

Abstract
Self-assembled coordination cages form host-guest complexes through weak non-covalent interactions. Knowledge of how these weak interactions affect the structure, reactivity, and dynamics of guest molecules is important to further the design principles of current systems and optimize their specific functions. In this manuscript, we apply ultrafast mid-IR polarization dependent pump-probe spectroscopy to probe the effects of two Pd6L4 self-assembled nanocages on the properties and dynamics of fluxional group VIII metal carbonyl guest molecules. We find that the interactions between the Pd6L4 nanocages and guest molecules act to alter the ultrafast dynamics of the guests; restricting rotational diffusional motion and decreasing the vibrational lifetime.
Keywords: Ultrafast spectroscopy, host-guest complexes, vibrational spectroscopy, anisotropy, fluxional metal-carbonyls
Graphical Abstract
Metal directed self-assembled coordination cages can form nanosized cavities that can act to trap and stabilize guest molecules through non-covalent interactions.1-5 The physical and chemical properties of these host-guest complexes are dictated by the interplay of many factors including steric restrictions, electrostatics, and weak interactions such as π-stacking.1-3, 6 One such example of the self-assembled coordination cages is the octahedral Pd6L4 nanocage first synthesized by Fujita and coworkers.7 This nanocage has been successfully employed over the last two decades to carry out selective ground and excited reactions and trap and stabilize reactive species.8-11 Recently, Fujita and co-workers have demonstrated that the Pd6L4 nanocages can trap group VIII di-ruthenium carbonyl complexes.9, 11 The group VIII di-metal carbonyl complexes are well known for their interesting photochemistry12-14 and catalytic properties15 in addition to their fluxional behavior where they exist as multiple isomers in dynamic equilibrium.15-19 When encapsulated by the Pd6L4 nanocage, the di-ruthenium carbonyl complexes were stabilized in their cis-bridging isomeric form9 and were shown to undergo carbonyl photosubstitution without cleavage of the metal-metal bond11. From analysis of the X-ray crystal structures the stabilization was attributed to π-stacking interactions between the triazine walls of the cage and the cyclopentadienyl ligands of the metal carbonyl complexes9 and the photochemical products were suggested to arise from the restricted dynamics of the guests within the cage11. Even though extensive information can be extracted from the X-ray crystal structure, the crystal structure does not resolve the ultrafast dynamics of the encapsulated guest. A better understanding of how the ultrafast dynamics of the guest molecules are altered by the Pd6L4 hosts could lead to further insight into the rational design of Pd6L4 nanocages to stabilize and optimize various properties of the guests.20 Though there has been many studies focusing on Pd6L4 host-guest complexes there have been limited studies characterizing the ultrafast dynamics of the encapsulated guests.8, 21-22 Ultrafast vibrational spectroscopy has proven to be a powerful tool for elucidating structural changes and dynamics of structurally confined systems; including solvent trapped in metal organic frameworks,21 cyclodextrin host-guest complexes,23 and micelles containing trapped water molecules24-27 . In this manuscript, we apply ultrafast vibrational spectroscopy to two Pd6L4 nanocages that encapsulate fluxional group VIII di-metal carbonyl guest complexes 1 and 2 (Figure 1). From the polarization dependent pump-probe spectra, we decipher how the ultrafast dynamics of the trapped guests are altered upon encapsulation. We find that encapsulation results in a decrease in the vibrational lifetimes of the metal carbonyl complexes and a restriction in their diffusional rotational motion. The change in rotational diffusion confirms that the guests are tethered to the nanocage over the course of the vibrational lifetime. Applying a wobbling-in-a-cone analysis we extract the maximum possible cone angles sampled by each metal carbonyl complex, finding that the larger di-ruthenium complex samples a smaller volume than the di-iron complex. As the restricted dynamics and volume sampled by the encapsulated guests are reported to be important for their reactivity11, the measurements and characterizing of the restricted dynamics presented in this manuscript could lead to further insight into how to modify the Pd6L4 hosts to alter the reactivity of the metal carbonyl guests.
Figure 1.

A schematic representation of the coordination cages (blue, grey) with a trapped guest (pink). The structure of the two guests 1 and 2 are shown in pink. The components of the Pd6L4 nanocage are shown in blue and grey. The blue triangles (L) form the tetrahedral faces of the nanocage and the grey spheres represent the Pd2+ ion with different chelating ligands for cages A and B.
