

Abstract
Sertoli cells (SeC) are responsible for the immunoprivileged status of the testis thanks to which allogeneic or xenogeneic engraftments can survive without pharmacological immune suppression if co‐injected with SeC. This peculiar ability of SeC is dependent on secretion of a plethora of factors including maturation factors, hormones, growth factors, cytokines and immunomodulatory factors. The anti‐inflammatory and trophic properties of SeC have been largely exploited in several experimental models of diseases, diabetes being the most studied. Duchenne muscular dystrophy (DMD) is a lethal X‐linked recessive pathology in which lack of functional dystrophin leads to progressive muscle degeneration culminating in loss of locomotion and premature death. Despite a huge effort to find a cure, DMD patients are currently treated with anti‐inflammatory steroids. Recently, encapsulated porcine SeC (MC‐SeC) have been injected ip in the absence of immunosuppression in an animal model of DMD resulting in reduction of muscle inflammation and amelioration of muscle morphology and functionality, thus opening an additional avenue in the treatment of DMD. The novel protocol is endowed with the advantage of being potentially applicable to all the cohort of DMD patients regardless of the mutation. This mini‐review addresses several issues linked to the possible use of MC‐SeC injected ip in dystrophic people.
Keywords: Duchenne muscular dystrophy, encapsulation, muscle inflammation, Sertoli cell, therapeutic approaches
1. INTRODUCTION
Sertoli cells (SeC) are major component of the seminiferous tubules of the testis where they contribute to the development of germ cells and protect germ cells from the attack by the host immune system.1, 2 Indeed, newly synthesized markers on germ cell surface would be recognized as non‐self by the immune system, as this latter becomes mature before spermatogenesis starts. SeC exert their double role (a) by creating a physical barrier (the blood‐testis barrier, BTB) made of adjacent SeC linked together with tight junctions, isolating the lumen of seminiferous tubules from the interstitial fluid, and (b) by secreting a plethora of trophic and immunomodulatory factors.3, 4 This latter ability of SeC has prompted researchers to use them in many experimental models of diseases in which supplying trophic factors, abating inflammation or modulating the immune system activity might result in reversion or attenuation of the pathology.2, 5
2. SERTOLI CELLS
The immunoprivileged status of the testis is long known. In 1767, during his studies on transplantation, John Hunter observed that rooster testes transplanted into the abdominal cavity of a hen maintained normal structure over time.6 Subsequent investigation identified SeC as the cell type mainly responsible for the immunological properties of the testis. Indeed, allogeneic and/or xenogeneic pancreatic islets,7, 8 adrenal chromaffin cells9 and dopaminergic neurons10 were successfully protected by SeC in the absence of pharmacological immune suppression in different experimental models. Similarly, skin11 and heart12 grafts showed prolonged survival when co‐injected with SeC. Moreover, grafts of SeC alone exerted trophic effects in the central nervous system in animal models of Huntington’s disease and amyotrophic lateral sclerosis.13, 14
Intriguing results about SeC derive from their use as encapsulated cells in several pre‐clinical studies. Alginate‐based microcapsules containing SeC (MC‐SeC) have been successfully employed as a single intraperitoneal (ip) injection in experimental models of type 1 and type 2 diabetes, acute hepatic failure, skin graft and Huntington’s disease.15, 16, 17, 18, 19, 20, 21 In an animal model reproducing the human Laron syndrome, in which mutations in the growth hormone receptor (GHR) lead to reduced production of IGF‐1 and subsequent dwarfism, intraperitoneally injected MC‐SeC promoted body growth through the release of IGF‐1 into the circulation.22
The trophic and immunomodulatory properties of SeC are dependent on the complex SeC secretory activity resulting in a cocktail of factors whose formulation is difficult to dissect, being affected by the biological status of the cells and likely by environmental cues. While the composition of SeC’ secretory product still awaits to be identified, it includes maturation factors, hormones, growth factors, cytokines and immunomodulatory factors (Table 1).
Table 1.
