Outcome 1 Pain.
1.2. Analysis.
Comparison 1 Tramadol versus placebo, Outcome 2 Physical function.
1.3. Analysis.
Comparison 1 Tramadol versus placebo, Outcome 3 Number of participants experiencing any adverse events.
1.4. Analysis.
Comparison 1 Tramadol versus placebo, Outcome 4 Number of participants who withdrew due to adverse events.
1.5. Analysis.
Comparison 1 Tramadol versus placebo, Outcome 5 Number of participants experiencing any serious adverse events.
Comparison 2. Tramadol versus active treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pain | 10 | Std. Mean Difference (Fixed, 95% CI) | 0.12 [0.01, 0.22] | |
| 1.1 Tramadol alone vs acetaminophen | 1 | Std. Mean Difference (Fixed, 95% CI) | 0.13 [‐0.80, 1.06] | |
| 1.2 Tramadol alone vs NSAIDs | 3 | Std. Mean Difference (Fixed, 95% CI) | 0.21 [0.07, 0.36] | |
| 1.3 Tramadol alone vs other opioids | 4 | Std. Mean Difference (Fixed, 95% CI) | ‐0.11 [‐0.33, 0.12] | |
| 1.4 Tramadol in combination with acetaminophen vs NSAIDs | 2 | Std. Mean Difference (Fixed, 95% CI) | 0.12 [‐0.16, 0.39] | |
| 1.5 Tramadol in combination with acetaminophen vs other opioids | 1 | Std. Mean Difference (Fixed, 95% CI) | 0.06 [‐0.31, 0.43] | |
| 2 Physical function | 4 | Std. Mean Difference (Fixed, 95% CI) | 0.23 [0.09, 0.36] | |
| 2.1 Tramadol alone vs NSAIDs | 3 | Std. Mean Difference (Fixed, 95% CI) | 0.23 [0.09, 0.37] | |
| 2.2 Tramadol in combination with acetaminophen vs NSAIDs | 1 | Std. Mean Difference (Fixed, 95% CI) | 0.2 [‐0.21, 0.61] | |
| 3 Function: overall assessment | 1 | 190 | Risk Ratio (IV, Fixed, 95% CI) | 1.32 [1.04, 1.68] |
| 3.1 Tramadol alone vs other opioids | 1 | 190 | Risk Ratio (IV, Fixed, 95% CI) | 1.32 [1.04, 1.68] |
| 4 Number of participants experiencing any adverse even | 5 | 663 | Risk Ratio (IV, Fixed, 95% CI) | 1.07 [0.96, 1.19] |
| 4.1 Tramadol alone vs NSAIDs | 1 | 128 | Risk Ratio (IV, Fixed, 95% CI) | 1.31 [1.03, 1.67] |
| 4.2 Tramadol alone vs other opioids | 3 | 438 | Risk Ratio (IV, Fixed, 95% CI) | 0.99 [0.87, 1.13] |
| 4.3 Tramadol in combination with acetaminophen vs NSAIDs | 1 | 97 | Risk Ratio (IV, Fixed, 95% CI) | 1.17 [0.87, 1.57] |
| 5 Number of participants who withdrew due to adverse events | 6 | 1387 | Risk Ratio (IV, Fixed, 95% CI) | 2.07 [1.57, 2.73] |
| 5.1 Tramadol alone vs acetaminophen | 1 | 20 | Risk Ratio (IV, Fixed, 95% CI) | 5.0 [0.27, 92.62] |
| 5.2 Tramadol alone vs NSAIDs | 2 | 929 | Risk Ratio (IV, Fixed, 95% CI) | 1.88 [1.27, 2.76] |
| 5.3 Tramadol alone vs other opioids | 3 | 438 | Risk Ratio (IV, Fixed, 95% CI) | 2.26 [1.52, 3.37] |
| 6 Number of participants experiencing any serious adverse events | 7 | 698 | Risk Ratio (IV, Fixed, 95% CI) | 1.31 [0.16, 10.79] |
| 6.1 Tramadol alone vs NSAID | 2 | 188 | Risk Ratio (IV, Fixed, 95% CI) | 0.21 [0.01, 4.34] |
| 6.2 Tramadol alone vs other opioids | 4 | 495 | Risk Ratio (IV, Fixed, 95% CI) | 7.42 [0.39, 141.00] |
| 6.3 Tramadol in combination with acetaminophen vs NSAIDs | 1 | 15 | Risk Ratio (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7 Symptoms of opioid dependence: propensity for abuse | 1 | 97 | Risk Ratio (IV, Fixed, 95% CI) | 1.03 [0.62, 1.74] |
| 7.1 Tramadol alone vs NSAIDs | 1 | 97 | Risk Ratio (IV, Fixed, 95% CI) | 1.03 [0.62, 1.74] |
2.1. Analysis.
Comparison 2 Tramadol versus active treatment, Outcome 1 Pain.
2.2. Analysis.
Comparison 2 Tramadol versus active treatment, Outcome 2 Physical function.
2.3. Analysis.
Comparison 2 Tramadol versus active treatment, Outcome 3 Function: overall assessment.
2.4. Analysis.
Comparison 2 Tramadol versus active treatment, Outcome 4 Number of participants experiencing any adverse even.
2.5. Analysis.
Comparison 2 Tramadol versus active treatment, Outcome 5 Number of participants who withdrew due to adverse events.
2.6. Analysis.
Comparison 2 Tramadol versus active treatment, Outcome 6 Number of participants experiencing any serious adverse events.
2.7. Analysis.
Comparison 2 Tramadol versus active treatment, Outcome 7 Symptoms of opioid dependence: propensity for abuse.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Babul 2004.
| Methods | 12‐week multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group study Setting: clinic |
|
| Participants | Inclusion criteria: > 50 years of age, < 30 minutes of morning stiffness with OA of knee or crepitus (or both); Participants met the ACR diagnostic criteria. Number of participants: 246; tramadol ER group: 124; control group: 122 Mean age: 61 years % women: tramadol ER group: 66.1% women; control group: 56.6% women |
|
| Interventions | Active group: tramadol ER 100 mg once/day, up to 400 mg/day in the morning Control group: identical appearing placebo in the morning 2–7‐day washout period during which all analgesics were discontinued. Tramadol ER initiated at 100 mg once/day and increased to 200 mg once/day by the end of 1 week of treatment. After the first week, further increases to tramadol ER 300 mg or 400 mg once/day were allowed. Mean tramadol ER dose 276 mg once daily. Treatment continued for 12 weeks. |
|
| Outcomes | Analgesia evaluated by Arthritis Pain Intensity VAS, and WOMAC Osteoarthritis Index Pain VAS subscale (5 questions rating overall OA pain) Physical function and stiffness evaluated using WOMAC Osteoarthritis Index and WOMAC Osteoarthritis Index Physical Function subscale Pain and physical function evaluated with VAS and WOMAC Osteoarthritis Index Sleep evaluated using CPSI Global Assessment of Therapy Withdrawals due to adverse events Adverse events; only number of adverse events reported Extracted pain outcome: Arthritis Pain Intensity VAS (0–100 mm) at 12 weeks, with lower values indicating benefit Extracted physical function outcome: WOMAC Physical Function subscale (0–1700 mm) at 12 weeks, with lower values indicating benefit |
|
| Notes | We contacted the author to clarify how the WOMAC Osteoarthritis Index was reported, and the author provided all the information requested. For the pooling, we normalized the WOMAC Total score. Study managed by SCIREX Corporation, Horsham (PA) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A list of randomization numbers based on a computer‐generated randomization schedule was prepared." (p.61) |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Patients who fulfilled all study selection criteria were assigned a randomization number sequentially with an equal likelihood of being assigned to either treatment group." (p.61) No mention of allocation concealment |
| Blinding of personnel | Unclear risk | Authors did not explicitly report blinding of personnel. |
| Blinding of participants | Low risk | Quote: "…identical appearing placebo, also given once a day." (p.61) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Efficacy assessments were performed throughout the course of the study using a daily patient diary and clinic visit‐based assessments." (p.61) Since this was a participant‐reported outcome and participants were blinded, the outcome assessment was considered blinded. |
| Incomplete outcome data for pain and physical function | High risk | Quote: "Efficacy analyses were conducted on an intent‐to‐treat (ITT) population." (p.62) Withdrawals due to adverse events were 27% (33/124) with tramadol vs 7% (9/122) with placebo. The percentage of withdrawals for reasons other than adverse events (lack of efficacy and others) were 23.4% (29/124) with tramadol vs 40.9% (50/122) with placebo. There was a high number of withdrawals for reasons other than adverse events so although benefits analyses were done on ITT, the true outcome data for half of the participants were unavailable. |
| Incomplete outcome data for adverse effects All outcomes | High risk | Quote: "Safety analyses were conducted for the safety population that included all randomized patients who received at least one dose of study medication." Withdrawals due to adverse events were 27% (33/124) with tramadol vs 7% (9/122) with placebo. The percentage of withdrawals for reasons other than adverse events (lack of efficacy and other) were 23.4% (29/124) with tramadol vs 40.9% (50/122) with placebo. There was a high number of withdrawals for reasons other than adverse events so although benefits analyses were done on ITT, the true outcome data for half of the participants were unavailable. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (pain, physical function and stiffness, and sleep quality) appeared to have been reported in the results, but we did not have access to the protocol for verification. |
| Other biases | Low risk | No other sources of bias detected. |
Beaulieu 2008.
| Methods | Randomized, double‐blind, parallel‐group comparison of the benefits, safety and clinical benefits of CR tramadol vs SR diclofenac over a 6‐week treatment period Setting: clinic |
|
| Participants | Number of participants: 129: CR tramadol: 62; SR diclofenac: 66 Men and non‐pregnant women ages 35–75 years with chronic pain due to primary knee or hip OA of at least moderate severity |
|
| Interventions | Active group: CR tramadol titrated to optimal dose (200 mg/day, 300 mg/day or 400 mg/day) Control group: SR diclofenac packaged and labeled in the same way as the treatment and titrated to their optimal dose (75 mg or 100 mg once daily, or 75 mg twice a day), unless adequate pain control was achieved or adverse effects prevented the dose from being titrated further. All participants were randomly assigned an initial dose of either active CR tramadol 200 mg and placebo SR diclofenac 75 mg each morning, or active SR diclofenac 75 mg and placebo CR tramadol 200 mg each morning. Treatment lasted 6 weeks |
|
| Outcomes | The overall PI over the preceding 2 weeks assessed with 100‐mm VAS. The WOMAC Physical Function subscale used to report physical function and pain. Impact of pain on quality and quantity of sleep using the Pain and Sleep Questionnaire Withdrawals due to adverse events Other outcomes included PI while walking on a flat surface, going up or down stairs, at night while in bed, sitting or lying, standing upright, effect of pain on quality and quantity of sleep, global assessment of clinical benefits (participant and investigator) and Drug Liking Index. The effect sizes were assessed in terms of change over the study period. Extracted pain outcome: WOMAC Pain subscale (0–100 mm) during the last 2 weeks of treatment, with lower values indicating benefit Extracted physical function outcome: WOMAC Physical Function subscale (0–1700 mm) at 8 weeks, with lower values indicating benefit |
|
| Notes | Study funded by Purdue Pharma, Canada | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Study medication was prepackaged with an assigned randomization number, according to a computer‐generated code, in blocks of four." (p.105) |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Study medication was prepackaged with an assigned randomization number, according to a computer‐generated code, in blocks of four." (p.105) Not mentioned if packaging was opaque and sealed. Techniques used to implement the sequence not addressed. |
| Blinding of personnel | Unclear risk | Authors did not explicitly report blinding of personnel |
| Blinding of participants | Low risk | Quote: "Blinding was maintained using the double‐dummy technique." (p.105) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Patients recorded their pain intensity in a diary." (p.105) Quote: "At the completion of the study, the patient and investigator each provided a global assessment of clinical effectiveness." (p.105) Since this was a participant‐reported outcome and participants were blinded, the outcome assessment was considered blinded. |
| Incomplete outcome data for pain and physical function | Low risk | Quote: "Safety data are presented using the ITT population and efficacy data are presented using the per protocol population." (p.106) Quote: "All patients who completed the study were included in the per protocol population, with the exception of one patient with a protocol violation." (p.106) Quote: "one patient was excluded from all analyses due to lack of evidence of OA." (p.106) Quote: "Ninety‐seven patients were evaluated for efficacy; 45 in the CR tramadol group and 52 in the SR diclofenac group." (p.106) Only 97/129 (75%) participants were included in the per‐protocol population and evaluated for benefits. The missing 31 people withdrew from the study before completion due to reasons including adverse events and inadequate pain control. Withdrawals due to adverse events were 16.1% (10/62) with tramadol vs 15.2% (10/66) with diclofenac. Percentage of withdrawals for reasons other than adverse events (inadequate pain control, intermittent illness, voluntary withdrawal, etc.) were 11.3% (7/62) with tramadol vs 6.1% (4/66) with diclofenac. Despite the overall withdrawal rates being > 20%, most withdrawals were due to reasons other than adverse events. |
| Incomplete outcome data for adverse effects All outcomes | Low risk | Quote: "Safety data are presented using the ITT population and data are presented using the per protocol population." (p.106) Quote: "All randomly assigned patients were included in the intent‐to‐treat (ITT) population, with the exception of one patient who did not meet the eligibility criteria." (pp.105–106) 128/129 participants evaluated for safety since they were included in ITT population. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (pain, physical function and stiffness, and sleep quality) appeared to have been reported in results but we did not have access to the protocol for verification. |
| Other biases | Low risk | No other sources of bias detected. |
Bianchi 2003.
| Methods | Prospective, randomized, double‐blind, between‐patient study Setting: Rheumatology Unit of the Ospedale di Circolo e Fondazione Macchi, Varese (Italy) |
|
| Participants | 20 adults (2 men, 18 women), ages ≥ 38 years (mean age 67.5 years in tramadol group and 71 years in control group) with OA of the knee and a minimum VAS score of 40 mm. Any treatments for pain were discontinued ≥ 24 hours before study. Intake of any other analgesic medications suspended during study. Number of participants: tramadol group: 10; control group (acetaminophen): 10. |
|
| Interventions | Active group: tramadol 50 mg 3 times/day for 7 days: n = 10 Control group: acetaminophen 500 mg 3 times/day for 7 days: n = 10 All treatments were masked by administration of identical capsules containing the drugs. Any treatment for pain was discontinued ≥ 24 hours before study. |
|
| Outcomes | PI: recorded 120 minutes after taking drug on a 10‐cm line VAS with endpoints 'no pain' and 'worst pain' Synovial fluid and blood plasma: concentrations of tramadol, O‐desmethyl‐tramadol, substance P and IL‐6 Withdrawals due to adverse events: tramadol: 2/10 (due to nausea and vomiting); acetaminophen: 0/10 Extracted pain outcome: PI (0–10 cm VAS) 2 hours after the medication administration, with lower values indicating benefit No physical function outcome reported |
|
| Notes | We contacted the author to request the percentage of participants with pain relief. We obtained no response. Funding source not mentioned |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Enrolled patients were assigned by computer‐generated random numbers." (p.1902) |
| Allocation concealment (selection bias) | Unclear risk | No mention of allocation concealment. |
| Blinding of personnel | Unclear risk | Authors did not explicitly report blinding of personnel. |
| Blinding of participants | Low risk | Quote: "All treatments were masked by administration of identical capsules containing the drugs." (p.1902) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Data regarding the efficacy of the treatments were collected in the presence of the investigator before the first drug administration, and on the last day of each drug treatment." (p.1903) Since this was a participant‐reported outcome and participants were blinded, the outcome assessment was considered blinded. |
| Incomplete outcome data for pain and physical function | Low risk | Data on participant baseline pain assessment provided. However, 2/10 participants withdrew from treatment due to adverse events. It is not mentioned if the 2 dropouts were included in the benefits assessment. |
| Incomplete outcome data for adverse effects All outcomes | Low risk | Data on participant baseline pain assessment provided. However, 2/10 participants withdrew from treatment due to adverse events. It is not mentioned if the 2 dropouts were included in the benefits assessment. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (pain, synovial fluid, plasma, IL‐6 concentrations, O‐desmethyl‐tramadol concentrations) appeared to have been reported in the results but no access to protocol for verification. |
| Other biases | Low risk | No other sources of bias detected. |
Bird 1995.