The series investigated is schematically depicted in Figure 1 and consists of two variants of the octahedral Pd6L4 nanocage, A and B, that trap the fluxional metal carbonyl complexes (Bis(cyclopentadienyl ruthenium dicarbonyl) dimer) 1 and (Bis(cyclopentadienyl iron dicarbonyl) dimer) 2 as guests to form 1⊂A, 1⊂B, 2⊂A, and 2⊂B. The choice of 1 and 2 was motivated by the ability of the Pd6L4 nanocages to non-covalently trap the hydrophobic molecules in aqueous solutions8-10 and the potential for the guests to serve as probes of structural changes, the local environment, and ultrafast dynamics23, 28-32. To investigate the effect of the host structure on the guest’s dynamics we subtly alter the volume of the nanocavity by using different chelating groups having different bite angles to form nanocages A and B.33
The synthesis of the Pd6L4 nanocages A and B, and incarceration of the metal carbonyls 1 and 2 by the nanocages A and B was carried out according to previous protocols8-9 where we have implemented slight modifications to the procedures that are detailed in sections 2 and 3 of the Supporting Information (SI). Formation of the nanocages and host-guest complexes was confirmed through 1H-NMR (see section 2, 3, and Figures Sl-7 in SI) in addition to comparison of UV/VIS and FTIR spectra.
Previous studies have shown that encapsulation of di-ruthenium metal carbonyl complexes by a similar Pd6L4 nanocage results in changes to the UV/VIS spectra, with the formation of the host-guest complex leading to the appearance of a bright charge transfer band in the visible spectral region.9 Comparing the UV/VIS spectra of the guests (1 and 2) and host-guest complexes (1⊂A, 1⊂B, 2⊂A, and 2⊂B) we observe the appearance of a charge transfer band, further confirming the formation of the host-guest complex. The UV/VIS spectra are displayed in Figure 2. As 1 and 2 are not soluble in D2O, their spectra are reported in 2 chloroform, consistent with previous studies9.
Figure 2.

UV/VIS of 1 in chloroform (cyan) ; 1⊂A. (purple) ; 1⊂B (green) ; 2 in chloroform (pink) ; 2⊂A. (red) ; 2⊂B (orange) . Inset shows UV/VIS normalized at 440 nm.
Previous studies performed on 1 and 2 have assigned the transitions beyond 400 nm to metal-to-ligand charge transfer (MLCT) bands of the different isomeric forms.12, 14 Upon insertion of 1 and 2 into cages A and B, a change in this spectral region is observed, with a loss of the structured MLCT features and appearance of a broad spectral feature that extends beyond the local absorption of 1 and 2. As the steady state UV/VIS spectra of cages A and B do not extend beyond 400 nm (see figure S8-9 in SI), the change in the UV/VIS spectrum is assigned to an electronic state associated with the host-guest complex and is attributed to the formation of a host-guest charge transfer state observed in previous studies.8-9 The UV/VIS spectra confirm that the changes in the chelating ligands of the cages and the smaller size of the di-iron metal carbonyl complex still result in the formation of the host-guest charge transfer state.
Along with changes to the electronic excited state, previous studies have observed that the Pd6L4 nanocage alters the dynamic equilibrium of the di-ruthenium complex (1).9 When free in solution metal carbonyl complexes 1 and 2 exist as different isomers in dynamic equilibrium with 1 existing in 4 forms, a cis-bridging, trans-bridging, gauche-non-bridging and trans-non-bridging, and 2 existing in two forms, a cis-bridging and trans-bridging.16, 18-19, 32 Each isomer has a distinct vibrational spectrum in the carbonyl stretching region which can be used as a spectroscopic reporter for a given isomer.12, 18-19, 32 Fujita and coworkers have demonstrated that this dynamic equilibrium is altered when the diruthenium metal carbonyl complex (1) is encapsulated by a Pd6L4 nanocage, with the Pd6L4 nanocage acting to stabilize the cis-bridging form.9 Here we expand on these studies through comparing FTIR spectra of 1 and 2 with their encapsulated forms, 1⊂A, 1⊂B, 2⊂A, and 2⊂B, to determine if altering the size of the nanocage cavity and the metal carbonyl guest will result in the stabilization of the cis-bridging isomer. Figure 3 displays the FTIR spectra of the free and encapsulated metal-carbonyl complexes as dashed and solid lines, respectively. It is important to note that different solvents were employed with a polar and non-polar solvent chosen for the free metal carbonyl complexes, and D2O employed for the encapsulated host-guest complexes. Ideally one would desire to employ the same solvent for the free in solution and encapsulated measurements. However, as the metal carbonyl complexes are not soluble in D2O we are unable to perform the free in solution measurements in D2O. In fact, the insolubility ensures that the host-guest complexes are not contaminated by free metal carbonyl signals. Given the issues with solubility we compare the FTIR spectra of the encapsulated complexes to that of the chloroform (a polar solvent), hexane (a nonpolar solvent), DFT calculations, and previous results9.