Factors known to be secreted by SeC
| Maturation factors and hormones |
Activins116 Dhh117 |
Oestrogens118 Inhibins116 |
MIS/AMH121 |
| Growth factors and cytokines |
BDNF122 bFGF123 BP4124 EGF125 GDNF126 Heregulin‐β194 |
IFN‐γ127 IGF‐2130 SCSGF133 |
PDGF135 SGP‐1/Prosaposin, SGP‐2136, 137 VEGF140 |
| Immunomodulatory factors |
Activin A116 BCL‐w141 Clusterin142 Complement cascade inhibitors26 |
IDO15 IL‐2 suppressor factors23 JAG129 |
MIF145 Serpins27 TGF‐β139 |
AMH, anti‐Müllerian hormone; BDNF, brain‐derived neurotrophic factor; bFGF, basic fibroblast growth factor; BMP4, bone morphogenetic protein 4; Dhh, desert hedgehog; EGF, epidermal growth factor; FasL, Fas ligand; GDNF, glial cell–derived neurotrophic factor; IDO, indoleamine 2,3‐dioxygenase; IFN, interferon; IGF, insulin‐like growth factor; IL, interleukin; JAG1, soluble JAGGED1; KL, kit ligand; MIF, macrophage inhibitory factor; MIS, Müllerian‐inhibiting substance; NT, neurotrophin; PDGF, platelet‐derived growth factor; SCF, stem cell factor; SCSGF, SeC‐secreted growth factor; SGP, sulphated glycoprotein; TGF, transforming growth factor; VEGF, vascular endothelial growth factor.
However, the immunomodulatory effect of SeC is obtained by a multimodal mechanism. SeC secrete (still unidentified) factors thatblock T lymphocyte proliferation and interleukin (IL)‐2 production,23, 24 and SeC induce apoptosis of lymphocytes mediated by the interaction of FasL expressed on their surface and Fas receptor (CD95) expressed on T cells.25 SeC secrete inhibitors of the complement cascade and granzyme, a cytolytic molecule released by cytotoxic T cells.26, 27 Moreover, the secretion of specific factors, such as TGF‐β, IDO (indoleamine 2,3‐dioxygenase), activin A and JAG1, concurs to immunomodulation favouring the emergence of tolerogenic cells, including M2 (anti‐inflammatory) macrophages, and Th2 and Tregs.15, 28, 29
3. DUCHENNE MUSCULAR DYSTROPHY AND RELATED THERAPEUTIC APPROACHES
Duchenne muscular dystrophy (DMD) is an X‐linked recessive disease due to mutations in the dystrophin gene (DMD), the biggest gene of the human genome for which about 4700 different mutations have been reported.30, 31, 32, 33 Dystrophin is an essential component of the dystrophin‐associated protein complex (DAPC), a multiprotein complex located at the sarcolemma and responsible for the mechanical link between the intracellular cytoskeleton and the extracellular matrix; dystrophin ensures the structural and functional integrity of myofibres during contraction. DMD gene mutations translating into absence of dystrophin or expression of functionally inefficient protein lead to the Duchenne phenotype, in which loss of the integrity of DAPC causes myofibre degeneration and progressive loss of muscle efficiency, wheelchair dependency before teenage years, and premature death by cardiac and respiratory failure.31, 34 Morphologically, DMD muscles are characterized by infiltration with immune cells and chronic activation of inflammatory signalling pathways due to continuous degeneration/regeneration cycles, with the final result that fibrous and fatty tissues progressively overtake functional myofibres.35
Therapeutic approaches to DMD have been experiencing multiple obstacles against their success (Table 2). Firstly, DMD gene is a too large gene (2.4 Mb and 79 exons, corresponding to about 0.1% of the human genome)30 to be delivered using classical recombinant adeno‐associated viruses (rAAVs), which has led to investigation of the use of parts of the gene translating into shorter but still efficient proteins, that is, mini‐ and micro‐dystrophins.36, 37, 38, 39, 40 Although AAV vectors carrying mini‐dystrophin have given encouraging results in mdx mice (the most used experimental model for DMD) and GRMD (golden retriever muscular dystrophy) dogs,41, 42, 43 phase I clinical trial revealed only a limited dystrophin expression and irrelevant muscle improvements due to the host immune response.44
Table 2.