| Methods | Cross‐over, double‐blind RCT Setting: clinic |
|
| Participants | 40 participants with radiologically confirmed diagnosis of OA of hip or knee within 12 months of starting the study; 19 participants completed both treatment periods. % women: active group: 65%; control group: 70% Active group: 13 participants had moderate to severe OA; control group: 14 participants had moderate to severe OA |
|
| Interventions | Cross‐over trial Initial visit: active group received tramadol 50 mg 4 times/day; control group received pentazocine 50 mg 4 times/day) for 2‐week period. Second visit: participants crossed over to the other drug for another 2 weeks. No washout period |
|
| Outcomes | Major: Pain severity (4‐point Likert scale: none, mild, moderate, severe) recorded on diary cards completed daily Duration of morning stiffness (minutes) and severity (same 4‐point Likert scale) Number of acetaminophen tablets consumed Minor: Duration of inactivity stiffness (minutes) Pain during daily activities and walking (none, mild, moderate, severe) Pain during sleep (normal sleep, some interruption of sleep, moderate interruption of sleep, no sleep) Functional impairment, e.g. climbing stairs, getting out of bed and rising from a chair (no difficulty, a little difficulty, moderate difficulty, great difficulty or impossible) Participant's assessment of treatment (very good, good, fair, poor or very poor) Extracted pain outcome: severity of pain (4‐point Likert scale) during the last 7 days of treatment, with lower values indicating benefit Extracted physical function outcome: no extractable data |
|
| Notes | Grünenthal GmbH supplied the drugs | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were then randomly allocated to either tramadol 50 mg qds or pentazocine 50 mg qds." (p.182) No mention of how the allocation sequence was randomly generated. |
| Allocation concealment (selection bias) | Unclear risk | No mention of allocation concealment. |
| Blinding of personnel | Unclear risk | Authors did not explicitly report blinding of personnel |
| Blinding of participants | Unclear risk | Authors did not explicitly report blinding of participants |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Authors did not explicitly report blinding of outcome assessors |
| Incomplete outcome data for pain and physical function | Unclear risk | Quote: "analyses of these total scores were (…) carried out on two patient cohorts. Cohort 1 comprised those patients who took at least one dose of the study medication in each period and who had pain scores for at least four days. Cohort 2 comprised patients who took at least one dose of study medication in each period and recorded pain scores on less than four days unless they withdrew due to lack of efficacy." (p.183) Quote: "The single patient withdrawing for treatment failure was taking pentazocine and experienced a flare in OA." (p.184) Quote: "the study was somewhat compromised by a high withdrawal rate due to adverse events." (p.187) Only 19/40 (47.5%) participants completed the study. Total withdrawals: 45% with tramadol vs 60% with pentazocine. Percentage for withdrawals due to adverse events: 45% with tramadol vs 55% with pentazocine. In cohort 2, unknown how many people withdrew due to lack of benefits so it is unknown how many participants were excluded from the analyses. Unknown how many people were in each cohort. |
| Incomplete outcome data for adverse effects All outcomes | Unclear risk | Quote: "analyses of these total scores were (…) carried out on two patient cohorts. Cohort 1 comprised those patients who took at least one dose of the study medication in each period and who had pain scores for at least four days. Cohort 2 comprised patients who took at least one dose of study medication in each period and recorded pain scores on less than four days unless they withdrew due to lack of efficacy." (p.183) Quote: "The single patient withdrawing for treatment failure was taking pentazocine and experienced a flare in OA." (p.184) Quote: "the study was somewhat compromised by a high withdrawal rate due to adverse events." (p.187) Only 19/40 (47.5%) participants completed the study. Total withdrawals: 45% with tramadol vs 60% with pentazocine. Percentage of withdrawals due to adverse events: 45% with tramadol vs 55% with pentazocine. In cohort 2, unknown how many people withdrew due to lack of benefits so it is unknown how many participants were excluded from the analyses. Unknown how many people were in each cohort. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (pain, stiffness, sleep quality, and physical function) appeared to have been reported in the results but we did not have access to the protocol for verification. |
| Other biases | High risk | 4/18 (22%) participants in the pentazocine group used ≥ 80% of their medication but 13/19 (68.4%) participants in the tramadol group used ≥ 80% of their medication. Results on treatment benefits may have been biased because participants in the pentazocine group were not as compliant with taking their medication. Was use of a cross‐over design appropriate? Yes, OA was stable. Was it clear that the order of receiving treatments was randomized? Unclear, randomized but no details on randomization procedure. Can it be assumed that the trial was not biased from carry‐over effects? No, no washout period. Are unbiased data available? No, used a Wilcoxon Rank Sum test which is used for independent, not dependent samples. |
Burch 2007.
| Methods | Multicenter, randomized, consisting of open‐label and double‐blind phase. Setting: clinic |
|
| Participants | 1028 participants ages 40–80 years with pain due to OA (646 of whom were randomized to double‐blind treatment). % women: active group: 62%; control group: 64% |
|
| Interventions | 7‐day washout period between the open‐label and double‐blind treatments included. During double‐blind treatment, active group received tramadol titrated to final dose of 200 mg or 300 mg, which was maintained for 12 weeks. Control group received placebo that was packaged and labeled in the same way as the treatment. |
|
| Outcomes | PI rated on an 11‐point PI‐NRS. Assessments of the Patient and Physician Global Impressions of Change were both based on the overall change in status from the beginning of the study using a 7‐point categorical scale ranging from 1 (very much improved) to 7 (very much worse). The Patient and Physician Global Impressions of Change integrated the effect of treatment on pain, adverse effects and the participant's expectation of pain relief. Safety assessed by physical exam, clinical laboratory tests, vital signs, adverse events and concomitant medication at all study visits. Withdrawals due to adverse effects reported. The effect sizes were assessed in terms of change over the study period. Extracted pain outcome: PI‐NRS (0–11 point) at 12 weeks, with lower values indicating benefit No physical function outcome reported |
|
| Notes | Funding source not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "At the beginning of the double‐blind phase, eligible patients were randomized (…) in blocks of six according to a previously established randomization schedule computer‐generated" (p.330) Quote: "patients were assigned study medication by means of a central interactive voice‐response system" (p.330) |
| Allocation concealment (selection bias) | Low risk | Quote: "…patients were assigned study medication by means of a central interactive voice‐response system. (p.330) Quote: "…inactive placebo tablets identical to the different dose forms of tramadol Contramid OAD were packaged and labelled in the same way as the active treatment" (p.330) |
| Blinding of personnel | Low risk | Quote: "Participants and site personnel were blinded to treatment assignments" (p.330) |
| Blinding of participants | Low risk | Quote: "Participants and site personnel were blinded to treatment assignments" (p.330) Quote: "To maintain the double blind, inactive placebo tablets identical to the different dose forms of Tramadol Contramid OAD were packaged and labelled in the same way as the active treatment." (p.330) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Participants were asked to evaluate the intensity of their pain." (p.330) Because participants and site personnel described as blinded, risk of bias in outcome assessment deemed low. |
| Incomplete outcome data for pain and physical function | Low risk | Quote: "The efficacy analysis was conducted on the full‐analysis population, defined as all patients who received at least one dose of the randomized study medication regardless of the status of the post‐dosing assessment." (p.331) 645/646 randomized participants were included in the full analysis population. Withdrawals due to adverse events were 10.2% (44/431) with tramadol vs 5.1% (11/214) with placebo. Withdrawals due to reasons other than adverse events were 14.4% (62/431) with tramadol vs 17.8% (38/214) with placebo |
| Incomplete outcome data for adverse effects All outcomes | Low risk | Quote: "The safety population included all patients who received at least one dose of study medication." (p.331) All 646 randomized participants included in safety population. Data from participants who did not discontinue due to lack of benefits were censored at time of their last dose of study medication. Withdrawals due to adverse events: 10.2% (44/431) with tramadol vs 5.1% (11/214) with placebo. Withdrawals due to reasons other than adverse events: 14.4% (62/431) with tramadol vs 17.8% (38/214) with placebo. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (pain and physical function) appeared to have been reported in the results, but we did not have access to the protocol for verification. |
| Other biases | Low risk | No other sources of bias |
DeLemos 2011.
| Methods | Phase III, 12‐week, multicenter, randomized, double‐blind, placebo‐controlled, dose‐ranging trial | |
| Participants | 1011 participants randomized, 1001 analyzed for safety/benefits; 555 completed study treatment Adults with knee or hip (or both) OA and baseline PI ≥ 40/100 on 100‐mm VAS. Safety/ITT population for 5 groups (number of participants): tramadol 100 mg: 201; 200 mg: 199; 300 mg: 199; celecoxib: 202; placebo: 200 % women: tramadol 100 mg: 58.2%; tramadol 200 mg: 62.3%; tramadol 300 mg: 61.8%; celecoxib: 64.9%; placebo: 68.5% |
|
| Interventions | Eligible participants underwent a 2–7‐day washout of prior analgesic therapy. Double‐blind treatment period (12 weeks) Active group 1: tramadol 100 mg Active group 2; tramadol 200 mg Active group 3: tramadol 300 mg Active group 4: celecoxib Control: placebo |
|
| Outcomes | Arthritis PI assessed over entire study using 100‐mm VAS (0 = no pain, 100 = extreme pain) WOMAC Pain, Physical Functioning and Stiffness subscales Participant and physician global assessment of disease activity on 100‐mm VAS Sleep assessed with the CPSI (which included 100‐mm VAS) At baseline and at weeks 6 and 12, participants completed the SF‐36 Health Survey Safety assessments included reports of adverse events, either spontaneously or in response to non‐directed questioning, and results of physical exams, vital signs, clinical laboratory tests and electrocardiograms at study visits. Participants completed 49‐item Short Form ARCI questionnaire at baseline and week 12. Participants also completed 16‐item PDQ at baseline, week 12 (or early discontinuation), and week 13 (or 1 week after early discontinuation) Extracted pain outcome: WOMAC Pain subscale (0–100 mm) at 12 weeks, with lower values indicating benefit Extracted physical function outcome: WOMAC Physical Function subscale (0–1700 mm) at 12 weeks, with lower values indicating benefit |
|
| Notes | Supported by Biovail Corporation and Ortho‐McNeil Janssen Scientific Affairs, LLC | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized" (p.217) Quote: "Patients (…) were randomly assigned to 12 weeks of study treatment in a 1:1:1:1:1 ratio." (p.217) No mention of how the randomization process was carried out. |
| Allocation concealment (selection bias) | Unclear risk | No mention of allocation concealment |
| Blinding of personnel | Unclear risk | Authors did not explicitly report blinding of personnel |
| Blinding of participants | Low risk | Quote: "placebo tablets and capsules were matched in size and color to the active tramadol ER tablets and the over encapsulated celecoxib capsules, respectively." (p.218) Quote: "The double‐blind was maintained during the study by each patient taking 3 tablets and 1 capsule once daily." (p.218) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Since this was a participant‐reported outcome and participants were blinded, the outcome assessment was considered blinded. |
| Incomplete outcome data for pain and physical function | High risk | Quote: "Patients who discontinued early had their last observation carried forward for efficacy analyses" (p.224) 10/1011 (9.9%) randomized participants were not in the safety/ITT population due to "no dose documented" (p.219). Although only 555/1011 (54.9%) participants completed the study, 1001 participants were included in the safety/ITT analysis. Withdrawals due to adverse events: 18.9% with tramadol vs 7.5% with placebo. Withdrawals for reasons other than adverse events (lack of efficacy, participant choice, other): 24.6% with tramadol vs 41% with placebo. The overall high withdrawal rate due to reasons other than adverse events was likely to lead to biased outcome data despite ITT analysis. |
| Incomplete outcome data for adverse effects All outcomes | High risk | 10/1011 (9.9%) randomized participants were not in the safety/ITT population due to "no dose documented" (p.219). Although only 555/1011 (54.9%) participants completed the study, 1001 participants were included in the safety/ITT analysis. Withdrawals due to adverse events: 18.9% with tramadol vs 7.5% with placebo. Withdrawals for reasons other than adverse events (lack of efficacy, participant choice, other): 24.6% with tramadol vs 41% with placebo. The overall high withdrawal rate due to reasons other than adverse events was likely to lead to biased outcome data despite ITT analysis. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (pain, physical function and stiffness, and sleep quality) appeared to have been reported in the results, but we did not have access to the protocol for verification. |
| Other biases | Low risk | No other sources of bias. |
Emkey 2004.
| Methods | Multicenter, randomized, double‐blind, placebo‐controlled trial | |
| Participants | Randomized 307 participants with ≥ 1 year of OA of hip or knee, experiencing at least moderate OA pain. ITT population (number of participants): tramadol/acetaminophen: 153; control: 153. % women: active group: 65%; control group: 71.2% |
|
| Interventions | Participants randomized after 3‐week screening and washout period of all non‐COX‐2 analgesics Active group: tramadol 37.5 mg/acetaminophen 325 mg combination pills for a total of 13 weeks. Medication titrated by 1 pill every 3 days to a total of 4 pills/day on day 10, and thereafter as needed to a maximum of 8 pills/day Control group: matching placebo for a total of 13 weeks |
|
| Outcomes | Major benefits variable was VAS scores, which participants rated from 'no pain' (0 mm) to 'extreme pain' (100 mm).
Minor outcomes included pain relief rating scores (scale of 4 to –1: 4 = complete, 3 = a lot, 2 = moderate, 1 = slight, 0 = none, –1 = worse), overall medication assessment by both physicians and participants at final visit, cumulative distribution of time to discontinuation due to lack of benefits, proportion of participants discontinuing due to lack of benefits, WOMAC Osteoarthritis Index questionnaire scores, and SF‐36 Health Survey scores. Extracted pain outcome: pain on VAS (0–100 mm) at 91 days, with lower values indicating benefit Extracted physical function outcome: WOMAC Physical Function subscale (0–10) at 91 days, with lower values indicating benefit |
|
| Notes | Supported by Ortho‐McNeil Pharmaceutical, Inc., Raritan (NJ) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized" Quote: "Subjects were recruited from within investigator's medical practices and through advertising." (p.151) No description of randomization process. |
| Allocation concealment (selection bias) | Unclear risk | No mention of allocation concealment. |
| Blinding of personnel | Low risk | Quote: "All subjects, investigators, and clinical personnel were blinded to treatment assignments until the trial was complete and the database had been finalized." (p.151) |
| Blinding of participants | Low risk | Quote: "Tramadol/APAP [acetaminophen] or matching placebo was titrated…" (p.150) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All subjects, investigators, and clinical personnel were blinded to treatment assignments until the trial was complete and the database had been finalized." (p.151) |
| Incomplete outcome data for pain and physical function | High risk | Quote: "Efficacy analyses were performed on the intent‐to‐treat population, defined as all randomized subjects who took at least one dose of study medication and for whom a post‐randomization efficacy measurement was available." (p.151) 306/307 randomized participants were included in the ITT and evaluable‐for‐safety populations. 227/307 randomized participants completed treatment. Withdrawal due to insufficient pain relief: 13/153 (8.5%) with tramadol vs 26/154 (16.9%) with placebo. Withdrawals due to adverse events: 13.1% with tramadol vs 3.9% with placebo. Withdrawals for reasons other than adverse events (insufficient pain relief, participant choice, intercurrent illness, other): 21/153 (13.7%) with tramadol vs 54/153 (35.3%) with placebo. There was an imbalance between the groups regarding reasons for withdrawal and an important (> 20%) overall withdrawal rate. |
| Incomplete outcome data for adverse effects All outcomes | High risk | Quote: "Safety assessments were performed on randomized subjects who took at least one dose of study medication and had at least one available post baseline safety measurement." Withdrawals due to adverse events: 13.1% with tramadol vs 3.9% with placebo. Withdrawals for reasons other than adverse events (insufficient pain relief, participant choice, intercurrent illness, other): 21/153 (13.7%) with tramadol vs 54/153 (35.3%) with placebo. There was an imbalance between the groups regarding reasons for withdrawal and an important (> 20%) overall withdrawal rate. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (pain, WOMAC Osteoarthritis Index, SF‐36 Health Survey) appeared to have been reported in the results, but we did not have access to the protocol for verification. |
| Other biases | Low risk | No other sources of bias. |
Fishman 2007.