Figure 3.

FTIR spectra of 1 in chloroform (cyan); 1⊂A. (purple); 1⊂B (green) ; 2 in chloroform (pink) ; 2⊂A. (red) ; 2⊂B (orange) ; 1 and 2 in hexane are shown as black dashed lines.
For 1 and 2, free in solution, it is known that their different isomeric forms are stabilized by different solvents and changing the solvent leads to a shift in the relative population of the different isomers, resulting in a change in the amplitudes of the peaks in the FTIR spectra associated with the different isomeric forms .18-19 This can be seen when comparing the FTIR spectra of 1 and 2 in chloroform (dashed line in cyan and pink respectively) and hexane (dashed line in black) displayed in Figure 3.
The FTIR spectrum of the encapsulated metal carbonyls (1⊂A, 1⊂B, 2⊂A, and 2⊂B) in D2O are displayed in Figure 3 as solid lines. The spectral changes along with comparison to previous results9 indicate that 1 and 2 exist in their cis-bridging isomeric forms when encapsulated in A and B. The cis-bridging isomer exhibits three peaks in the carbonyl stretching region: a strong transition in the bridging carbonyl region ~1700 cm−1, and in the terminal carbonyl region, a weak lower-frequency transition at ~1960 cm−1 and a strong higher-frequency transition at ~2000 cm−1 (see section 5, Figure S10, 11 and Table SI in the SI for DFT calculated spectra) . The FTIR spectra of 1⊂A, 1⊂B, 2⊂A, and 2⊂B indicate the cis-bridging isomer is stabilized when either 1 or 2 is encapsulated by either of the nanocages A or B. This observation is consistent with the previous results of 1 in a similar Pd6L4 cage9 and demonstrates that the smaller di-iron complex is also stabilized in its cis-bridging form when encapsulated. Our findings exemplify that changes in the size of nanocage cavity and the metal carbonyl complexes result in a similar stabilization of the cis-bridging isomer for 1 and 2.
The FTIR spectra confirm that the cages A and B stabilize the cis-bridging isomeric form of both 1 and 2, but steady state measurements lack the dynamic information of the guests encapsulated by the nanocages. To investigate the impact of the Pd6L4 nanocages on the dynamics of the cis-bridging isomer we applied ultrafast mid-IR pump-probe spectroscopy to 1 and 2 free in solution and encapsulated by the cage. Ultrafast mid-IR pump-probe measurements were obtained under two different polarizations schemes, parallel (I∥) and perpendicular I┴, to extract the population relaxation, P(t) (Eq. 1), and anisotropy decay, r(t) (Eq. 2) , (see section 1 and 6, Figure S12-23 of SI) .34-36
| Eq. 1 |
| Eq. 2 |
Note that the population and anisotropy decays reported in the SI and Figure 4 are extracted from the highest frequency transition to ensure minimal contributions from additional excited state absorptions and ground state bleaches (see SI for additional information)
Figure 4 .

Population and anisotropy decays are plotted for the highest frequency transition of 1 in chloroform (cyan); 1⊂A (purple); 1⊂B (green); 2 in chloroform (pink); 2⊂A (red); 2⊂B (orange).
The population relaxation reports on the decay of the population in the excited vibrational state, serving as a measure of the vibrational lifetime. Figure 4(a) and 4(c) display the population relaxation for the metal carbonyl complexes 1 and 2 in chloroform and cages A and B. As noted above, due to issues with solubility the same solvents cannot be used for the free in solution and encapsulated measurements. As the spectral broadening and spectral shift of the metal carbonyl in the positively charged cage is more akin to the chloroform solvent compared to hexane (see Figure 3), we compare the encapsulated forms to the free forms of 1 and 2 in chloroform. This choice is also motivated by previous studies.9 We note that for both 1 and 2 we observe that the lifetime in chloroform solution is longer than their cage encapsulated forms. As the cages can be thought of as isolating the metal carbonyls from the bulk D2O solvent, one may expect the lifetime of the metal carbonyls to be longer when encapsulated. However, the cage also introduces additional pathways by which vibrational energy can be dissipated due to the increase in vibrational degrees of freedom. We attribute the faster population relaxation of the encapsulated metal carbonyls to additional vibrational degrees of freedom introduced by the cage. We note that the cage may also alter the intramolecular vibrational energy redistribution among the carbonyl modes of the metal carbonyl complexes leading to changes in the population relaxation. Future work employing 2DIR spectroscopy will focus on investigating the influence of cage on intramolecular vibrational energy redistribution of the metal carbonyl complexes.