Advantages and disadvantages of the main therapeutic approaches to DMD
| Approach | Advantages | Disadvantages | References |
|---|---|---|---|
| Gene therapy |
Potential rescue of functional dystrophin in cardiac and skeletal muscles Independent from DMD gene mutation AAV vectors are already approved for other pathologies |
Need to use truncated forms of dystrophin due to the high size of DMD gene Requirement of high doses of vectors Possible immune reaction against vectors Potential need for immunosuppressive therapy |
[37, 38, 39, 40] |
| Exon skipping |
Restoration of expression of partially functional dystrophin Some AON are well tolerated Eteplirsen received conditional approval by USA FDA |
DMD gene mutation‐dependent Scarce tissue uptake Large doses and repeated injections are required Significant side effects are reported in some cases Controversial efficacy |
[47, 48, 49] |
| Cell therapy |
Restoration of functional dystrophin Possibility to reprogramme adult somatic cells to iPSC Possibility to correct mutations ex vivo in patient cells Low risk of immune reaction in autologous transplantations |
Short lifespan and low migration ability of injected cells Immune reaction when cells come from healthy donors Requirement of immunosuppression in allogeneic transplantations |
[47, 55, 56, 57] |
| Utrophin induction |
Independent from DMD gene mutation Oral administration Well‐tolerated compounds No requirement of immunosuppressive treatment |
Ezutromid (SMT C1100) failed to reach its objectives | [53, 69] |
AAV, adeno‐associated viruses; AON, antisense oligonucleotides; FDA, Food and Drug Administration; iPSC, induced pluripotent stem cells.
Exon skipping is an approach to overcome specific regions with deletions, duplications or small mutations in the DMD gene pointing to recovery of the reading frame and production of truncated but functional forms of dystrophin, and translating into a switch from the DMD pathology to the milder phenotype known as Becker muscular dystrophy (BMD).45, 46 Specifically designed antisense oligonucleotides (AON), which are 20‐30 nucleotides in length, are used to obtain skipping of different exons resulting in truncated but in‐frame transcripts.47, 48, 49 The modified AON, 2′‐O‐methyl‐p phosphorothioate oligonucleotides (PS) and phosphorodiamidate morpholino oligomers (PMO) have shown high stability and efficacy, and low toxicity. One major limit of AON is that they are only useful for DMD patients with specific mutations and not applicable to the remaining cohort of patients. The PS, Drisapersen and the more promising PMO, Eteplirsen, both of which are in clinical trials, are specific for skipping of exon 51, which applies to 14% of patients, that is, the largest cohort of DMD subjects who may benefit from single exon skipping.39, 50 Other limitations inherent to the use of AON are represented by their scarce tissue uptake and low rescue of dystrophin expression in muscles.
Cell therapy represents a second front of therapeutic approaches to DMD. It tries to use different cell types (especially, satellite cells/myoblasts, mesoangioblasts and induced pluripotent stem cells [iPSC]) from healthy donors or genetically engineered cells from the patients themselves to obtain the re‐expression of dystrophin in muscle tissue and recovery of muscle performance.51, 52, 53, 54 Cells obtained from patients are corrected ex vivo and re‐implanted in the donors, whereas cells from healthy donors are used in allogeneic transplantations in dystrophic patients.55 This kind of approach is finding limits in the low survival of injected cells and their inability to migrate for long distances, so that repeated local injections are required.56 The reasons why injected cells show low survival and scarce ability to migrate are not fully understood, but deficiency of specific growth factors might play an important role. Immune rejection of transplanted cells is another relevant concern in the case of allogeneic approaches, which require concomitant pharmacological immunosuppression.57
Healthy human satellite cells and myoblasts (ie, muscle precursor cells) induce dystrophin expression in DMD patients to a certain extent when injected intramuscularly58; a phase I/II clinical trial (NCT02196467) is still ongoing. iPSC are somatic cells (including fibroblasts, hepatocytes, pancreatic beta cells, lymphocytes and neural progenitor cells) reprogrammed in vitro to a pluripotent state by ectopic expression of Oct4 and Sox2 in combination with either Klf4 and c‐Myc or Lin28 and Nanog.59 iPSC show similarity to embryonic stem cells, can be directed to mesenchymal differentiation, and patient‐derived iPSC can be genetically engineered for potential autologous therapies. However, iPSC have found employment only in up to pre‐clinical studies so far.60 Mesoangioblasts (ie, cells associated with the walls of large vessels) represent one of the most promising cell types for cell therapy in DMD patients. Mesoangioblasts are endowed with myogenic potential and ability to cross the blood vessel wall, and their use has resulted in improvement of muscle morphology in several experimental models of muscular dystrophy.45, 61, 62 Intra‐arterial injection of allogeneic human mesoangioblasts isolated from adult skeletal muscle is currently under phase I clinical trial (EudraCT #2011‐000176‐33).