| Methods | Randomized, multicenter, double‐blind, parallel arm trial; 3 phases: baseline, run‐in and maintenance | |
| Participants | 552 participants ages 40–75 years with pain from OA of the knee, with WOMAC Pain score > 150 mm at baseline. % women: tramadol 100 mg: 60.2%; tramadol 200 mg: 59.8%; tramadol 300 mg: 65.7%; placebo: 61.6% |
|
| Interventions | Baseline period included washout of prior analgesics. Active group 1: tramadol 100 mg Active group 2: tramadol 200 mg Active group 3 tramadol 300 mg Control group: placebo Run‐in: 6 days during which the dose was titrated by 100 mg increments every 2–3 days until the randomized dose reached. Maintenance: benefits evaluations were performed at end of run‐in and 3, 6 and 12 weeks of maintenance treatment at the randomized dose (of either tramadol or placebo). Duration of treatment in all groups: 12 weeks |
|
| Outcomes | WOMAC Pain score Global rating of pain relief Adverse events Serious adverse events Extracted pain outcome: WOMAC Pain subscale (0–500 mm) at 12 weeks, with lower values indicating benefit No physical function outcome was extracted as end‐of‐study data were not available. |
|
| Notes | Funded by Labopharm. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A centralized computer‐generated randomization list produce by Aptuit, Allendale, NJ, assigned the three different doses of study medication and placebo to individual randomization numbers in a ratio of 1:1:1:2 and in blocks of five." (p.275) |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients at each center were assigned a sequential patient number that corresponded to one of the random medication supplies in the block provided to the center." (p.275) Used double‐dummy technique |
| Blinding of personnel | Low risk | Quote: "A double‐blind, double‐dummy technique was used to ensure that patients and study personnel remained blinded to both treatment assignment and dose. Treatment assignments remained blinded until the database was locked." (p.275) |
| Blinding of participants | Low risk | Quote: "A double‐blind, double‐dummy technique was used to ensure that patients and study personnel remained blinded to both treatment assignment and dose. Treatment assignments remained blinded until the database was locked." (p.275) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "A double‐blind, double‐dummy technique was used to ensure that patients and study personnel remained blinded to both treatment assignment and dose. Treatment assignments remained blinded until the database were locked." (p.275) |
| Incomplete outcome data for pain and physical function | High risk | Quote: "The efficacy analyses were conducted on the full‐analysis population: all randomized patients who received at least one dose of study medication and who had at least one post baseline assessment of any functional scale." (p.275) Quote: "Thirteen patients who were randomized were not included in the full analysis population because they did not have a post‐baseline efficacy assessment." (p.277) Withdrawals due to adverse events: 20.9% (68/325) in combined tramadol treatment groups vs 7.5% (17/227) in placebo group. Withdrawals for reasons other than adverse events (treatment failure, participant request, investigator initiated): 24.6% (80/325) in the combined tramadol treatments groups vs 33.5% (76/227) in placebo group. This led to an imbalance between groups. The reason for missing outcome data was likely related to true outcome. |
| Incomplete outcome data for adverse effects All outcomes | High risk | Quote: "The safety population included all patients who received at least one dose of randomized study medication." (p.275) All 552 randomized participants were included in the safety population. Withdrawals due to adverse events: 20.9% (68/325) in combined tramadol treatment groups vs 7.5% (17/227) in placebo group. Withdrawals for reasons other than adverse events (treatment failure, participant request, investigator initiated): 24.6% (80/325) in the combined tramadol treatments groups vs 33.5% (76/227) in placebo group. This led to an imbalance between groups. The reason for missing outcome data was likely related to true outcome. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (pain, physical function, and stiffness) appeared to have been reported in the results, but we did not have access to the protocol for verification. |
| Other biases | Low risk | No other sources of bias |
Fleischmann 2001.
| Methods | Multicenter, outpatient, randomized, double‐blind, placebo‐controlled, parallel‐group clinical trial | |
| Participants | 129 participants ages 35–75 years, with symptomatic (painful) OA of knee for ≥ 1 year, who had used NSAIDS ≥ 3 months before study entry and were otherwise in good health. Participants were required to have at least moderate pain (PI ≥ 2 on a scale of 0 to 4, with 0 being the least and 4 being the greatest PI) in the target knee when their current analgesic was discontinued. Number of participants: tramadol group: 63; control (placebo) group: 66 % women: active group: 65.1%; control group: 59.1% |
|
| Interventions | 10‐day analgesic washout period Active group: tramadol in 50 mg increments every 2 days, titrated to a target 200 mg after 7 days (1 capsule 4 times daily). Participants were permitted to increase their dose up to 400 mg/day if needed for 84 days. 91‐day treatment period. Control group: placebo identical in appearance for 91 days. |
|
| Outcomes | PI (5‐point Likert scale) Pain relief: measure of change in pain relative to the end of the washout phase (7‐point Likert Scale Overall WOMAC score; subscores for pain, stiffness and physical function Global Assessment of Efficacy Number of participants in each group with who reported adverse events Withdrawals due to adverse events We reported on: WOMAC Pain and Physical Function subscales, adverse events and withdrawals due to adverse events Extracted pain outcome: PI score (5‐point Likert scale) at 91 days, with lower values indicating benefit Extracted physical function outcome: WOMAC Disability score (0–10) at 91 days, with lower values indicating benefit |
|
| Notes | Funded by OrthoMcNeil Pharmaceutical, Raritan (NJ) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients (…) were randomly assigned in a 1:1 ratio to receive tramadol or placebo. Study medications were randomly assigned by a computer to a numerical list for each site, and patients were enrolled sequentially using the list" (p.117) |
| Allocation concealment (selection bias) | Unclear risk | No mention of allocation concealment |
| Blinding of personnel | Unclear risk | Authors did not explicitly report blinding of personnel |
| Blinding of participants | Low risk | Quote: "tramadol 50‐mg capsules were identical in appearance to the placebo capsules." (p.117) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Since this was a participant‐reported outcome and participants were blinded, the outcome assessment was considered blinded. |
| Incomplete outcome data for pain and physical function | High risk | Quote: "All analyses included all randomized patients who took ≥ 1 dose of study medication and for whom an efficacy measurement was available (the intent‐to‐treat population)." (p.118) Unknown how many of the 129 randomized participants were included in the analyses. Withdrawals due to adverse events: 22.2% with tramadol vs 15.2% with placebo. Total withdrawals from study: 68.3% with tramadol vs 74.2% with placebo. The high withdrawal rate impacts the validity of the imputed data used for the ITT analysis. |
| Incomplete outcome data for adverse effects All outcomes | High risk | Quote: "All analyses included all randomized patients who took ≥ 1 dose of study medication and for whom an efficacy measurement was available (the intent‐to‐treat population)." (p.118) Unknown how many of the 129 randomized participants were included in the analyses. Total withdrawals from study: 68.3% with tramadol vs 74.2% with placebo. The high withdrawal rate impacts the validity of the imputed data used for the ITT analysis. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (PI, WOMAC Osteoarthritis Index) appeared to have been reported in the results, but we did not have access to the protocol for verification. |
| Other biases | Low risk | No other sources of bias |
Fujii 2014.
| Methods | Randomized, prospective, parallel group, active‐controlled study. Participants randomized according to minimization method for 3 groups. Authors employed sex and age as stratification factors. Setting: hospital |
|
| Participants | 200 participants (148 female, 52 male) who attended authors' hospital for knee or hip pain, mean age 71.0 (SD 7.0) years who had had knee or hip pain originating from OA for ≥ 1 month, were admitted into the study. Number of participants: tramadol/acetaminophen: 65; loxoprofene: 70; transdermal fentanyl: 65. % women: tramadol/acetaminophen: 45%; loxoprofen: 56%; transdermal fentanyl: 47% |
|
| Interventions | Participants were randomized 1:1:1 to tramadol/acetaminophen, loxoprofen or transdermal fentanyl. Active group 1: tramadol/acetaminophen (tramadol 37.5 mg/acetaminophen 325 mg combination pills) starting dose 2 pills/day. If this dose was not effective, it was increased to 8 pills/day. Maximum dose 8 pills. Active group 2: loxoprofen sodium 60 mg 3 times/day, or a total of 180 mg/day. Active group 3: transdermal fentanyl starting dose 12.5 μg/hour. If this dose was not effective, it was increased sequentially to 25 μg/hour, 37.5 μg/hour and 50 μg/hour. Maximum dose 50 μg/hour. Study medications administered for 12 weeks. Other drugs and injections into the knee or hip joints were not allowed. |
|
| Outcomes | VAS evaluation of pain on movement before randomization, and after 1, 4 and 12 weeks of randomized therapy. Extracted pain outcome: pain on VAS (0–10) at 12 weeks, with lower values indicating benefit No physical function outcome reported |
|
| Notes | Authors' hospital not specified in article, only academic affiliation: Department of Orthopaedic Surgery, Graduate School of Medicine, Chiba University, Chiba, Japan. Funding source not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Two hundred patients were selected from 210 knee or hip pain patients who matched the following criteria." (p.1380) Quote: "The patients were randomized according to the minimization method for three groups." (p.1380) Minimization method used; author confirmed that 10/210 knee or hip participants were excluded because they did not match the observation of OA of the knee or hip joint on examination of an anterior‐posterior X‐ray image in the supine position. |
| Allocation concealment (selection bias) | Unclear risk | No mention of allocation concealment. |
| Blinding of personnel | Unclear risk | Blinding not confirmed or described for outcome of interest of this review (i.e. pain). |
| Blinding of participants | Unclear risk | Blinding not confirmed or described for outcome of interest of this review (i.e. pain). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Blinding not confirmed or described for outcome of interest of this review (i.e. pain). |
| Incomplete outcome data for pain and physical function | Unclear risk | Quote: "Thirty patients dropped out of this study." (p.1381) Author confirmed that reasons for withdrawal of 15% of randomized participants (15% with tramadol vs 13% with loxoprofen vs 17% with transdermal fentanyl) unknown; appeared that per‐protocol approach to analysis was used (see Figure 1, p.1381). Note: physical function not reported on in this study. |
| Incomplete outcome data for adverse effects All outcomes | Unclear risk | Adverse effects not reported on in this study. |
| Selective reporting (reporting bias) | Unclear risk | Protocol not found online. All prespecified outcomes (pain and X‐ray examinations) appeared to have been reported in the results, but we did not have access to the protocol for verification. |
| Other biases | Low risk | No other sources of bias. |
Gana 2006.
| Methods | Multicenter, randomized, double‐blind, placebo‐controlled, fixed‐dose, parallel‐group clinical trial *We also included data from another publication by Kosinski which was a secondary analysis of the trial conducted by Gana and coworkers (Gana 2006). |
|
| Participants | 1011 men/women ages 18–74 years with radiographically confirmed ACR Functional Class I–III OA of the knee or hip. Number of participants: tramadol 100 mg: 202; tramadol 200 mg: 201; tramadol 300 mg: 201; tramadol 400 mg: 202; control group: 205 Participants were required to have baseline index joint pain of at least 40 mm on 100‐mm pain VAS (0 = no pain, 100 = extreme pain) after the washout period. % women: tramadol 100 mg: 62.4%; tramadol 200 mg: 63.7%; tramadol 300 mg: 59.2%; tramadol 400 mg: 57.9%; placebo: 68.8% |
|
| Interventions | Active group 1: tramadol 100 mg once daily Active group 2: tramadol 200 mg once daily Active group 3: tramadol 300 mg once daily Active group 4: tramadol 400 mg once daily Control group: placebo once daily Treatment for 12 weeks. |
|
| Outcomes | From the Gana publication: PI: WOMAC Osteoarthritis Index for index and non‐index joints in the past 48 hours using 100‐mm VAS (0 'no pain' to 100 'extreme pain'), overall pain rated daily at approximately 8:00 p.m. using a 100‐mm VAS in response to the question "Overall, how much pain have you experienced in your study joint today?" Other: WOMAC Physical Function subscale (0–1700 mm), participant and physician global assessments of disease activity on a 100‐mm VAS, participants responded to the following sleep‐related questions using a 100‐mm VAS: (trouble falling asleep, need for sleep medication, how often they were awakened by pain during the night and how often they were awakened by pain in the morning). Participants assessed the overall quality of sleep using a 100‐mm VAS in response to the question, "Over the past week, how would you rate the overall quality of your sleep?"; SF‐36 Health Survey; adverse events (either spontaneously or in response to non‐directed questioning; results of physical exams, vital signs, clinical laboratory tests and electrocardiograms at study visits); and 16‐item questionnaire to record the presence or absence of common symptoms of physical dependence. Extracted physical function outcome: WOMAC Physical Function subscale (0–1700 mm) at 12 weeks, with lower values indicating benefit From the Kosinski publication: PI: arthritis PI during the past 48 hours in the index joint using a 100‐mm VAS with anchors of 0 (no pain) and 100 (extreme pain). Other: CPSI, which consists of 5 questions about severity of sleep impairment during previous week; adverse events, withdrawal symptoms, and physical dependence after abrupt discontinuation of study treatment. Extracted pain outcome: Arthritis Pain Intensity scale (0–100 mm) at 12 weeks, with lower value indicating less pain |
|
| Notes | From the Gana publication: Supported by Biovail Laboratories International SRL From the Kosinski publication: Supported by a grant from Ortho‐McNeil Janssen Scientific Affairs, LLC |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A randomization schedule was generated with permuted blocks of 10 subjects. Each site received study medication kits that were marked with the randomization numbers. Investigators used an interactive voice‐response system to assign randomization numbers to subjects." (p.1392) |
| Allocation concealment (selection bias) | Low risk | Quote: "Each site received study medication kits that were marked with the randomization numbers. Investigators used an interactive voice‐response system to assign randomization numbers to subjects. Eligible subjects were randomly assigned to a 1:1:1:1:1 ratio." (p.1392) |
| Blinding of personnel | Unclear risk | Authors did not explicitly report blinding of personnel. |
| Blinding of participants | Low risk | Quote: "To preserve blinding, study medication tablets were similar in appearance and size." (p.1392) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Since this was a participant‐reported outcome and participants were blinded, the outcome assessment can be considered blinded. |
| Incomplete outcome data for pain and physical function | High risk | Quote: "Analyses were conducted on an intent‐to‐treat (ITT) population, defined as all randomized subjects who took at least one dose of study medication, using the last‐observation‐carried‐forward approach to replace missing post‐baseline efficacy data." (p.1393) 558/1020 (55.2%) of participants completed 12 weeks of treatment. However, 1011/1020 participants were included in the ITT population. Those omitted from ITT did not receive treatment. Withdrawals due to adverse events: 22.7% with tramadol vs 10.2% with placebo. Withdrawals for reasons other than adverse events: 22.4% with tramadol vs 15.3% with placebo. The imbalance in withdrawals due reasons other than adverse events was likely to also impact the outcome data despite an ITT analysis. *Gana 2006 provided outcome data for physical function while Kosinski 2007 provided outcome data for pain. |
| Incomplete outcome data for adverse effects All outcomes | High risk | Quote: "Analyses were conducted on an intent‐to‐treat (ITT) population, defined as all randomized subjects who took at least one dose of study medication, using the last‐observation‐carried‐forward approach to replace missing post‐baseline efficacy data." (p.1393) 558/1020 (55.2%) of participants completed 12 weeks of treatment. However, 1011/1020 participants were included in the ITT population. Those omitted from ITT did not receive treatment. Withdrawals due to adverse events: 22.7% with tramadol vs 10.2% with placebo. Withdrawals for reasons other than adverse events: 22.4% with tramadol vs 15.3% with placebo. The imbalance in withdrawals due reasons other than adverse events was likely to also impact the outcome data despite an ITT analysis. *Gana 2006 provided outcome data for any adverse events, withdrawals due to adverse events and serious adverse events. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (pain, physical function and stiffness, and sleep quality) appeared to have been reported in the results, but we did not have access to the protocol for verification. |
| Other biases | Low risk | No other sources of bias. |
Jensen 1994.