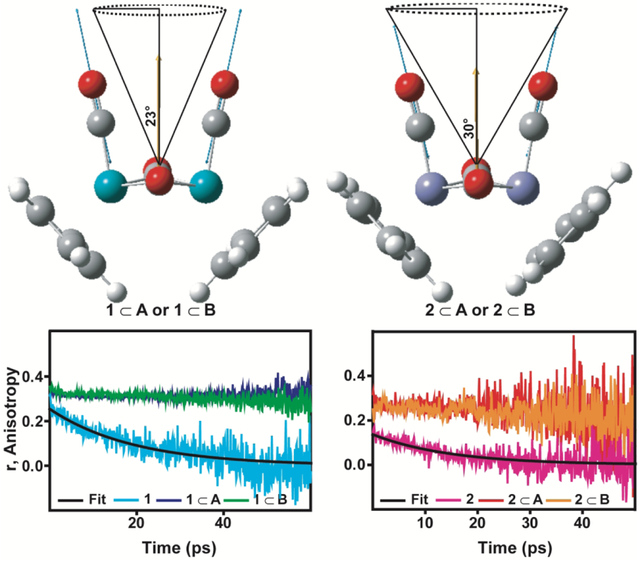
To further exemplify changes in the structural and dynamical nature of the metal carbonyls resulting from encapsulation, we compared their anisotropy decays. The anisotropy is a measure of the diffusional rotational motion of the molecule. The anisotropy decays for 1 and 2 in chloroform and cages A and B are plotted in Figure 4 (b) and 4 (d) . For all the systems, the anisotropy has initial drop from the theoretical value of 0.4 at zero time for an isotropic system, 37-40 indicating an unresolved ultrafast component. After the initial drop in anisotropy, the metal carbonyls in solution decay to zero with a mono-exponential timescale of ~20 ps for 1, the larger metal carbonyl complex, and ~12 ps for 2, the smaller metal carbonyl complex. We note that the anisotropy decay will vary in different solvents, depending on viscosity, temperature, and solute-solvent interactions. 34-35, 41-43 Nonetheless, the measurements in chloroform demonstrate that the metal carbonyls decay as a mono-exponential to zero indicating that the metal carbonyls can freely rotate in solution on the picosecond timescale. When the metal carbonyls are encapsulated in cages A and B the anisotropies do not decay to zero over the course of the vibrational lifetime. This indicates a restriction of the metal carbonyl’s angular motion. We attribute this to the previously observed π-stacking interactions9 between the host and guest, in addition to favorable electrostatic interactions between the cyclopentadienyl groups of the metal carbonyl and the triazine walls of the cage. These interactions effectively tether the metal carbonyl complex to the triazine wall of the host over the course of the vibrational lifetime, indicating that the interactions are not breaking/reforming on the timescale of the vibrational lifetime.
To extract additional information we apply a wobbling-in-a-cone model to the anisotropy data.38-39 Previous mid-IR pump-probe experiments performed on various probe molecules in confined environments such as membranes, polymers, or MOFs have employed a wobbling-in-a-cone model to interpret the anisotropy decay where angular motion of the probe molecule is influenced by the local environment.34-35, 38-39 According to the wobbling-in-a-cone model the anisotropy is given by the following expression (Eq. 3) where r0 is the anisotropy at zero time, r∞ accounts for the non-decaying anisotropy at long times, and τθ is the timescale associated with the reorientational motion as the probe decays from r0 to r∞.38-39
| Eq. 3 |
With this model it is possible to calculate the orientational diffusion of the probe molecule within the boundary of a cone with a half angle given by θC. The initial and final anisotropy values are related to the cone semi-angle (θC) according to the following expression.38-39
| Eq. 4 |
The cone semi-angle (θC) reports on the extent by which the nanocages restrict the motion of the metal carbonyls. To determine (θC) we fit the anisotropy data using Eq. 3 to obtain r∞. The fits are plotted as black lines in Fig. S24 in the supporting information and the optimized parameters are given in Table S3 in the SI. From the optimized parameters, we extract a final anisotropy of r∞ ~0.31 for 1⊂ (A, B) and r∞~0.25 for 2⊂(A, B) . We find that 1 has a larger final anisotropy, which corresponds to a smaller region of space sampled during restricted rotational motion. To quantify the differences in the samples, we extract an upper limit for the cone semi-angle sampled as the metal carbonyls undergo restricted rotational diffusion by employing Eq. 4. As we do not completely capture the initial ultrafast decay, due to the 120 fs temporal width of the incoming mid-IR pulses, we employ Eq. 4 to extract an upper limit for the semi-cone angle. The largest possible semi-cone angle provides insight into the restricted motion of the encapsulated metal carbonyl complexes and enables a comparison of guests 1 and 2. To extract the upper limit, we assume a single exponential decaying component with an initial anisotropy of 0.4. Within this limit we extract an angle of θc = 23.2°± 0.1° for 1⊂A., θc = 22.8°± 0.3° for 1⊂B, θc = 30.0°± 0.7°for 2⊂A, and θc = 32.3 °± 0.5° for 2⊂B.