| Methods | Parallel, multicenter double‐blind RCT | |
| Participants | Participants with radiologically confirmed diagnosis of OA of hip or knee Number of participants: tramadol group: 135; control group: 129 % women: active group: 76%; control group: 82% |
|
| Interventions | 3–7‐day washout period: participants received up to 4 mg of acetaminophen. 2‐week double‐blind phase: participants with moderate to severe pain despite the acetaminophen were randomly allocated to: Active group 1: tramadol 100 mg 3 times/day Active group 2: dextropropoxyphene 100 mg 3 times/day Drugs were administered in capsules identical in appearance. |
|
| Outcomes | Assessment of pain/pain relief: pain during walking/daily activities, and pain during sleep (4‐point Likert scale) Assessment of functional impairment: climbing stairs, getting out of bed, or rising from a chair (4‐point Likert scale) Overall assessment of therapy at the last visit Adverse effects: signs and symptoms Withdrawals due to adverse events Extracted pain outcome: pain relief on VAS (0–100 mm) at 2 weeks, with lower values indicating benefit Extracted disability outcome: participant overall assessment (proxy for physical function) |
|
| Notes | Funded by Grünenthal GmBH, Aachen, Germany | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were given a patient number in the order that they were enrolled, and received their allocated treatment according to a computer‐generated assignment schedule." (p.213) |
| Allocation concealment (selection bias) | Unclear risk | No mention of allocation concealment |
| Blinding of personnel | Unclear risk | Authors did not explicitly report blinding of personnel. |
| Blinding of participants | Low risk | Quote: "Both drugs were administered as capsules, and were identical in appearance." (p.213) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "To quantify pain relief, patients made daily recordings." (p.213) Quote: "At the last visit, the patient and the investigator were asked to give an overall assessment of the therapy." (p.213) |
| Incomplete outcome data for pain and physical function | Low risk | Quote: "Patients who provided information on different efficacy parameters, i.e. who attended all visits, were evaluated for efficacy (evaluable cohort, EVAL)." (p.213) Quote: "The results in the EVAL cohort were consistent with the ITT cohort." (p.215) For tramadol (135 ITT, 81 EVAL, 54 [40%] not included in benefits assessment). For dextropropoxyphene (129 ITT, 109 EVAL, 20 [16%] not included in benefits assessment). Since only participants who attended all their visits were included in the benefits assessment, other participant data would have been missed. Withdrawals due to adverse events: 35.6% with tramadol vs 10.9% with dextropropoxyphene. The reason for missing outcome data was likely to be related to true outcome. |
| Incomplete outcome data for adverse effects All outcomes | Low risk | Quote: "All patients randomized (intent‐to‐treat cohort, ITT) were included in the analysis of safety." (p.213) Quote: "The results in the EVAL cohort were consistent with the ITT cohort" (p.215) Quote: "A significantly larger number of withdrawals in the tramadol group occurred as a result of adverse events." (p.215) Data on safety analysis were presented in table III for 135 tramadol participants and 129 dextropropoxyphene participants which was the amount of people included in the ITT cohort. However, withdrawals due to adverse events were 35.6% with tramadol vs 10.9% with dextropropoxyphene. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (pain status, physical impairment, adverse signs/symptoms) appeared to have been reported in the results, but we did not have access to the protocol for verification. |
| Other biases | Low risk | No other sources of bias. |
Karlsson 2009.
| Methods | Randomized, open‐label, controlled, parallel‐group non‐inferiority study | |
| Participants | Participants ages > 18 years with clinical diagnosis of OA of the hip or knee (or both), based on ACR and radiographic criteria. % women: active group: 59.4%; control group: 53.8% |
|
| Interventions | Active group 1: tramadol pills (75, 100, 150 and 200 mg) titrated as needed to achieve stable pain control over 12 weeks Active group 2: 7‐day buprenorphine patches (5, 10 and 20 μg/hour) |
|
| Outcomes | PI: mean weekly BS‐11 pain score, calculated from the scores recorded in the participant diaries every evening. Global assessment of pain relief obtained by asking participants and investigators to rate the study medication in terms of pain relief (very poor, poor, fair, good or very good). Investigators assessed participants' pain, stiffness and ability to perform daily activities using WOMAC Other: participant‐recorded number of acetaminophen pills (rescue medication) taken daily. Sleep disturbance and quality of sleep assessed by asking participants the following questions: "How many nights have you woken due to pain in the past 7 nights?" and "Please rate the quality of sleep over the past 7 nights" (response options: very poor, poor, fair, good and very good). Participants' quality of life using the EuroQol EQ‐5D Health Status Index and EQ‐VAS. Adverse events reported. At visits 2 and 8, physical exam performed, and systolic and diastolic blood pressure and heart rate measured. Extracted pain outcome: scores on 11‐point box scale at 12 weeks, with lower values indicating benefit Physical function outcomes not reported |
|
| Notes | Sponsored and designed by Mundipharma AB, Goteborg, Sweden | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "a computer‐generated randomization schedule was used to allocate patients." (p.505) Quote: "Patients were randomized in a 1:1 ratio." (p.503) Quote: "Eligible patients were randomized to the lowest available patient number at their site." (p.506) |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Sealed envelopes with the treatment codes were forwarded to investigators at each site." (p.506) Insufficient information about allocation concealment. |
| Blinding of personnel | High risk | Quote: "When the study was designed, it was felt that the potential benefits of an open‐label design outweighed those of a blinded design." (p.511) |
| Blinding of participants | High risk | Quote: "When the study was designed, it was felt that the potential benefits of an open‐label design outweighed those of a blinded design." (p.511) |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "When the study was designed, it was felt that the potential benefits of an open‐label design outweighed those of a blinded design." (p.511) |
| Incomplete outcome data for pain and physical function | High risk | Quote: "The full analysis set (FAS) included all patients who were randomized and received at least 1 dose of study medication. The per‐protocol analysis set (PPAS) included patients who were in the FAS and had no major protocol violations." (p.507) The FAS did not include all randomized participants who completed the treatment. 20/69 (28.9%) participants in the patches group were not included in FAS. 25/66 (37.8%) participants in the tramadol group were not included in the FAS. Withdrawals due to adverse events: 28.8% with tramadol vs 14.5% with patches. Total withdrawals: 31.8% with tramadol vs 20.3% with patches. |
| Incomplete outcome data for adverse effects All outcomes | Low risk | Quote: "The full analysis set (FAS) included all patients who were randomized and received at least 1 dose of study medication. The per‐protocol analysis set (PPAS) included patients who were in the FAS and had no major protocol violations." (p.507) The per‐protocol analysis set does not include all randomized participants who completed the treatment. 20/69 (28.9%) participants in the patches group were not included in FAS. 25/66 (37.8%) participants in the tramadol group were not included in the FAS. Withdrawals due to adverse events: 28.8% with tramadol vs 14.5% with patches. Total withdrawals: 31.8% with tramadol vs 20.3% with patches. |
| Selective reporting (reporting bias) | Low risk | Quote: "There were changes from baseline to study completion on all WOMAC Osteoarthritis Index subscale scores in both treatment groups, with no significant differences between treatment groups." (p.508) WOMAC Osteoarthritis Index subscale scores not reported but stated to have no significant differences between treatments. |
| Other biases | Low risk | No other sources of bias. |
Kean 2009.
| Methods | 2 parallel, multicenter, double‐blind, double‐dummy, placebo‐controlled, phase III clinical trials | |
| Participants | 685 women ages 40–75 years with moderate‐to‐severe pain associated with OA of the knee. Conducted from January to August 2003 across 149 active centers in the US included in analysis. Participants were required to have a WOMAC Pain subscale VAS score > 150 mm at baseline. |
|
| Interventions | Active group 1: tramadol 100 mg for up to 12 weeks Active group 2: tramadol 200 mg for up to 12 weeks Active group 3: tramadol 300 mg for up to 12 weeks Control group: placebo. Washout period ≥ 2 days or a minimum of 5 half‐lives. All participants who were randomized to active treatment started by taking 100 mg/day. Over the next 6 days, participants randomized to 200 mg/day and 300 mg/day treatment arms were titrated in a double‐blind manner by 100 mg/day increments every 2–3 days until they reached their randomized dosages. Randomized dosage or placebo maintained for a maximum of 12 weeks. |
|
| Outcomes | PI: WOMAC Pain and Physical Function subscale scores at baseline and at end of study, participant global rating of pain relief was assessed using a Likert‐type scale with the possible responses of 'very effective,' 'effective' and 'ineffective.' This evaluation was completed at all study visits during the maintenance phase. Other: treatment compliance (number of dispensed pills taken relative to the number of dispensed pills planned) and adverse events, physical examination, laboratory assessments, and concomitant medications was used to assess safety. Extracted pain outcome: WOMAC Pain subscale (0–500 mm) at 12 weeks, with lower values indicating benefit Extracted physical function outcome: WOMAC Physical Function subscale (0–1700 mm) at 12 weeks, with lower values indicating benefit |
|
| Notes | Funded by Labopharm Inc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomized to a predetermined fixed dose." (p.1003) Quote: "Once randomized, patients entered the run‐in phase." (p.1003) No mention as to how the randomization process was carried out. |
| Allocation concealment (selection bias) | Unclear risk | No mention of allocation concealment. |
| Blinding of personnel | Low risk | Quote: "Treatment assignments remained blinded until the clinical trial database was locked." (p.1003) |
| Blinding of participants | Low risk | Quote: "double‐blind, double‐dummy" (p.1003) Quote: "Treatment assignments remained blinded until the clinical trial database was locked." (p.1003) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Treatment assignments remained blinded until the clinical trial database was locked." (p.1003) |
| Incomplete outcome data for pain and physical function | High risk | Quote: "The primary analysis population for this post hoc efficacy analyses was the female full‐analysis (FA) population, defined as all randomized women who received at least one dose of the assigned study medication and had at least one post‐baseline assessment of any functional scale." (p.1004) Quote: "Sixteen women from both studies who were randomized were not included in the full‐analysis population because they did not have a post‐baseline efficacy assessment." (p.1005) Quote: "A total of 309 (45.1%) discontinued the study prior to week 12 of the maintenance phase." (p.1004) 685 women randomized. Withdrawals due to adverse events: 25.2% (102/405) with tramadol vs 5% (14/280) with placebo. Withdrawals for reasons other than adverse events (treatment failure, participant request, investigators initiated, administrative, death): 24.9% (101/405) with tramadol vs 32.9% (92/280) with placebo. Withdrawals between the groups were not balanced and it was likely that missing outcome data were related to true outcome. |
| Incomplete outcome data for adverse effects All outcomes | High risk | Quote: "The safety population was comprised of all women from each study who received at least one dose of the assigned study medication within each of the treatment arms used in the analysis." (p.1004) Quote: "A total of 685 women comprised the safety population." (p.1004) Table 3 presented data regarding adverse events for the total randomized population of 685 women. Withdrawals due to adverse events: 25.2% (102/405) with tramadol vs 5% (14/280) with placebo. Withdrawals for reasons other than adverse events (treatment failure, participant request, investigators initiated, administrative, death): 24.9% (101/405) with tramadol vs 32.9% (92/280) with placebo. Withdrawals between the groups were not balanced and it was likely that missing outcome data were related to true outcome. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (pain, physical function, and stiffness) appeared to have been reported in the results, but we did not have access to the protocol for verification. |
| Other biases | Low risk | No other sources of bias. |
Malonne 2004.
| Methods | Parallel, multicenter, double‐blind RCT | |
| Participants | 231 adults ages 45–80 years with OA of hip or knee diagnosis made with the European League Against Rheumatism criteria. Participants were included if they had a pain score ≥ 35 mm on 100‐mm Huskisson horizontal VAS scale (scale 0 = no pain to 100 = worst pain) and a functional discomfort score ≥ 4 on the Lequesne Functional Discomfort Index (total score 0 = absence of pain to 20 = most intense pain). % women: active group: 72.1%; control group: 73.1% |
|
| Interventions | 14‐day treatment period. Active group: tramadol LP SR 200 mg/day Control group: placebo Concomitant treatment with acetaminophen as a rescue medication |
|
| Outcomes | PI evaluated with VAS Patient Global Assessment and use of rescue medication. Extracted pain outcome: Huskisson VAS for pain (0–100 mm) at 14 days, with lower values indicating benefit. Physical function data not extractable for purposes of this review. |
|
| Notes | Funding source not mentioned. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Study drugs were allocated to patients based on a center randomization list." (p.1776) No mention as to how the randomization was carried out. |
| Allocation concealment (selection bias) | Unclear risk | No mention of allocation concealment. |
| Blinding of personnel | Unclear risk | Authors did not explicitly report blinding of personnel |
| Blinding of participants | Low risk | Quote: "Capsules of identical appearance containing either inactive ingredients or tramadol LP 200 mg were prepared and dispensed in blister packs." (p.1776) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Since this was a participant‐reported outcome and participants were blinded, the outcome assessment was considered blinded. |
| Incomplete outcome data for pain and physical function | Low risk | Quote: "Two hundred thirty‐one patients were randomized to treatment, and 230 (…) were evaluable for efficacy and safety." (p.1776) Quote: "A separate analysis was conducted in the patients who did not take rescue medication." Quote: "92 patients were included in the assessment of those who did not take rescue medication." (p.1776) 230/231 analyzable participants used in safety analysis but only 197 completed treatment and were used in the benefits analysis. The missing 33 participants were omitted due to no VAS at day 14. Total withdrawals: 23.4% with tramadol vs 5.9% with placebo. Withdrawals due to adverse events: 21.6% with tramadol vs 1.7% with placebo. |
| Incomplete outcome data for adverse effects All outcomes | Low risk | Quote: "Two hundred thirty‐one patients were randomized to treatment, and 230 (…) were evaluable for efficacy and safety." (p.1776) Quote: "A separate analysis was conducted in the patients who did not take rescue medication." Quote: "92 patients were included in the assessment of those who did not take rescue medication." (p.1776) 230/231 analyzable participants used in safety analysis but only 197 completed treatment and were used in the benefits analysis. The missing 33 participants were omitted due to no VAS at day 14. Total withdrawals: 23.4% with tramadol vs 5.9% with placebo. Withdrawals due to adverse events: 21.6% with tramadol vs 1.7% with placebo. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (global pain score, Lequesne Functional Discomfort Index, safety/adverse events) appeared to have been reported in the results, but we did not have access to the protocols for verification. |
| Other biases | Low risk | No other sources of bias. |
Park 2012.
| Methods | Randomized, multicenter, open comparative study in outpatients at 6 sites Setting: outpatient clinic at 6 sites. |
|
| Participants | Ages 40–75 years. 143 participants (women 121, men 22) enrolled, mean age 61.15 (SD 7.80). With symptomatic moderate knee OA pain (≥ 5 on NRS) for > 1 year despite treatment with stable doses of NSAIDs (meloxicam 7.5 mg or 15 mg once daily or aceclofenac 100 mg twice daily) for ≥ 4 weeks. Investigators used the 1986 ACR clinical and radiographic criteria for classification of idiopathic knee OA and checked standing anteroposterior view of knee joints. Participants with moderate knee joint pain (≥ 5 on NRS) in last 48 hours of the screening/washout phase eligible to enter study. % women: active group: 84%; control group: 87%. |
|
| Interventions | During the 14‐day screening/washout phase, participants discontinued cyclobenzaprine, antidepressant or anticonvulsant therapy and underwent clinical and radiologic exam. During the 4‐week tramadol/acetaminophen add‐on period, participants maintained their existing NSAID dose and tramadol/acetaminophen was titrated from 1 pill at bedtime for 3 days, 1 pill twice/day for 4 days, 1 pill 3 times/day for 3 days, and thereafter as needed from 3 to 8 pills per day. On day 29, participants with reduced pain (< 4 on NRS) were randomized to continue with either tramadol/acetaminophen or NSAID for a further 8 weeks. | |
| Outcomes | Major benefits measure was the Korean version of the WOMAC Osteoarthritis Index score; minor outcome measures included PI on NRS, pain relief score, and overall medication assessment by participants and investigators. Benefits evaluations were performed on days 29 and 57 during monotherapy. Safety assessments comprised adverse event monitoring, changes from baseline in vital signs, physical examination at every visit and clinical laboratory tests at the end of study. Extracted pain outcome: PI on NRS (0–8) for 4 weeks, with lower values indicating benefit Extracted physical function outcome: WOMAC Physical Function subscale (0–1700 mm) for 4 weeks, with lower values indicating benefit |
|
| Notes | Specific sites involved were not listed in the paper. All authors based in South Korea. Supported by a grant from Janssen Korea, Ltd, Seoul, Korea. ClinicalTrials.gov identifier: NCT00635349. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study described as randomized, but randomization procedure not described by authors. |
| Allocation concealment (selection bias) | High risk | No mention of allocation concealment. |
| Blinding of personnel | High risk | Quote: "This was a randomized, multicenter, open comparative study in out‐patients at six sites." (p.318) Study described as "open," author confirmed that personnel were not blinded. |
| Blinding of participants | High risk | Quote: "This was a randomized, multicenter, open comparative study in out‐patients at six sites." (p.318) Study described as "open," author confirmed that participants were not blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "This was a randomized, multicenter, open comparative study in out‐patients at six sites." (p.318) Study described as "open," author confirmed that outcome assessors were not blinded. |
| Incomplete outcome data for pain and physical function | High risk | Quote: "Efficacy analyses were performed on the intent‐to‐treat population, defined as patients who took at least one dose of study medication and had available efficacy measurements." (p.319) Quote: "Ninety‐one of the 97 randomized subjects were included in the ITT population (44 in tramadol/APAP [acetaminophen] group; 47 in NSAID group)…" (p.319) For ITT population, 3 excluded in each group. Total discontinued: 19% (23% with tramadol vs 14% with NSAID); reasons for discontinuation differed between 2 groups. |
| Incomplete outcome data for adverse effects All outcomes | Unclear risk | Quote: "The population evaluable for safety was used for above safety analyses…" (p.319) Safety population not specified. |
| Selective reporting (reporting bias) | Unclear risk | Outcome measures listed in 'Methods' were adequately reported, with 1 exception: secondary measure "overall medication assessment by patients and investigators" (p.319). This measure was reported as follows: "Although NSAID monotherapy tended to be superior to tramadol/APAP with respect to pain relief score and overall assessment by participants and investigators, the differences failed to reach statistical significance" (p.320), with no data to support this conclusion. ClinicalTrials.gov identifier: NCT00635349. |
| Other biases | Low risk | No other sources of bias. |
Pavelka 1998.
| Methods | Cross‐over, double‐blind RCT | |
| Participants | 60 adults (8 men, 52 women) ages > 18 years with radiologically confirmed diagnosis of OA of hip or knee and at least moderate pain on a one‐off 4‐point verbal rating scale assessment (0 = none to 3 = severe). | |
| Interventions | Cross‐over trial Participants randomized to tramadol (50–100 mg up to 3 times/day on demand), then diclofenac (25–50 mg up to 3 times/day on demand) for 28 days. 1‐week washout period before the first course of trial medication and again between the first and second courses. |
|
| Outcomes | Pain and physical function were evaluated with WOMAC Index Pain Intensity scores, WOMAC Composite Index and global assessment were similar in both treatment phases Extracted pain outcome: WOMAC Osteoarthritis Index Pain subscore (0–100 mm) at 28 days, with lower values indicating benefit Extracted physical function outcome: WOMAC Total score |
|
| Notes | Sponsored by Grünenthal GmbH, Aachen, Germany. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomized via a computer‐generated code." (p.423) |
| Allocation concealment (selection bias) | Unclear risk | No mention of allocation concealment. |
| Blinding of personnel | Unclear risk | Authors did not explicitly report blinding of personnel |
| Blinding of participants | Low risk | Quote: "The appearance of the tramadol and diclofenac medication (…) was identical ('capsule‐in‐a‐capsule' technique)." (p.423) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Since this was a participant‐reported outcome and participants were blinded, the outcome assessment was considered blinded. |
| Incomplete outcome data for pain and physical function | Low risk | Quote: "Six patients terminated the study prematurely, three from group one and three from group two, all because of adverse events." (p.425) Quote: "sample size of 30 per group." (p.423) Quote: "Only patients with all measurements at all visits were evaluated." (p.423) Table I provided WOMAC questionnaire scores for 54/60 participants in total so there were missing data for 6 participants who dropped out due to adverse events. The 6 participants were not included in the benefits analysis. However, there was only a 10% withdrawal rate due to adverse events for the tramadol and diclofenac groups, and there was an equal amount of withdrawals in each group. |
| Incomplete outcome data for adverse effects All outcomes | Low risk | Quote: "Six patients terminated the study prematurely; three from group one and three from group two, all because of adverse events." (p.425) Quote: "sample size of 30 per group." (p.423) Quote: "Only patients with all measurements at all visits were evaluated." (p.423) Those who terminated the study early would not have had all measurements taken so they were not included in the analyses. However, there was only a 10% withdrawal rate due to adverse events for the tramadol and diclofenac groups, and there was an equal amount of withdrawals in each group. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (pain, physical function and stiffness, and sleep quality) appeared to have been reported in the results, but we did not have access to the protocol for verification. |
| Other biases | High risk | Quote: "There was comparability between groups with regard to age, weight, Broca index and vital parameters." (p.424) There was no figure showing the distribution of participants in each study group and the group demographics. Was use of a cross‐over design appropriate? Yes, OA was stable. Was it clear that the order of receiving treatments was randomized? Yes, randomized "computer generated code." Can it be assumed that the trial was not biased from carry‐over effects? Yes, 1‐week washout period; "there were only slight period effects." Were unbiased data available? No, used Chi2 (independent test). |
Peeva 2010.
| Methods | Randomized, double‐blind, placebo‐controlled, 3‐period cross‐over study | |
| Participants | Participants ages ≥ 45 years with knee OA > 6 months based on clinical and radiographic criteria and had an American Rheumatological Association functional class of I–III. Eligible participants had PI while standing ≤ 5 on a 0–10 NRS. % women (active and control group combined): 63.6%. |
|
| Interventions | Cross‐over trial Comprised 3 × 3‐day periods. Participants randomized to naproxen, tramadol/acetaminophen and placebo for 3 days with a 4–7‐day washout period between the 3 phases of treatment. Tramadol/acetaminophen period: participants received total daily doses of tramadol 75 mg/acetaminophen 650 mg on day 1 and tramadol 112.5 mg/acetaminophen 975 mg on day 2 and tramadol 75 mg/acetaminophen 650 mg administered as a single dose on day 3. Naproxen period: participants received 1000 mg total daily dose of naproxen on days 1 and 2 and 500 mg on the morning of day 3. |
|
| Outcomes | Pain: change from baseline in TWA PI for both postdose self‐pace walks on day 3, with TWA reflective of pain across the entire walk. Key secondary endpoints included TWA PI for all self‐pace walks on day 1 and for each individual walk on days 1 and 3, TTMP using a 4‐point Likert scale (none, slight, moderate, severe), and if applicable, TTSP. WOMAC questionnaire VAS 3.0 (100‐mm VAS) collected at end of days 1 and 3 after the completion of the last timed walks, with participants reporting pain, physical function and stiffness results for the preceding 24 hours. Other: the incidence of overall adverse events, serious adverse events, drug‐related adverse events and discontinuation due to adverse events collected to evaluate tolerability and safety. No extractable pain or physical function outcomes as authors combined time periods for this cross‐over study. |
|
| Notes | ClinicalTrials.gov Identifiers: NCT00772967 and NCT00565084. sponsored by Merck & Co., Inc. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Allocation to balanced treatment sequences was determined according to a computer‐generated schedule by the study statistician." (p.647) |
| Allocation concealment (selection bias) | Low risk | Quote: "Numbered containers were used to implement allocation." (p.647) |
| Blinding of personnel | Low risk | Quote: "All study personnel, including investigators, study site personnel, patients, monitors, and central laboratory personnel, were blinded to treatment allocation throughout the study; the code was revealed to the researchers once recruitment, data collection, and laboratory analyses were complete." (p.647) |
| Blinding of participants | Low risk | Quote: "Study medication was administered in double‐dummy fashion with over‐encapsulated pills." (p.647) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All those involved in study were blinded, as noted above. |
| Incomplete outcome data for pain and physical function | Low risk | Quote: "Only observed data were analyzed; no data were imputed." (p.648) Quote: "Nineteen patients (86.4%) completed the study, and all 19 were included in both the primary efficacy analysis and safety analysis. Two patients discontinued due to protocol violations, and one patient discontinued due to an adverse event (acute gouty attack), which was not considered to be drug‐related." (p.648) Unknown what treatment (naproxen 500 mg twice daily, tramadol/acetaminophen or placebo) the participant was receiving when they withdrew. ClinicalTrials.gov indicated that the 22 participants were randomized to 6 groups corresponding to a different order of treatment administration (e.g. placebo, naproxen, tramadol/acetaminophen; or naproxen, tramadol/acetaminophen, placebo, etc.). However, the percentage of total withdrawals in both groups combined was only 13.6% and reasons for withdrawal were included. % of withdrawals due to adverse events in both groups combined was 4.5%. |
| Incomplete outcome data for adverse effects All outcomes | Low risk | Quote: "Only observed data were analyzed; no data were imputed." (p.648) Quote: "Nineteen patients (86.4%) completed the study, and all 19 were included in both the primary efficacy analysis and safety analysis. Two patients discontinued due to protocol violations, and one patient discontinued due to an adverse event (acute gouty attack), which was not considered to be drug‐related." (p.648) Unknown what treatment (naproxen 500 mg twice daily, tramadol/acetaminophen or placebo) the participant was receiving when they withdrew. ClinicalTrials.gov indicated that the 22 participants were randomized to 6 groups corresponding to a different order of treatment administration (e.g. placebo, naproxen, tramadol/acetaminophen; or naproxen, tramadol/acetaminophen, placebo, etc.). However, the percentage of total withdrawals in both groups combined was only 13.6% and reasons for withdrawal were included. % of withdrawals due to adverse events in both groups combined was 4.5%. |
| Selective reporting (reporting bias) | Low risk | Outcomes listed in earliest iteration of protocol were consistent with those reported as results in the article (ClinicalTrials.gov Identifier: NCT00772967). |
| Other biases | Low risk | No other sources of bias. Was use of a cross‐over design appropriate? Yes, OA was stable. Was it clear that the order of receiving treatments was randomized? Yes, randomized "computer generated schedule." Can it be assumed that the trial was not biased from carry‐over effects? Yes, 4–7‐day washout period; "there were only slight period effects." Were unbiased data available? Yes, analysis of variance model for a 3‐period cross‐over design. |
Schnitzer 1999.
| Methods | Parallel, multicenter, double‐blind RCT Setting: clinic |
|
| Participants | Participants ages ≥ 45 years with symptomatic OA of knee. % women: naproxen responders tramadol group: 55.6%; naproxen responders placebo group: 57.4%; naproxen non‐responders tramadol group: 61.5%; naproxen non‐responders placebo group: 70.6% |
|
| Interventions | 2 phases, and we evaluated the 8‐week double‐blind phase. Participants whose pain did not resolve with 500 mg of naproxen were randomized. Randomization was stratified based on response to naproxen 1000 mg (responders and non‐responders). Active group: tramadol plus naproxen for 54 days. Control group: placebo plus naproxen for 54 days. During the double‐blind phase the naproxen dose was reduced to 250 mg every 2 weeks. Dosage of tramadol (200 mg/day) or placebo remained constant during the double‐blind phase. |
|
| Outcomes | Primary aim to determine whether tramadol decreased naproxen requirements. No data on PI during the double‐blind phase. Number of participants who discontinued therapy due to adverse events was reported Pain data not reported for double‐blind phase Physical function data not reported for double‐blind phase |
|
| Notes | Supported by research grant from Ortho‐McNeil Pharmaceutical, Inc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The randomization was stratified based on the patient's baseline VAS score" (p.1371) Unknown if the sequence was computer‐generated. No mention of sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No mention of allocation concealment. |
| Blinding of personnel | High risk | Quote: "During the 8‐week double‐blind phase, the initial dosage of naproxen in the double‐blind phase was 750 mg/day. This dosage was reduced by 250 mg every two weeks. The naproxen dosage reduction was accomplished in a single‐blind manner (i.e. the patients did not know what dosage of naproxen they were receiving). The dosage of tramadol or placebo remained constant during the double‐blind phase." (p.1372) Authors did not describe blinding of personnel, and the quote provided above suggests that they may have known which participants were taking naproxen once the dosage started being reduced. |
| Blinding of participants | Low risk | Quote: "patients were randomly assigned to treatment with tramadol 200 mg/day or matching placebo" (p.1372) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Personnel interacting with participants with regard to outcome assessment were not explicitly reported to be blinded. |
| Incomplete outcome data for pain and physical function | Low risk | Quote: "Four patients (3 taking tramadol, 1 taking placebo) were randomized but were not included in the efficacy analysis because they did not have an efficacy assessment or they did not take the study medication. A total of 236 patients were evaluated." (p.1373) 236/240 randomized participants were analyzed for benefits. % withdrawals due to adverse events: 22% with tramadol vs 13% with placebo. Total number of withdrawals in each treatment group unknown. |
| Incomplete outcome data for adverse effects All outcomes | Low risk | Quote: "Twenty‐two percent of tramadol patients and 13% of placebo patients discontinued due to an adverse event during the double‐blind phase." (p.1374) Outcome data for withdrawals due to adverse events were likely included in the analysis since there were only 4 participants in total not included in the benefits analysis. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (pain) appeared to have been reported in the results, but we did not have access to the protocol for verification. |
| Other biases | Low risk | No other sources of bias. |
Silverfield 2002.
| Methods | Parallel, multicenter, double‐blind RCT | |
| Participants | Participants ages 35–75 years with symptomatic OA of hip or knee received stable doses of NSAID or COX‐2 Number of participants: tramadol/acetaminophen group: 197; control group: 111 % women: active group: 76.6%; control group: 63.1% |
|
| Interventions | Active group: tramadol 37.5 mg/acetaminophen 325 mg 1 or 2 pills QID Control group: placebo Treatment for 10 days in addition to ongoing NSAID or COX‐2‐selective inhibitor therapy. Number of pills/day increased up to 8. Participants continued receiving NSAID or COX‐2 at the same doses taken before study entry. |
|
| Outcomes | PI and pain relief evaluated using a 4‐point adjective scale (none, mild, moderate, severe) % participants in each relief category WOMAC Index score Participants who received tramadol had less pain than participants who received placebo (data not used in the pooling because of 4‐point scale) Extracted pain outcome: WOMAC Pain subscale (0–5) at 10 days, with lower values indicating benefit Extracted physical function outcome: WOMAC Physical Function subscale (0.5) at 10 days, with lower values indicating benefit |
|
| Notes | We contacted 1 of the coauthors and obtained the requested information (percentage of participants with moderate pain relief). Funding source not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were assigned sequentially according to a randomization schedule" (p.286) Quote: "A list of unique medication code numbers was prepared using a computerized random‐number generator to ensure that any given patient was assigned randomly to 1 of 3 initial treatment groups." (p.286) |
| Allocation concealment (selection bias) | Unclear risk | No mention of allocation concealment. |
| Blinding of personnel | Low risk | Quote: "Treatment assignments were not revealed to patients, investigators, clinical staff, or monitors until all patients had completed treatment and database was finalized." (p.286) |
| Blinding of participants | Low risk | Quote: "Study medication consisted of identical‐appearing tablets containing tramadol/acetaminophen or matching placebo." (p.286) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Treatment assignments were not revealed to patients, investigators, clinical staff, or monitors until all patients had completed treatment and database was finalized." (p.286) |
| Incomplete outcome data for pain and physical function | High risk | Quote: "Efficacy summaries were based on the intent‐to‐treat population, defined as all patients randomized to receive study medication who took at least 1 dose and had post randomization efficacy data. Safety summaries were based on all randomized patients who took at least 1 dose of study medication." (p.287) Quote: "If a patient dropped out within the first 4 hours after taking the first dose, the baseline‐observation‐carried forward approach was used to impute missing pain assessments. All other missing assessments were imputed using the last‐observation‐carried‐forward approach." (p.287) All randomized participants were included in ITT analysis and were evaluable for safety. Withdrawals due to adverse events: 12.7% (25/197) with tramadol vs 5.4% (6/111) with placebo. Withdrawals for reasons other than adverse events (discontinued prematurely, lack of efficacy, protocol violations, other): 7.6% (15/197) with tramadol vs 0% with placebo. Last or baseline observations carried forward are hardly adequate imputation techniques. In addition, there was differential dropout since the adverse events predominantly occurred in the tramadol group. |
| Incomplete outcome data for adverse effects All outcomes | Low risk | Quote: "Efficacy summaries were based on the intent‐to‐treat population, defined as all patients randomized to receive study medication who took at least 1 dose and had post randomization efficacy data. Safety summaries were based on all randomized patients who took at least 1 dose of study medication." (p.287) Quote: "If a patient dropped out within the first 4 hours after taking the first dose, the baseline‐observation‐carried forward approach was used to impute missing pain assessments. All other missing assessments were imputed using the last‐observation‐carried‐forward approach." (p.287) All randomized participants were included in ITT analysis and were evaluable for safety. Withdrawals due to adverse events: 12.7% (25/197) with tramadol vs 5.4% (6/111) with placebo. Withdrawals for reasons other than adverse events (discontinued prematurely, lack of efficacy, protocol violations, other): 7.6% (15/197) with tramadol vs 0% with placebo. Last or baseline observations carried forward are hardly adequate imputation techniques. In addition, there was differential dropout since the adverse events predominantly occurred in the tramadol group. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (pain, physical function, and WOMAC Index score) appeared to have been reported in the results, but we did not have access to the protocol for verification. |
| Other biases | Low risk | No other sources of bias. |
Thorne 2008.