The difference in the observed cone angles is attributed to the structural differences in the metal carbonyl complexes 1 and 2. This is schematically depicted in Figure 5. Fig. 5 displays the DFT optimized structures of 1 and 2 with the nuclear displacements of the ~2000 cm−1 terminal carbonyl symmetric stretching vibrational mode (blue arrows) and the corresponding dipole derivate unit vector (yellow arrow). Within the wobbling-in-a-cone model we can think of the molecular reorientation probed by monitoring the symmetric carbonyl stretching mode as rotation about the derivative dipole vector of the symmetric mode, with the center of the cone lying along the derivative dipole vector. The cone angles are schematically depicted in Fig. 5. The larger cone angle associated with 2⊂A or B is attributed to the smaller size of 2 with a Fe-Fe bond length of 2.566 Å and molecular volume of 371.45 Å3/molecule for the cis-bridging isomer. The smaller size of the guest leads to less spatial constraints and more accessible volume for the metal carbonyl complex 2 to sample, hence the larger cone angle. The larger size of 1, with a Ru-Ru bond length of 2.785 Å and molecular volume of 411.35 Å3/molecule for the cis-bridging isomer, renders it more sterically hindered when encapsulated by the nanocage with less accessible free volume for rotation. For a given metal carbonyl complex, the anisotropies for cage A and B are very similar, indicating that structural differences in the cage that lead to changes in the electronic spectrum (see Figure 2), do not have a large influence on the restricted rotational diffusion of the encapsulated metal carbonyl complexes.
Figure 5.

The DFT optimized structure for the cis-bridging isomers of 1 and 2 are shown. Nuclear displacements associated with the symmetric stretching mode of the terminal carbonyl groups are indicated with blue arrows and the corresponding dipole derivative unit vector is shown in yellow. Semi-cone angles (θC) extracted from the anisotropy decays of 1 and 2 in nanocages A and B are indicated schematically.
To summarize, we demonstrated the influence of π-stacking, electrostatics, and steric constraints imposed by Pd6L4 self-assembled nanocages (A and B) on the structure and dynamics of the fluxional metal carbonyl guests (1 and 2) . Previous studies have demonstrated that Pd6L4 nanocages can trap and stabilize the cis-bridging isomer of 1.9 We have expanded on these studies by performing a systematic study, varying the cavity size of the nanocage and the metal carbonyl guest, to investigate the ultrafast dynamics of the trapped metal carbonyl complexes. Comparing UV/VIS spectra we find that the presence of different coordination ligands for cages A and B results in the formation of charge transfer states with different spectral properties for the four different host-guest complexes (1⊂A, 1⊂B, 2⊂A, and 2⊂B) . From FTIR spectra, we find that both cages act to primarily stabilize the cis-bridging isomer of both guest molecules. Probing the cis-bridging isomer with ultrafast mid-IR polarization dependent pump-probe spectroscopy, we find that encapsulation leads to a faster population relaxation. We attribute this to the accessibility of additional bath modes resulting from the presence of the cage. The anisotropy decays show that the metal carbonyl complexes are free to rotate in solution; however, when encapsulated by the cage their angular motion is restricted, with the larger metal carbonyl complex sampling less volume when encapsulated. The non-decay anisotropy indicates that the π-stacking and electrostatic interactions act to effectively tether the metal carbonyl complexes to the cage over the course of the vibrational lifetime. The group VIII metal carbonyl complexes are well known for their fluxional behavior,16-19 photophysics, and photochemistry16,19. Recent studies have shown that the photochemistry of the group VIII di-metal carbonyl complexes can be altered through encapsulation by Pd6L4 based nanocages.11 This was attributed to a restriction in the dynamics of the metal carbonyl complexes.11 Here we have characterized the ultrafast dynamics of the metal carbonyls when encapsulated by Pd6L4 nanocages, which could lead to further insight into how to modify Pd6L4 nanocages to alter the structural properties and photochemistry of the metal carbonyl complexes, and guests in general44.
Supplementary Material
ACKNOWLEDGMENT/FUNDING SOURCES:
The authors acknowledge support for this work by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under award number DE-SC-0016043 and startup funds from the University of Pennsylvania. S.L.M. acknowledges support from NIH T32 predoctoral training grant GM008275 and the University of Pennsylvania School of Arts and Science Mitchell Fellowship.
Footnotes
Supporting Information: Synthetic protocol, details of steady state and time-resolved measurements, pump-probe experimental apparatus, comparison of UV/VIS spectra, DFT calculations, pump-probe spectra, wobbling-in-a-cone analysis
Notes: The authors declare no competing financial interests.
REFERENCES
- (1).Inokuma Y; Kawano M; Fujita M Crystalline molecular flasks. Nat. Chem 2011, 3, 349. [DOI] [PubMed] [Google Scholar]
- (2).McConnell AJ; Wood CS; Neelakandan PP; Nitschke JR Stimuli-Responsive Metal-Ligand Assemblies. Chem. Rev 2015, 115, 7729–7793. [DOI] [PubMed] [Google Scholar]
- (3).Brown CJ; Toste FD; Bergman RG; Raymond KN Supramolecular Catalysis in Metal-Ligand Cluster Hosts. Chem. Rev 2015, 115, 3012–3035. [DOI] [PubMed] [Google Scholar]
- (4).Johnstone MD; Schwarze EK; Ahrens J; Schwarzer D; Holstein JJ; Dittrich B; Pfeffer FM; Clever GH Desymmetrization of an Octahedral Coordination Complex Inside a Self-Assembled Exoskeleton. Chem-Eur J. 2016, 22, 10791–10795. [DOI] [PubMed] [Google Scholar]
- (5).Cullen W; Metherell AJ; Wragg AB; Taylor CGP; Williams NH; Ward MD Catalysis in a Cationic Coordination Cage Using a Cavity-Bound Guest and Surface-Bound Anions: Inhibition, Activation, and Autocatalysis. J. Am. Chem. Soc 2018, 140, 2821–2828. [DOI] [PubMed] [Google Scholar]
- (6).Frank M; Johnstone MD; Clever GH Interpenetrated Cage Structures. Chem-Eur J. 2016, 22, 14104–14125. [DOI] [PubMed] [Google Scholar]
- (7).Fujita M; Oguro D; Miyazawa M; Oka H; Yamaguchi K; Ogura K Self-assembly of ten molecules into nanometer-sized organic host frameworks. Nature 1995, 378, 469. [Google Scholar]
- (8).Gera R; Das A; Jha A; Dasgupta J Light-Induced Proton-Coupled Electron Transfer Inside a Nanocage. J. Am. Chem. Soc 2014, 136, 15909–15912. [DOI] [PubMed] [Google Scholar]
- (9).Horiuchi S; Murase T; Fujita M Noncovalent Trapping and Stabilization of Dinuclear Ruthenium Complexes within a Coordination Cage. J. Am. Chem. Soc 2011, 133, 12445–12447. [DOI] [PubMed] [Google Scholar]
- (10).Yoshizawa M; Miyagi S; Kawano M; Ishiguro K; Fujita M Alkane Oxidation via Photochemical Excitation of a Self-Assembled Molecular Cage. J. Am. Chem. Soc 2004, 126, 9172–9173. [DOI] [PubMed] [Google Scholar]
- (11).Horiuchi S; Murase T; Fujita M A Remarkable Organometallic Transformation on a Cage-Incarcerated Dinuclear Ruthenium Complex. Angew. Chem. Int. Edit 2012, 51, 12029–12031. [DOI] [PubMed] [Google Scholar]
- (12).Jaworska M; Macyk W; Stasicka Z Structure, Spectroscopy and Photochemistry of the [M(η5-C5H5)(CO)2]2 Complexes (M=Fe, Ru) In Optical Spectra and Chemical Bonding Inorganic Compounds: Special Volume dedicated to Professor Jørgensen I, Mingos DMP; Schönherr T, Eds. Springer Berlin Heidelberg: Berlin, Heidelberg, 2004; pp 153–172. [Google Scholar]
- (13).Bitterwolf TE Photochemistry and reaction intermediates of the bimetallic Group VIII cyclopentadienyl metal carbonyl compounds, (η5-C5H5)2M2(CO)4 and their derivatives. Coordin. Chem. Rev 2000, 206–207, 419–450. [Google Scholar]