| Methods | Randomized, double‐blind, cross‐over RCT Setting: clinic |
|
| Participants | 100 participants (45 men and 55 non‐pregnant, non‐nursing women) ages ≥ 18 years, diagnosed with OA and requiring the use of acetaminophen, anti‐inflammatory agents or combination opioid and non‐opioid analgesics for ≥ 3 months. 77/100 randomly assigned participants (36 men and 41 women with a mean age 59.4 (SD 9.6) years were evaluable for efficacy of the 8‐week cross‐over study. |
|
| Interventions | Active group: conventional release tramadol 150 mg/day Control group: placebo Treatment was titrated weekly to 200 mg, 300 mg or a maximum of 400 mg once daily over 4 weeks, at which point participants were crossed over to the alternate treatment for another 4 weeks. Analgesic washout for 2–4 days except acetaminophen before start of randomly selected treatment. |
|
| Outcomes | Pain: PI in a diary, twice per day (08:00 and 20:00), using 5‐point ordinal scale (0 = none, 1 = mild, 2 = moderate, 3 = severe, 4 = excruciating) and 100‐mm VAS bounded by 'no pain' and 'excruciating pain.' PI over the previous 24 hours and over the previous week was assessed using the 100‐mm VAS and 5‐point ordinal scales. WOMAC Pain, Stiffness and Physical Function subscales. Other: pain‐related disability using the PDI, which consists of 7 × 11‐point ordinal subscales. Impact of pain on sleep (since the last evaluation) assessed with 8‐item Pain and Sleep Questionnaire. SF‐36 benefits of treatment assessed by participant and investigator using a 4‐point categorical scale (not effective, slightly effective, moderately effective, highly effective). Overall treatment phase preference assessed by participant and investigator at end of study, without unblinding the treatment allocation, clinical benefit and adverse events. No extractable pain or physical function outcomes as it appeared that authors combined time periods for this cross‐over study |
|
| Notes | Funding source not reported. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized treatment phase." (p.95) Quote: "All patients were randomly assigned to receive either active or placebo CR tramadol." (p.95) No mention of how the randomization process was carried out. |
| Allocation concealment (selection bias) | Unclear risk | No mention of allocation concealment. |
| Blinding of personnel | Low risk | Quote: "Both patients and investigators rated CR tramadol in a blinded manner…" (p.100) |
| Blinding of participants | Low risk | Quote: "Medications included oral CR tramadol 150 mg, 200 mg, 300 mg and 400 mg tablets and matching placebo tablets." (p.95) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Overall treatment phase preference was assessed by the patient and the investigator at the end of the study, without unblinding the treatment allocation, by answering the question: 'Which treatment period did you prefer in the management of your pain?' (treatment period 1; treatment period 2; no preference)." (p.95) |
| Incomplete outcome data for pain and physical function | Low risk | Quote: "The full analysis set (intent‐to‐treat [ITT]) was used to confirm the results of the primary efficacy variables, the WOMAC and overall treatment preference." (p.96) Quote: "Seventy seven patients (36 men, 41 women) were evaluable for efficacy (Figure 1), with an average age of 59.4 ± 9.6 years and a mean weight and height of 91.0 ± 21.4 kg and 167 ± 10.9 cm, respectively." (p.96) Composite scores for pain and physical function of the WOMAC Osteoarthritis Index reported. 75/100 participants completed full 8 weeks of treatment. In Phase 1, % withdrawals due to reasons other than adverse events: 8% with tramadol vs 6% with placebo. |
| Incomplete outcome data for adverse effects All outcomes | Low risk | Quote: "The full analysis set (intent‐to‐treat [ITT]) was used to confirm the results of the primary efficacy variables, the WOMAC and overall treatment preference." (p.96) Quote: "Seventy seven patients (36 men, 41 women) were evaluable for efficacy (Figure 1), with an average age of 59.4 ± 9.6 years and a mean weight and height of 91.0 ± 21.4 kg and 167 ± 10.9 cm, respectively." (p.96) Composite scores for pain and physical function of the WOMAC Osteoarthritis Index reported. 75/100 participants completed full 8 weeks of treatment. In Phase 1, % withdrawals due to reasons other than adverse events: 8% with tramadol vs 6% with placebo. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (pain, physical function and stiffness, and sleep quality) appeared to have been reported in the results, but we did not have access to the protocol for verification. |
| Other biases | Low risk | No other sources of bias detected. Was use of a cross‐over design appropriate? Yes, OA was stable. Was it clear that the order of receiving treatments was randomized? Unclear, randomized but no details on randomization procedure. Can it be assumed that the trial was not biased from carry‐over effects? Yes, 2–7‐day washout (tramadol can take about a day and a half for the drug to completely exit the body). Are unbiased data available? Yes, paired t‐test and tested for carry‐over effect which was not statistically significant. *We did not combine the data for pain or physical function outcomes since the authors combined time periods for this cross‐over study. |
Wilder‐Smith 2001.
| Methods | Open‐label, randomized, parallel‐group study Setting: Groote Schuur Hospital Rheumatology Department outpatient clinic |
|
| Participants | Over 6 months, investigators recruited 95 participants with OA awaiting hip or knee replacement surgery. 8 of these dropped out. Data from 29 participants with dihydrocodeine, 28 with tramadol and 30 with control with NSAIDs only were completely evaluable. Ages 55–65 years. % women: dihydrocodeine group: 31%; tramadol group: 29%; NSAID‐only group: 37% |
|
| Interventions | Participants were hospitalized for dose titration for the first 4 days. Active group 1: tramadol 100 mg Active group 2: dihydrocodeine 60 mg every 12 hours Control group: NSAID control The corresponding immediate‐release drug solution (tramadol: 100 mg/mL and dihydrocodeine: 10 mg/mL) was used for dose titration and breakthrough pain. Adaptations of study drug doses during the 1‐month treatment period were performed as required. |
|
| Outcomes | PI at rest and with movement using a 4‐point adjective scale. Extracted pain outcome: pain on movement (0–3 scale) for 28 days, with lower values indicating benefit Physical function outcomes not reported |
|
| Notes | We contacted the author to determine the percentage of participants with minor and major adverse events, but obtained no response. Supported by research funds from Grűnenthal AG, Switzerland and Grűnenthal GmbH, Germany. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "randomised using a computer‐generated code list kept by the hospital pharmacy." (p.24) Quote: "Sixty successive patients (…) were recruited for the opioid trial." (p.24) Quote: "Thirty additional successive patients from the same department with osteoarthritis and NSAID treatment, but mean pain intensity below 3 in the VRS [verbal rating scale] in the run‐in period were included for comparison (NSAID‐only control arm)." (p.24) Only the treatment group was randomized. Control group was specifically chosen for their low disease activity to compare against the treatment group. |
| Allocation concealment (selection bias) | High risk | No mention of allocation concealment. |
| Blinding of personnel | High risk | Quote: "Due to open label design investigators were not blinded to treatment." (p.24) |
| Blinding of participants | High risk | Trial described as open. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Investigators interacting with participants were not blinded, and participant blinding was not described. |
| Incomplete outcome data for pain and physical function | Low risk | Quote: "95 patients were recruited for this study. There were eight drop‐outs. The reasons for drop‐out were: 'too busy' (n = 2), 'no transport to hospital' (2), 'poor compliance during dosing' (2), and two were lost to follow‐up." (pp.25–26) Unknown if missing outcome data for dropouts were imputed. Although the reason for participants lost to follow‐up was not mentioned, only 2/95 (2.1%) participants fell into this category so it is reasonable to assume that their missing data would not significantly alter the overall outcome assessment. |
| Incomplete outcome data for adverse effects All outcomes | Low risk | Quote: "95 patients were recruited for this study. There was eight drop‐outs. The reasons for drop‐out were: 'too busy' (n = 2), 'no transport to hospital' (2), 'poor compliance during dosing' (2), and two were lost to follow‐up." (pp.25–26) Unknown if missing outcome data for dropouts were imputed. Although the reason for participants lost to follow‐up was not mentioned, only 2/95 (2.1%) participants fell into this category so it is reasonable to assume that their missing data would not significantly alter the overall outcome assessment. |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes (pain, sleep quality, physiologic tests, and sensory tests) appeared to have been reported in the results, but we did not have access to the protocol for verification. |
| Other biases | Low risk | No other sources of bias. |
ACR: American College of Rheumatology; ARCI: Addiction Research Center Inventory; COX: cyclo‐oxygenase; CPSI: Chronic Pain Sleep Inventory; CR: controlled release; EQ‐VAS: EuroQol visual analog scale; ER: extended release; IL: interleukin; ITT: intention to treat; n: number of participants; NRS: numerical rating scale; NSAID: non‐steroidal anti‐inflammatory drug; OA: osteoarthritis; PDI: Pain and Disability Index; PDQ: Physical Dependence Questionnaire; PI: pain intensity; PI‐NRS: Pain Intensity – Numerical Rating Scale; RCT: randomized controlled trial; SF‐36: 36‐item Short Form; SR: sustained release; TTMP: time to develop moderate pain; TTSP: time to develop severe pain; TWA: time‐weighted average; VAS: visual analog scale; WOMAC: Western Ontario and McMaster Universities Arthritis Index.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Adams 2006 | Data not presented separately for people with osteoarthritis. |
| Argoff 2009 | Narrative review |
| Avouac 2007 | Meta‐analysis |
| Choi 2007 | Titration study |
| Di Lorenzo 2010 | Non‐randomized |
| Estrada 2006 | Osteoarthritis not evaluated. |
| Estrada 2007 | Osteoarthritis not evaluated. |
| Florete 2008 | Post‐hoc analyses of an included trial (Gana, 2006) |
| Grupo Empresarial Químico‐Farmacéutico 2010 | Not an RCT. |
| Mariconti 2008 | Pain not evaluated. |
| McMahon 2008 | Review of evidence, not single RCT |
| McMeniman 2010 | Osteoarthritis not evaluated. |
| Olaya 2011 | Osteoarthritis not evaluated. |
| Pascual 2007 | Data not presented separately for people with osteoarthritis. |
| Rauck 2006 | RCT but evaluated osteoarthritis and other pain syndromes and results not reported separately. |
| Stitik 2006 | Review of evidence, not single RCT |
| Turhanoğlu 2010 | Tramadol iontophoresis |
| Vorsanger 2007 | Post‐hoc analyses of an included trial (Gana, 2006) |
RCT: randomized controlled trial.
Characteristics of studies awaiting assessment [ordered by study ID]
EUCTR2014‐004718‐27‐PL.
| Methods | Title: a study comparing the effectiveness and safety of tramadol and micronized magnesium lactate as functional excipient to tramadol alone for the treatment of moderate to severe pain due to osteoarthritis (OA). Randomized, multicenter, single‐blinded, parallel‐group study that seeks to assess the efficacy, safety and tolerability of the combination of IR tramadol with micronized magnesium lactate in managing chronic pain in people with OA of the hip or knee, or both |
| Participants | Adults with clinical diagnosis of OA of hip or knee (or both), based on ACR and radiographic criteria (presence of knee or hip joint symptoms (pain, stiffness, disability) and signs (bony crepitus), and radiographic evidence of OA (functional class I–III)). Inclusion criteria
|
| Interventions | Non‐inferiority study. Main objective is to evaluate efficacy, safety and tolerability of the new formulation composed of tramadol 50 mg and micronized magnesium lactate 75 mg of magnesium ions in the application of daily dose tramadol 150 mg/magnesium lactate 225 mg for the management of chronic pain. Secondary objective is to assess acceptance during disease treatment and to collect data on quality of life and the impact on the economy (cost‐effectiveness analysis and cost utility analysis). |
| Outcomes | Major outcome
Minor outcome
|
| Notes | World Health Organization International Clinical Trials Registry Platform Main ID: EUCTR2014‐004718‐27‐PL URL: www.clinicaltrialsregister.eu/ctr‐search/search?query=eudract_number:2014‐004718‐27 Contact Information: Department of Pharmacodynamics Address: 1B Banacha Street, 02‐097, Warsaw, Poland Telephone: +4822116 61 26 Email: farmakodynamika@wum.edu.pl Affiliation: Medical University of Warsaw |
Krebs 2017.
| Methods | Title of publication: Design, recruitment outcomes, and sample characteristics of the Strategies for Prescribing Analgesics Comparative Effectiveness (SPACE) trial. Pragmatic randomized comparative effectiveness trial conducted in multiple VA primary care clinics within 1 VA healthcare system to compare benefits and harms of opioid therapy vs non‐opioid medication therapy over 12 months. |
| Participants | Participants with moderate‐to‐severe chronic back pain or hip/knee OA pain despite analgesic therapy; participants already receiving regular opioid therapy were excluded. |
| Interventions | Participants were randomized to receive opioid therapy or non‐opioid medication therapy, for 12 months. Opioid therapy: participants first received morphine IR, hydrocodone/acetaminophen or oxycodone IR. Opioid dosage was titrated, with adjustments made nearly every 4 weeks, to a maximum daily dosage of 100 ME mg. If participants did not have a clear response at a daily dosage of 60 ME mg, rotation to a different opioid was considered. Non‐opioid therapy: participants first received acetaminophen or an NSAID medication. Adjuvant oral therapies were usually added to an initial regimen, but were given if first‐line medications were not appropriate. Tramadol was included in the third‐line and considered only when participants did not respond to other medications or combinations of medications. Diclofenac 1% gel was added in the last year of the trial as a first line agent or adjuvant option when oral NSAIDs could not be used. In both groups, pain medication management was tailored to participant preferences. Outcomes evaluated by masked assessors at 3, 6, 9 and 12 months after enrollment. |
| Outcomes | Among the 265 enrolled participants, 25 withdrew before randomization. Of 240 randomized participants, 87.9% were men, 84.1% were white and age range was 21–80 years. Major outcomes
Minor outcomes
|
| Notes | Protocol outlined here: Krebs EE, Jensen AC, Nugent S, DeRonne B, Rutks I, Leverty D, Gravely A, Noorbaloochi S, Bair MJ, Kroenke K. Design, recruitment outcomes, and sample characteristics of the Strategies for Prescribing Analgesics Comparative Effectiveness (SPACE) trial. Contemporary Clinical Trials. 201;62:130‐9. |
NCT00426647.
| Methods | Official title: a randomised double‐blind multicentre equivalence study with active parallel comparator group to evaluate the efficacy and safety of Norspan® patches versus tramadol in subjects with chronic, moderate to severe osteoarthritis pain in the hip, knee &/or lumbar spine. Randomized, double‐blind, multicenter equivalence study with active comparator, parallel group, to evaluate the efficacy and safety of Norspan® patches vs tramadol in people with OA pain in hip, knee, lumbar spine, or a combination of these, currently receiving suboptimal analgesic treatment (defined as BS‐11 score > 4) when treated with acetaminophen 4000 mg/day or another analgesic at least comparable to this. 4 phases: run‐in, washout, double‐blind and follow‐up |
| Participants | Diagnosed with OA pain in hip, knee, lumbar spine, or a combination of these and aged ≥ 18 years. |
| Interventions | Tramadol Buprenorphine |
| Outcomes | Major outcome: efficacy of Norspan® Minor outcome: safety and general satisfaction |
| Notes | Study start date: February 2007 Primary completion date: July 2009 Study completion date: August 2009 Contact information: Address: GP, Noerretorv 10, DK‐7200 Grindsted, Denmark, Olavi, Airaksinen DM (principal investigator), Oma Lääkäri Oy, Vuorikatu 20, FIN‐70100 KUOPIO Email: olavi.airaksinen@kuh.fi Email: norpharma@norpharma.dk ClinicalTrials.gov identifier: NCT00426647 |
NCT00736853.
| Methods | Official title: a phase 3 study of JNS013 in patients with chronic pain Multicenter, double‐blind, placebo‐controlled, parallel‐group comparison study. Total duration will be 11 weeks and consists of 4 periods; a preobservation period (4 weeks), open‐label period (2 weeks), double‐blind period (4 weeks) and follow‐up period (1 week). Participants will receive tramadol hydrochloride plus acetaminophen pills orally 4 times/day for 2 weeks at ≥ 4‐hour intervals (up to 8 pills/day) during the open‐label period and the dose will be fixed for each participant in the latter 1 week. During the double‐period participants will receive tramadol hydrochloride plus acetaminophen pills or placebo at the same dose as used for the latter 1 week of the open‐label period for up to 4 weeks. Efficacy will be primarily evaluated by number of participants with insufficient pain relief after the start of double‐blind period. Participant's safety will be monitored throughout the study. |
| Participants | Participants with sustention of chronic pain associated with OA or LBP for ≥ 3 months and aged ≥ 20 years. |
| Interventions | Tramadol 37.5 mg and acetaminophen 325 mg 4 times/day or placebo, for 4 weeks. |
| Outcomes | Major outcome
Minor outcomes
|
| Notes | Study start date: June 2008 Primary completion date: January 2009 Study completion date: January 2009 Contact information: Address: Paranaque City, Metro Manila, Philippines, 1700 Email: info@janbe.jnj.com Phone: +32 14 60 21 11 ClinicalTrials.gov identifier: NCT00736853 |
NCT00743587.
| Methods | Official title: a randomized, double‐blind, placebo and active controlled methodology study investigating the effects of tramadol and naproxen on the pain thresholds of patients with severe pain due to osteoarthritis of the thumb Methods not provided in detail Allocation: randomized Intervention model: cross‐over assignment Blinding: double (participant, investigator) |
| Participants | Diagnosed with OA of the hand, ≥ 6 months' duration and aged ≥ 18 years |
| Interventions | Oxycodone 20 mg Tramadol 50 mg Naproxen 500 mg Placebo |
| Outcomes | Major outcome
Minor outcomes
|
| Notes | Study start date: September 2008 Primary completion date: March 2009 Study completion date: March 2009 Contact information: Address: Pfizer Investigational Site, Brussels, Belgium, 1070 Email: www.pfizer.com/contact/email_contact?inquiry=Clinical%20Research (to send an email) Phone: 1‐212‐733‐2323 ClinicalTrials.gov identifier: NCT00743587 |
NCT00832416.
| Methods | Study title: a four‐arm study comparing the analgesic efficacy and safety of tramadol once a day 100, 200 and 300 mg versus placebo for the treatment of pain due to osteoarthritis of the knee. Methods not provided. |
| Participants | Diagnosed with moderate to severe OA of the knee, consistent with the ACR clinical classification criteria for arthritis of the knee, and aged 40–75 years |
| Interventions | Tramadol 100 mg daily Tramadol 200 mg daily Tramadol 300 mg daily Placebo |
| Outcomes | Major outcomes
Minor outcomes
|
| Notes | Study start date: January 2003 Primary completion date: August 2003 Contact information: Address: 480 boulevard, Armand‐Frappier, Laval, Québec, H7V 4B4 Email: info@labopharm.com Phone: 450 680‐2444 ClinicalTrials.gov identifier: NCT00832416 |
NCT01019265.
| Methods | Official title: a randomised open label parallel group study comparing Norspan patch and oral tramadol. Primary objective of this non‐inferiority study with active, parallel control group is to compare and assess efficacy and safety of buprenorphine transdermal patch (Norspan® patch 5 mg, 10 mg and 20 mg) and tramadol (Tridol® SR (slow release) pill 100 mg) in people with moderate to severe pain due to OA. During the period of treatment for 8 weeks, titration and maintenance is kept up using 1:1 ratio randomization. |
| Participants | Diagnosed with OA of the hip or knee (or both) including fulfilling the ACR criteria L13 and aged ≥ 18 years |
| Interventions | Buprenorphine 5 mg for 8 weeks Buprenorphine 10 mg for 8 weeks Buprenorphine 20 mg for 8 weeks Tramadol 100 mg for 8 weeks |
| Outcomes | Major outcome:
Minor outcomes
|
| Notes | Study start date: March 2008 Primary completion date: March 2009 Study completion date: May 2009 Contact information: not provided ClinicalTrials.gov identifier: NCT01019265 |
NCT01728246.
| Methods | Official title: a randomized controlled trial on the efficacy, safety and quality of life effects of add‐on tramadol/acetaminophen combination in chronic osteoarthritis Open‐label, randomized controlled study to evaluate the efficacy, safety and effects on quality of life of tramadol/acetaminophen as an add‐on therapy in Filipino participants with chronic pain because of chronic OA. Participants will be randomly assigned to 2 groups: tramadol/acetaminophen group and non‐tramadol/acetaminophen group. Participants in tramadol/acetaminophen group will receive celecoxib 200 mg and fixed‐dose combination of tramadol 37.5 mg/acetaminophen 325 mg as add‐on therapy, and participants in non‐tramadol/acetaminophen group will receive celecoxib 200 mg only. Total duration of study will be 4 weeks. Participants in both groups will be given celecoxib 200 mg once daily for 4 weeks. In addition, participants in the tramadol/acetaminophen group will be given add‐on tramadol/acetaminophen doses 3 times/day for 4 weeks. Participants will be asked to return for follow‐up at weeks 2 and 4. Efficacy will be assessed using 100‐mm VAS while quality of life will be assessed using the Oswestry Disability Index. Participant safety will be monitored throughout the study. |
| Participants | Diagnosed with chronic OA of knee or hip for ≥ 1 year, and aged ≥ 18 years. |
| Interventions | Celecoxib 200 mg once daily and tramadol 37.5 mg and acetaminophen 325 mg, for 4 weeks Celecoxib once daily and non‐tramadol and acetaminophen, for 4 weeks |
| Outcomes | Major outcomes:
|
| Notes | Study start date: October 2007 Primary completion date: May 2008 Study completion date: May 2008 Contact information: Address: Paranaque City, Metro Manila, Philippines, 1700 Email: info@janbe.jnj.com Phone: +32 14 60 21 11 ClinicalTrials.gov identifier: NCT01728246 |
ACR: American College of Rheumatology; BPI: Brief Pain Inventory; BS‐11: Box Score‐11; IR: immediate release; LBP: low back pain; ME: morphine‐equivalent; NRS: numerical rating scale; NSAID: non‐steroidal anti‐inflammatory drug; OA: osteoarthritis; PI: pain intensity; PI‐NRS: pain intensity numerical rating scale; PID: pain intensity difference; VA: Veteran Affairs; VAS24: visual analog scale for the last 24 hours; WOMAC: Western Ontario and McMaster Universities Arthritis Index.
Differences between protocol and review
This is an update of a previous review published in 2008. This updated review has a new author team and there are some differences from the previous review.
We used the Cochrane 'Risk of bias' tool.
Our latest search did not include the LILACS database.
We created 'Summary of findings' tables according to the latest guidance in the Cochrane Handbook for Systematic Reviews of Interventions. We also added the percentage of people who reach the minimally clinically important difference in the 'Summary of findings' tables.
Our outcomes differed as we based our outcomes on the latest recommendations from the Cochrane Musculoskeletal Group.
We presented our results in terms of standardized mean difference instead of mean difference and used fixed‐effect models. We also used random‐effects models to verify if the two models gave consistent results.
Contributions of authors
KTA: conceived and designed the study, performed the literature search, participated in the appraisal of the methodologic quality of studies, contacted the authors of original clinical trials, performed the analysis and wrote the manuscript.
JB: participated in the appraisal of the methodologic quality of studies, performed the analysis and revised the final version of the manuscript.
VW: participated in the appraisal of the methodologic quality of studies, performed the analysis and revised the final version of the manuscript.
LM: participated in the appraisal of the methodologic quality of studies, performed the analysis and contributed to writing and revising the final version of the manuscript.
PJ: appraised the full‐text articles and revised the final version of the manuscript.
AWSR: appraised the full‐text articles and revised the final version of the manuscript.
EH: appraised the full‐text articles and revised the final version of the manuscript.
JV: participated in the appraisal of the methodologic quality of studies, performed the analysis and revised the final version of the manuscript.
TEH: participated in the appraisal of the methodologic quality of studies, performed the analysis and contributed to writing and revising the final version of the manuscript.
GW: conceived and designed the study, performed the analysis and revised the final version of the manuscript.
PT: conceived and designed the study and contributed to writing and revising the final version of the manuscript
Sources of support
Internal sources
No sources of support supplied
External sources
-
Ontario Early Researcher Award, Canada.
This support was provided to Drs. Toupin April and Welch to facilitate their research.
-
Canada Research Chair in Health Equity, Canada.
This support was provided to Dr. Tugwell to fund his research.
Declarations of interest
KTA: none.
JB: none.
VW: none.
LM: none.
PJ: Peter Jüni has received research grants from Astra Zeneca, Biotronik, Biosensors International, Eli Lilly and The Medicines Company, that has been paid to the institution. Peter serves as unpaid member of the steering group of trials funded by Astra Zeneca, Biotronik, Biosensors, St. Jude Medical and The Medicines Company.
AWSR: none.
EH: none.
JV: none.
TEH: none.
GW: none.
PT: Travel and accommodation for OMERACT meetings ‐ a registered non‐profit independent medical research organization, OMERACT, whose goal is to improve and advance the health outcomes for patients suffering from musculoskeletal conditions. OMERACT receives unrestriced educational grants from the American College of Rheumatology, European League of Rheumatology and several pharmaceutical companies listed below which is used to support fellows, international patient groups and support a major international bi‐annual conference which results in many peer reviewed publications; Amgen, Astra Zeneca, Bristol Myers Squibb, Celgene, EliLilly, Genentech/Roche, Genzyme/Sanofi, Horizon Pharma Inc, Merck, Novartis, Pfizer, PPD, Quintiles, Regeneron, Savient, Takeda Pharmaceutical, UCB Group, Vertex, Forest, Bioiberica Independent Committee Member for clinical trial Data Safety Monitoring Boards for FDA approved trials being conducted by UCB Biopharma GmbH & SPRL, Parexel International, and Prahealth Sciences. Independent medical consultation professional services for CHEOR Solutions (Canada) Ltd., Innovative Science Solutions LLC. An advisory committee member of the Canadian Reformulary Group Inc., a company that reviews the evidence for health insurance companies employer drug plans.
Edited (no change to conclusions)
References
References to studies included in this review
Babul 2004 {published data only}
- Babul N, Noveck R, Chipman H, Roth SH, Gana T, Albert K. Efficacy and safety of extended‐release, once‐daily tramadol in chronic pain: a randomized 12‐week clinical trial in osteoarthritis of the knee. Journal of Pain Symptom Management 2004;28(1):59‐71. [DOI] [PubMed] [Google Scholar]
Beaulieu 2008 {published data only}
- Beaulieu AD, Peloso PM, Haraoui B, Bensen W, Thomson G, Wade J, et al. Once daily, controlled‐release tramadol and sustained‐release diclofenac relieve chronic pain due to osteoarthritis: a randomized controlled trial. Pain Research & Management 2008;13(2):103‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bianchi 2003 {published data only}
- Bianchi M, Broggini M, Balzarini P, Baratelli E, Ferrario P, Panerai AE, et al. Effects of tramadol on synovial fluid concentrations of substance P and interleukin‐6 in patients with knee osteoarthritis: comparison with paracetamol. International Immunopharmacology 2003;3(13‐14):1901‐8. [DOI] [PubMed] [Google Scholar]
Bird 1995 {published data only}
- Bird HA, Hill J, Stratford ME, Fenn GC, Wright V, Fleischmann RM, et al. A double‐blind cross‐over study comparing the analgesic efficacy of tramadol with pentazocine in patients with osteoarthritis. Journal of Drug Development & Clinical Practice 1995;7(7):181‐8. [MEDLINE: ] [Google Scholar]
Burch 2007 {published data only}
- Burch F, Fishman R, Messina N, Corser B, Radulescu F, Sarbu A, et al. A comparison of the analgesic efficacy of tramadol Contramid OAD versus placebo in patients with pain due to osteoarthritis. Journal of Pain and Symptom Management 2007;34(3):328‐38. [DOI] [PubMed] [Google Scholar]
DeLemos 2011 {published data only}
- DeLemos BP, Xiang J, Benson C, Gana TJ, Pascual ML, Rosanna R, et al. Tramadol hydrochloride extended‐release once‐daily in the treatment of osteoarthritis of the knee and/or hip: a double blind, randomized, dose‐ranging Trial. American Journal of Therapeutics 2011;18:216‐26. [DOI] [PubMed] [Google Scholar]
Emkey 2004 {published data only}
- Emkey R, Rosenthal N, Wu SC, Jordan D, Kamin M. Efficacy and safety of tramadol/acetaminophen tablets (Ultracet) as add‐on therapy for osteoarthritis pain in subjects receiving a COX‐2 nonsteroidal antiinflammatory drug: a multicenter, randomized, double‐blind, placebo‐controlled trial. Journal of Rheumatology 2004;31(1):150‐6. [PubMed] [Google Scholar]
Fishman 2007 {published data only}
- Fishman R, Kistler CJ, Ellerbusch MT, Aparicio RT, Swami SS, Shirley ME, et al. Efficacy and safety of 12 weeks of osteoarthritic pain therapy with once‐daily tramadol (tramadol Contramid(R) OAD). Journal of Opioid Management 2007;5:273‐80. [DOI] [PubMed] [Google Scholar]
Fleischmann 2001 {published data only}
- Fleischmann RM, Caldwell JR, Roth SH, Tesser JR, Olson W, Kamin M. Tramadol for the treatment of joint pain associated with osteoarthritis: a randomized, double‐blind, placebo‐controlled trial. Current Therapeutic Research, Clinical & Experimental 2001;62(2):113‐28. [MEDLINE: ] [Google Scholar]
Fujii 2014 {published data only}
- Fujii T, Takana K, Orita S, Inoue G, Ochiai N, Kuniyoshi K, et al. Progressive change in joint degeneration in patients with knee or hip osteoarthritis treated with fentanyl in a randomized trial. Yonsei Medical Journal 2014;55(5):1379‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gana 2006 {published data only}
- Florete OG, Xiang J, Vorsanger GJ. Effects of extended‐release tramadol on pain‐related sleep parameters in patients with osteoarthritis. Expert Opinion Pharmacotherapy 2008;9(11):1817‐27. [DOI] [PubMed] [Google Scholar]
- Gana TJ, Pascual ML, Fleming RR, Schein JR, Janagap CC, Xiang J, Vorsanger GJ, 023 Study Group. Extended‐release tramadol in the treatment of osteoarthritis: a multicenter, randomized, double‐blind, placebo‐controlled clinical trial. Curr Med Res Opin 2006;22(7):1391‐1401. [DOI] [PubMed] [Google Scholar]
- Kosinski M, Janagap C, Gajria K, Schein J, Freedman J. Pain relief and pain‐related sleep disturbance with extended‐release tramadol in patients with osteoarthritis. Current Medical Research and Opinion 2007;23(7):1615‐26. [DOI] [PubMed] [Google Scholar]
- Vorsanger G, Xiang J, Jordan D, Farrell J. Post hoc analysis of a randomized, double‐blind, placebo‐controlled efficacy and tolerability study of tramadol extended release for the treatment of osteoarthritis pain in geriatric patients. Clinical Therapeutics 2007;29(Suppl):2520‐35. [DOI] [PubMed] [Google Scholar]
Jensen 1994 {published data only}
- Jensen EM, Ginsberg F. Tramadol versus dextropropoxyphene in the treatment of osteoarthritis: a short term double‐blind study. Drug Investigation 1994;8(4):211‐8. [Google Scholar]
Karlsson 2009 {published data only}
- Karlsson M, Berggren A. Efficacy and safety of low‐dose transdermal buprenorphine patches (5, 10, and 20 pg/h) versus prolonged‐release tramadol tablets (75, 100, 150, and 200 mg) in patients with chronic osteoarthritis pain: a 12‐Week, randomized, open label, controlled, parallel‐group noninferiority study. Clinical Therapeutics 2009;31(3):503‐13. [DOI] [PubMed] [Google Scholar]
Kean 2009 {published data only}
- Kean WF, Bouchard S, Gossen ER. Women with pain due to osteoarthritis: the efficacy and safety of a once‐daily formulation of tramadol. American Academy of Pain Medicine 2009;10(6):1001‐11. [DOI] [PubMed] [Google Scholar]
Malonne 2004 {published data only}
- Malonne H, Coffiner M, Sonet B, Sereno A, Vanderbist F. Efficacy and tolerability of sustained‐release tramadol in the treatment of symptomatic osteoarthritis of the hip or knee: a multicenter, randomized, double‐blind, placebo‐controlled study. Clinical Therapeutics 2004;26(11):1774‐82. [DOI] [PubMed] [Google Scholar]
Park 2012 {published data only}
- Park K‐S, Choi J‐J, Kim W‐U, Min J‐K, Park S‐H, Cho C‐S. The efficacy of tramadol/acetaminophen combination tablets (Ultracet©) as add‐on and maintenance therapy in knee osteoarthritis pain inadequately controlled by nonsteroidal anti‐inflammatory drug (NSAID). Clinical Rheumatology 2012;31:317‐23. [DOI] [PubMed] [Google Scholar]
Pavelka 1998 {published data only}
- Pavelka K, Peliskova Z, Stehlikova H, Ratcliffe S, Repas C. Intraindividual differences in pain relief and functional improvement in osteoarthritis with diclofenac or tramadol. Clinical Drug Investigation 1998;16(6):421‐9. [DOI] [PubMed] [Google Scholar]