- (14).Abrahamson HB; Palazzotto MC; Reichel CL; Wrighton MS Photochemistry and electronic structure of bis(dicarbonyl(.eta.5-cyclopentadienyl)ruthenium) and its iron analog. J. Am. Chem. Soc 1979, 101, 4123–4127. [Google Scholar]
- (15).Donovan ES; Felton GAN Electrochemical analysis of cyclopentadienylmetal carbonyl dimer complexes: Insight into the design of hydrogen-producing electrocatalysts. J. Organomet. Chem 2012, 111, 25–34. [Google Scholar]
- (16).Bullitt JG; Cotton FA; Marks TJ Structural dynamic properties of pentahaptocyclopentadienylmetal dicarbonyl dimers. Inorg. Chem 1972, 11, 671–676. [Google Scholar]
- (17).McArdle P; Manning AR The structures of dicarbonylcyclopentadienylruthenium dimer [{Ru([small pi]-C5H5)(CO)2}2] and some related complexes in solution. J. Chem. Soc. A 1970, 2128–2132. [Google Scholar]
- (18).Manning AR The structure of bis-[small pi]-cyclopentadienyldi-iron tetracarbonyl in solution. J. Chem. Soc. A 1968, 1319–1324. [Google Scholar]
- (19).Cotton FA; Yagupsky G Tautomeric changes in metal carbonyls. I. .pi.-Cyclopentadienyliron dicarbonyl dimer and .pi.-cyclopentadienyl-ruthenum dicarbonyl dimer. Inorg. Chem 1967, 6, 15–20. [Google Scholar]
- (20).Yoshizawa M; Klosterman JK; Fujita M Functional Molecular Flasks: New Properties and Reactions within Discrete, Self-Assembled Hosts. Angew. Chem. Int. Edit 2009, 48, 3418–3438 [DOI] [PubMed] [Google Scholar]
- (21).Nishida J; Fayer MD Guest Hydrogen Bond Dynamics and Interactions in the Metal-Organic Framework MIL-53(Al) Measured with Ultrafast Infrared Spectroscopy. J. Phys. Chem. C 2017, 121, 11880–11890. [Google Scholar]
- (22).Das A; Jha A; Gera R; Dasgupta J Photoinduced Charge Transfer State Probes the Dynamic Water Interaction with Metal-Organic Nanocages. J. Phys. Chem. C 2015, 119, 21234–21242. [Google Scholar]
- (23).Osborne DG; King JT; Dunbar JA; White AM; Kubarych KJ Ultrafast 2DIR probe of a host-guest inclusion complex: Structural and dynamical constraints of nanoconfinement. J. Chem. Phys 2013, 138, 144501. [DOI] [PubMed] [Google Scholar]
- (24).Yuan R; Yan C; Nishida J; Fayer MD Dynamics in a Water Interfacial Boundary Layer Investigated with IR Polarization-Selective Pump-Probe Experiments. J. Phys. Chem. B 2017, 121, 4530–4537. [DOI] [PubMed] [Google Scholar]
- (25).Groot CCM; Velikov KP; Bakker HJ Structure and dynamics of water molecules confined in triglyceride oils. Phys. Chem. Chem. Phys 2016, 18, 29361–29368. [DOI] [PubMed] [Google Scholar]
- (26).Lee J; Maj M; Kwak K; Cho M Infrared Pump-Probe Study of Nanoconfined Water Structure in Reverse Micelle. J. Phys. Chem. Lett 2014, 5, 3404–3407. [DOI] [PubMed] [Google Scholar]
- (27).Costard R; Greve C; Heisler IA; Elsaesser T Ultrafast Energy Redistribution in Local Hydration Shells of Phospholipids: A Two-Dimensional Infrared Study. J. Phys. Chem. Lett 2012, 3, 3646–3651. [DOI] [PubMed] [Google Scholar]
- (28).Marroux HJB; Orr-Ewing AJ Distinguishing Population and Coherence Transfer Pathways in a Metal Dicarbonyl Complex Using Pulse-Shaped Two-Dimensional Infrared Spectroscopy. J. Phys. Chem. B 2016, 120, 4125–4130. [DOI] [PubMed] [Google Scholar]