Peeva 2010 {published data only}
- Peeva E, Beals CR, Bolognese JA, Kivitz AJ, Taber L, Harman A, et al. A walking model to assess the onset of analgesia in osteoarthritis knee pain. Osteoarthritis and Cartilage 2010;18:646‐53. [DOI] [PubMed] [Google Scholar]
Schnitzer 1999 {published data only}
- Schnitzer TJ, Kamin M, Olson WH. Tramadol allows reduction of naproxen dose among patients with naproxen‐responsive osteoarthritis pain: a randomized, double‐blind, placebo‐controlled study. Arthritis & Rheumatism 1999;42(7):1370‐7. [DOI] [PubMed] [Google Scholar]
Silverfield 2002 {published data only}
- Silverfield JC, Kamin M, Wu SC, Rosenthal N. Tramadol/acetaminophen combination tablets for the treatment of osteoarthritis flare pain: a multicenter, outpatient, randomized, double‐blind, placebo‐controlled, parallel‐group, add‐on study. Clinical Therapeutics 2002;24(2):282‐97. [DOI] [PubMed] [Google Scholar]
Thorne 2008 {published data only}
- Thorne C, Beaulieu AD, Callaghan DJ, O'Mahony WF, Bartlett JM, Knight R, et al. A randomized, double‐blind, crossover comparison of the efficacy and safety of oral controlled‐release tramadol and placebo in patients with painful osteoarthritis. Pain Research & Management 2008;13(2):93‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wilder‐Smith 2001 {published data only}
- Wilder‐Smith CH, Hill L, Spargo K, Kalla A. Treatment of severe pain from osteoarthritis with slow‐release tramadol or dihydrocodeine in combination with NSAID's: a randomised study comparing analgesia, antinociception and gastrointestinal effects. Pain 2001;91(1‐2):23‐31. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Adams 2006 {published data only}
- Adams EH, Breiner S, Cicero TJ, Geller A, Inciardi JA, Schnoll SH, et al. A comparison of the abuse liability of tramadol, NSAIDs, and hydrocodone in patients with chronic pain. Journal of Pain and Symptom Management 2006;31(5):465‐76. [DOI] [PubMed] [Google Scholar]
Argoff 2009 {published data only}
- Argoff CE, Silvershein DI. A comparison of long‐ and short‐acting opioids for the treatment of chronic noncancer pain: tailoring therapy to meet patient needs. Mayo Clinic Proceedings 2009;84(7):602‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Avouac 2007 {published data only}
- Avouac J, Gossec L, Dougados M. Efficacy and safety of opioids for osteoarthritis: a meta‐analysis of randomized controlled trials. Osteoarthritis Cartilage 2007;15(8):957‐65. [DOI] [PubMed] [Google Scholar]
Choi 2007 {published data only}
- Choi CB, Song JS, Kang YM, Suh CH, Lee J, Choe JY, et al. A 2‐week, multicenter, randomized, double‐blind, double‐dummy, add‐on study of the effects of titration on tolerability of tramadol/acetaminophen combination tablet in Korean adults with knee osteoarthritis pain. Clinical Therapeutics 2007;29(7):1381‐9. [DOI] [PubMed] [Google Scholar]
Di Lorenzo 2010 {published data only}
- Lorenzo L, Foti C, Forte AM, Palmieri E, Formisano R, Vatakencherry A, et al. The addition of tramadol as a second opioid may improve pain relief in severe osteoarthritis: a prospective study. Pain Practice 2010;10(6):540‐7. [DOI] [PubMed] [Google Scholar]
Estrada 2006 {published data only}
- Estrada CdlP, Rodríguez IL, Rodríguez MR, Naranjo AM. Preemptive analgesia with tramadol and diclofenac in maxillofacial surgery [Analgesia preventiva con tramadol y diclofenaco en cirugía maxilofacial]. Revista Columbiana de Anestesiología 2006;34(1):15‐9. [Google Scholar]
Estrada 2007 {published data only}
- Estrada CdlP, Pérez MF. Comparative study of tramadol versus meperidine in knee surgery under regional anesthesia [Estudio comparativo entre tramadol vs meperidina en cirugía de rodilla bajo anestesia regional]. Revista Colombiana de Anestesiología 2007;35(4):273‐7. [Google Scholar]
Florete 2008 {published data only}
- Florete OG, Xiang J, Vorsanger GJ. Effects of extended‐release tramadol on pain‐related sleep parameters in patients with osteoarthritis. Expert Opinion on Pharmacotherapy 2008;9(11):1817‐27. [DOI] [PubMed] [Google Scholar]
Grupo Empresarial Químico‐Farmacéutico 2010 {published data only}
- Grupo Empresarial Químico‐Farmacéutico QUIMEFA. Tramadol, coated tablets [Tramadol, tabletas revestidas]. Revista Cubana de Farmacia 2010;44(2):276‐9. [Google Scholar]
Mariconti 2008 {published data only}
- Mariconti P, Collini R. Tramadol SR in arthrosic and neuropathic pain. Minerva Anestesiologica 2008;74(3):63‐8. [PubMed] [Google Scholar]
McMahon 2008 {published data only}
- McMahon K, Nelson M, Jones G. Treating the symptoms of osteoarthritis – oral treatments. Australian Family Physician 2008;37(3):133‐5. [PubMed] [Google Scholar]
McMeniman 2010 {published data only}
- McMeniman TJ, McMeniman PJ, Myers PT, Hayes DA, Cavdarski A, Wong MS, et al. Femoral nerve block vs fascia iliaca block for total knee arthroplasty postoperative pain control: a prospective, randomized controlled trial. Journal of Arthroplasty 2010;25(8):1246‐9. [DOI] [PubMed] [Google Scholar]
Olaya 2011 {published data only}
- Olaya HA, Hernández MA, Gómez UE, Valencia MH. Pharmacological management of neuropathic pain, current evidence [Manejo farmacológico del dolor neuropático, evidencia actual]. Acta Neurologica Colombiana 2011;27(Suppl 2):74‐86. [Google Scholar]
Pascual 2007 {published data only}
- Pascual ML, Fleming RR, Gana TJ, Vorsanger GJ. Open‐label study of the safety and effectiveness of long‐term therapy with extended‐release tramadol in the management of chronic nonmalignant pain. Current Medical Research and Opinion 2007;23(10):2531‐42. [DOI] [PubMed] [Google Scholar]
Rauck 2006 {published data only}
- Rauck RL, Ruoff GE, McMillen JI. Comparison of tramadol and acetaminophen with codeine for long‐term pain management in elderly patients. Current Therapeutic Research, Clinical & Experimental 2006;55(12):1‐15. [MEDLINE: ] [Google Scholar]
Stitik 2006 {published data only}
- Stitik TP, Altschuler E, Foye PM. Pharmacotherapy of osteoarthritis. American Journal of Physical Medicine and Rehabilitation 2006;85(11 Suppl):S15‐28. [DOI] [PubMed] [Google Scholar]
Turhanoğlu 2010 {published data only}
- Turhanoğlu AD, Güler H, İnanoğlu K, Turhanoğlu S. Tramadol iontophoresis added to treatment of knee osteoarthritis. Archives of Rheumatology 2010;25(4):174‐8. [Google Scholar]
Vorsanger 2007 {published data only}
- Vorsanger G, Xiang J, Jordan D, Farrell J. Post hoc analysis of a randomized, double‐blind, placebo‐controlled efficacy and tolerability study of tramadol extended release for the treatment of osteoarthritis pain in geriatric patients. Clinical Therapeutics 2007;29(Suppl):2520‐35. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
EUCTR2014‐004718‐27‐PL {unpublished data only}
Krebs 2017 {published data only}
NCT00426647 {unpublished data only}
NCT00736853 {unpublished data only}
NCT00743587 {unpublished data only}
NCT00832416 {unpublished data only}
NCT01019265 {unpublished data only}
NCT01728246 {unpublished data only}
Additional references
ACR 2000
- American College of Rheumatology (ACR) Subcommittee on Osteoarthritis Guidelines. Recommendations for the medical management of osteoarthritis of the hip and knee. Arthritis & Rheumatology 2000;43:1905‐15. [DOI] [PubMed] [Google Scholar]
Altman 1996
- Altman R, Brandt K, Hochberg M, Moskowitz R, Bellamy N, Bloch DA, et al. Design and conduct of clinical trials in patients with osteoarthritis: recommendations from a task force of the Osteoarthritis Research Society. Results from a workshop. Osteoarthritis and Cartilage 1996;4(4):217‐43. [DOI] [PubMed] [Google Scholar]
Bellamy 1997
- Bellamy N, Kirwan J, Boers M, Brooks P, Strand V, Tugwell P, et al. Recommendations for a core set of outcome measures for future phase III clinical trials in knee, hip, and hand osteoarthritis. consensus development at OMERACT III. Journal of Rheumatology 1997;24(4):799‐802. [PubMed] [Google Scholar]
Bjordal 2004
- Bjordal JM, Ljunggren AE, Klovning A, Slordal L. Non‐steroidal anti‐inflammatory drugs, including cyclo‐oxygenase‐2 inhibitors, in osteoarthritic knee pain: meta‐analysis of randomised placebo controlled trials. BMJ 2004;329(7478):1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Buckwalter 2004
- Buckwalter JA, Saltzman C, Brown T. The impact of osteoarthritis: implications for research [Review]. Clinical Orthopaedics & Related Research 2004;427 Suppl:S6‐S15. [DOI] [PubMed] [Google Scholar]
Cross 2014
- Cross M, Smith E, Hoy D, Nolte S, Ackerman I, Fransen M, et al. The global burden of hip and knee osteoarthritis: estimates from the Global Burden of Disease 2010 study. Annals of the Rheumatic Diseases 2014;73:1323‐30. [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177‐88. [DOI] [PubMed] [Google Scholar]
Dieppe 2005
- Dieppe PA, Lohmander LS. Pathogenesis and management of pain in osteoarthritis. Lancet 2005;365(9463):965‐73. [DOI] [PubMed] [Google Scholar]
Egger 2001
- Egger M, Smith GD. Principles of and procedures for systematic reviews. Systematic reviews in health care: meta‐analysis in context. BMJ Publishing Group, 2001:23‐42. [Google Scholar]
Fortin 2006
- Fortin M, Dionne J, Pinho G, Gignac J, Almirall J, Lapointe L. Randomized controlled trials: do they have external validity for patients with multiple comorbidities?. Annals of Family Medicine 2006;4(2):104‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro GDT [Computer program]
- McMaster University (developed by Evidence Prime). GRADEpro GDT. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015.
Green 2003
- Green CR, Anderson KO, Baker TA, Campbell LC, Decker S, Fillingim RB, et al. The unequal burden of pain: confronting racial and ethnic disparities in pain. Pain Medicine 2003;4(3):277‐94. [DOI] [PubMed] [Google Scholar]
Gøtzsche 2007
- Gøtzsche PC, Hróbjartsson A, Maric K, Tendal B. Data extraction errors in meta‐analyses that use standardized mean differences. JAMA 2007;298(4):430–7. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hochberg 2012
- Hochberg MC, Altman RD, April KT, Benkhalti M, Guyatt G, McGowan J, et al. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care & Research 2012;64(4):465‐74. [DOI] [PubMed] [Google Scholar]
Jett 1997
- Jett MF, McGuirk J, Waligora D, Hunter JC. The effects of mexiletine, desipramine and fluoxetine in rat models involving central sensitization. Pain 1997;69(1‐2):161‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Jevsevar 2013
- Jevsevar DS. Treatment of Osteoarthritis of the Knee: Evidence‐Based Guideline, 2nd Edition. Journal of the American Academy of Orthopaedic Surgeons 2013;21:571‐6. [DOI] [PubMed] [Google Scholar]
Jüni 2006
- Jüni P, Reichenbach S, Dieppe P. Osteoarthritis: rational approach to treating the individual. Best Practice & Research. Clinical Rheumatology 2006;20(4):721‐40. [DOI] [PubMed] [Google Scholar]
Kalso 2004
- Kalso E, Edwards JE, Moore RA, McQuay HJ. Opioids in chronic non‐cancer pain: systematic review of efficacy and safety. Pain 2004;112(3):372‐80. [DOI] [PubMed] [Google Scholar]
Kean 2004
- Kean WF, Kean R, Buchanan WW. Osteoarthritis: symptoms, signs and source of pain. Inflammopharmacology 2004;12(1):3‐31. [DOI] [PubMed] [Google Scholar]
Lammert 2014
- Lammert E, Zeeb M. Metabolism of Human Diseases: Organ Physiology and Pathophysiology. Vienna: Springer, 2014. [Google Scholar]
Lawrence 2008
- Lawrence RC, Felson DT, Helmick CG, Arnold LM, Choi H, Deyo RA, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States, Part II. Arthritis and Rheumatism 2008;58(1):26‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lesnoff‐Caravaglia 2007
- Lesnoff‐Caravaglia G. Health Aspects of Aging: the Experience of Growing Old. Springfield (IL): Charles C Thomas Publisher, 2007. [Google Scholar]
Murray 2012
- Murray CJ, Vos T, Lozano R, Naghavi M, Flaxman AD, Michaud C, et al. Disability‐adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380:2197‐223. [DOI] [PubMed] [Google Scholar]
NCCCC 2008
- National Collaborating Centre for Chronic Conditions. Osteoarthritis National Clinical Guideline for Care and Management in Adults. London: Royal College of Physicians, 2008. [PubMed] [Google Scholar]
Nissen 2016
- Nissen SE, Yeomans ND, Solomon DH, Lüscher TF, Libby P, Husni, ME, et al. Cardiovascular safety of celecoxib, naproxen, or ibuprofen for arthritis. New England Journal of Medicine 2016;375:2519‐29. [DOI] [PubMed] [Google Scholar]
Norman 2001
- Norman GR, Sridhar FG, Guyatt GH, Walter SD. Relation of distribution‐and anchor‐based approaches in interpretation of changes in health‐related quality of life. Medical Care 2001;39(10):1039‐47. [DOI] [PubMed] [Google Scholar]
Pelletier 2016
- Pelletier JP, Martel‐Pelletier J, Rannou F, Cooper C. Efficacy and safety of oral NSAIDs and analgesics in the management of osteoarthritis: evidence from real‐life setting trials and surveys. Seminars in Arthritis and Rheumatism 2016;45:S22‐7. [DOI] [PubMed] [Google Scholar]
Pendleton 2000
- Pendleton A, Arden N, Dougados M, Doherty M, Bannwarth B, Bijlsma JW, et al. EULAR recommendations for the management of knee osteoarthritis: report of a task force of the Standing Committee for International Clinical Studies Including Therapeutic Trials (ESCISIT). Annals of the Rheumatic Diseases 2000;59:936‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pham 2003
- Pham T, Heijde D, Lassere M, Altman RD, Anderson JJ, Bellamy N, et al. Outcome variables for osteoarthritis clinical trials: the OMERACT‐OARSI set of responder criteria. Journal of Rheumatology 2003;30(7):1648‐54. [PubMed] [Google Scholar]
Reginster 2002
- Reginster JY. The prevalence and burden of arthritis. Rheumatology (Oxford) 2002;41 Supp 1:3‐6. [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Richmond 2010
- Richmond J, Hunter D, Irrgang J, Jones MH, Snyder‐Mackler L, Durme D, et al. American Academy of Orthopaedic Surgeons clinical practice guideline on the treatment of osteoarthritis (OA) of the knee. Journal of Bone and Joint Surgery 2010;92(4):990‐3. [DOI] [PubMed] [Google Scholar]
Riley 2002
- Riley JL III, Wade JB, Myers CD, Sheffield D, Papas RK, Price DD. Racial/ethnic differences in the experience of chronic pain. Pain 2002;100(3):291‐8. [DOI] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JP, Deeks JJ, Glasziou P. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S, editor(s), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Tannenbaum 2000
- Tannenbaum H, Peloso PM, Russell AS, Marlow B. An evidence‐based approach to prescribing NSAIDs in the treatment of osteoarthritis and rheumatoid arthritis: the second Canadian Consensus Conference. Canadian Journal of Clinical Pharmacology 2000;7 (Suppl A):4A‐16A. [PubMed] [Google Scholar]
Towheed 2006
- Towheed T, Maxwell L, Judd M, Catton M, Hochberg M, Wells G. Acetaminophen for osteoarthritis. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD004257.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tubach 2005
- Tubach F, Ravaud P, Baron G, Falissard B, Logeart I, Bellamy N, et al. Evaluation of clinically relevant changes in patient reported outcomes in knee and hip osteoarthritis: the minimal clinically important improvement. Annals of Rheumatic Diseases 2005;64(1):29‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2004
- Zhang W, Jones A, Doherty M. Does paracetamol (acetaminophen) reduce the pain of osteoarthritis? A meta‐analysis of randomised controlled trials. Annals of the Rheumatic Diseases 2004;63(8):901‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2008
- Zhang W, Moskowitz RW, Nuki G, Abramson S, Altman RD, Arden N, et al. OARSI recommendations for the management of hip and knee osteoarthritis. Part II: OARSI evidence‐based, expert consensus guidelines. Osteoarthritis and Cartilage 2008;16:137‐62. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Cepeda 2006
- Cepeda MS, Camargo F, Zea C, Valencia L. Tramadol for osteoarthritis. Cochrane Database of Systematic Reviews 2006, Issue 3. [DOI: 10.1002/14651858.CD005522.pub2] [DOI] [PubMed] [Google Scholar]