- (29).Calabrese C; Vanselous H; Petersen PB Deconstructing the Heterogeneity of Surface-Bound Catalysts: Rutile Surface Structure Affects Molecular Properties. J. Phys. Chem. C 2016, 120, 1515–1522. [Google Scholar]
- (30).Nishida J; Tamimi A; Fei H; Pullen S; Ott S; Cohen SM; Fayer MD Structural dynamics inside a functionalized metal-organic framework probed by ultrafast 2D IR spectroscopy. P. Natl. Acad. Sci 2014, 111, 18442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Baiz CR; Kubarych KJ; Geva E; Sibert EL Local-Mode Approach to Modeling Multidimensional Infrared Spectra of Metal Carbonyls. J. Phys. Chem. A 2011, 115, 5354–5363. [DOI] [PubMed] [Google Scholar]
- (32).Anna JM; King JT; Kubarych KJ Multiple Structures and Dynamics of [CpRu(CO)2]2 and [CpFe(CO)2]2 in Solution Revealed with Two-Dimensional Infrared Spectroscopy. Inorg. Chem 2011, 50, 9273–9283. [DOI] [PubMed] [Google Scholar]
- (33).Fang Y; Murase T; Sato S; Fujita M Noncovalent Tailoring of the Binding Pocket of Self-Assembled Cages by Remote Bulky Ancillary Groups. J. Am. Chem. Soc 2013, 135, 613–615. [DOI] [PubMed] [Google Scholar]
- (34).Shin JY; Yamada SA; Fayer MD Dynamics of a Room Temperature Ionic Liquid in Supported Ionic Liquid Membranes vs the Bulk Liquid: 2D IR and Polarized IR Pump-Probe Experiments. J. Am. Chem. Soc 2017, 139, 311–323. [DOI] [PubMed] [Google Scholar]
- (35).Tamimi A; Fayer MD Ionic Liquid Dynamics Measured with 2D IR and IR Pump-Probe Experiments on a Linear Anion and the Influence of Potassium Cations. J. Phys. Chem. B 2016, 120, 5842–5854. [DOI] [PubMed] [Google Scholar]
- (36).Sebastian S; Andreas S; Johannes B; Patrick N; Tobias B Generalized magic angle for time-resolved spectroscopy with laser pulses of arbitrary ellipticity. J. Phys. B – At. Mol. Opt 2014, 47, 124014. [Google Scholar]
- (37).Moilanen DE; Fenn EE; Lin Y-S; Skinner JL; Bagchi B; Fayer MD Water inertial reorientation: Hydrogen bond strength and the angular potential. P. Natl. Acad. Sci 2008, 105, 5295–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kinosita K Jr.; Ikegami A; Kawato S On the wobbling-in-cone analysis of fluorescence anisotropy decay. Biophys. J 1982, 37, 461–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kinosita K Jr.; Kawato S; Ikegami A A theory of fluorescence polarization decay in membranes. Biophys. J 1977, 20, 289–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Gordon RG Molecular Collisions and the Depolarization of Fluorescence in Gases. J. Chem. Phys 1966, 45, 1643–1648. [Google Scholar]
- (41).Shin JY; Yamada SA; Fayer MD Carbon Dioxide in a Supported Ionic Liquid Membrane: Structural and Rotational Dynamics Measured with 2D IR and Pump-Probe Experiments. J. Am. Chem. Soc 2017, 139, 11222–11232. [DOI] [PubMed] [Google Scholar]
- (42).Nicodemus RA; Ramasesha K; Roberts ST; Tokmakoff A Hydrogen Bond Rearrangements in Water Probed with Temperature-Dependent 2D IR. J. Phys. Chem. Lett 2010, 1, 1068–1072. [Google Scholar]
- (43).Anna JM; Kubarych KJ Watching solvent friction impede ultrafast barrier crossings: A direct test of Kramers theory. J. Chem. Phys 2010, 133, 174506. [DOI] [PubMed] [Google Scholar]
- (44).Michito Y; K. KJ; Makoto F Functional Molecular Flasks: New Properties and Reactions within Discrete, Self-Assembled Hosts. Angew. Chem. Int. Edit 2009, 48, 3418–3438. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.