The existence of the dystrophin paralogue, utrophin, has fostered another approach to rescue homeostasis in the muscles of DMD patients. Utrophin shares a very high degree of sequence identity with dystrophin and even associates with members of the DAPC, thus mimicking the role of dystrophin in dystrophin‐negative myofibres.30, 63 In healthy adult muscle fibres, dystrophin and utrophin show different expression patterns, with dystrophin being expressed along the entire sarcolemma and utrophin confined to the myotendinous and the neuromuscular junctions (NMJs).64, 65 However, utrophin is expressed at high levels at the sarcolemma during development, when dystrophin is not expressed yet.66 Indeed, necrosis of mdx limb muscles begins only when the high neonatal levels of utrophin become reduced to adult levels.64 Since forced expression of utrophin in dystrophic myofibres can restore assembly of DAPC members at the sarcolemma and prevent the dystrophic pathology,67, 68 up‐regulation of utrophin in muscles represents a still active field of investigation in DMD treatment.69 In this regard, the small molecule, SMT C1100 (Ezutromid), which has shown promising results in a phase I clinical trial,70 was stopped after a phase II clinical trial (NCT02858362) since it failed to reach its primary and secondary objectives.
Several alternative approaches to treat DMD are currently under investigation (Table 3). They include use of the histone deacetylase (HDAC) inhibitor, Givinostat71, 72, 73; the phosphodiesterase‐5 (PDE5) inhibitors, Tadalafil and Sildenafil74, 75, 76; the benzoquinone, Idebenone (Catena/Raxone)77, 78, 79; the aminoglycoside, Ataluren (Translarna, former PTC124).80, 81 The anti‐fibrotic molecule, Halofuginone (HT‐100),82 and the anti‐myostatin monoclonal antibody, Domagrozumab,83, 84 have been blocked in phase II clinical trial due to death of a patient receiving the highest dose and no significant therapeutic effects, respectively. While treatment with Ataluren points to re‐expression of dystrophin in muscles, the majority of alternative approaches are aimed at restraining pathogenic mechanisms secondary to lack of dystrophin, particularly muscle inflammation and fibrosis.
Table 3.
Principal currently ongoing alternative approaches to treat DMD
| Drug | Description/Activity | Effects | Limitations | Clinical trial | References |
|---|---|---|---|---|---|
|
Ataluren (PTC124) PTC Therapeutics |
Small chemical compound that induces ribosomal read‐through of premature stop codons | Restoration of expression of full‐length dystrophin | Use limited to patients with nonsense mutations (nmDMD) |
Phase III completed Conditional approval in Europe |
[63, 64] |
|
Givinostat Italfarmaco |
Inhibitor of HDAC (enzymes that prevent gene activity), which are constitutively active in DMD muscles | Reduction of necrosis and fibrotic and adipose tissue deposition | No restoration of dystrophin expression | Phase III ongoing | [54, 55, 73] |
|
Idebenone (Catena/Raxone) Santhera Pharmaceuticals |
Chemical short‐chain benzoquinone; potent antioxidant and lipid peroxidation inhibitor at mitochondrial level | Expected cardioprotection and improvement of muscle performance and respiratory functions | No restoration of dystrophin expression | Phase III ongoing | [59, 60, 61] |
|
Tadalafil and Sildenafil Eli Lilly and Company |
PDE5 inhibitor induces vasodilatation through cGMP signalling activation | Expected improvement of muscle blood flow during physical exercise |
No restoration of dystrophin expression Little evidence of benefits |
Phase III completed | [56, 57, 76] |
|
Vamorolone (VBP15) ReveraGen BioPharma |
Glucocorticoid‐like oral drug with anti‐inflammatory and membrane‐stabilizing properties |
Reduction of muscle inflammation No glucocorticoid‐associated side effects |
No restoration of dystrophin expression | Phase II ongoing | [72] |
cGMP, cyclic guanosine monophosphate; HDAC, histone deacetylase; nmDMD, nonsense mutation Duchenne muscular dystrophy; PDE5, phosphodiesterase‐5.
4.
More recently, researchers are trying to useCRISPR/Cas9 genome editing system to remove mutated exons from the DMD gene.85, 86 This method uses small guide RNAs coupled with target‐specific double‐strand DNA endonuclease making possible targeted gene disruption, replacement or modification.87 The CRISPR/Cas9 approach has shown ability to rescue dystrophin expression in DMD patient‐derived iPSC in vitro, and in muscles of experimental models of DMD in vivo.88 Similar to AON, application of CRISPR/Cas9 method to DMD patients requires a personalized setting depending on the specific mutation. Other limitations for the use of CRISPR/Cas9 as a DMD treatment are represented by possible off‐targeting and activation of the host immune response.89
Such an extremely varying scenario in the therapeutic approaches to DMD is the result of the difficulty and, at the same time, the intense effort to find a cure for this pathology. Thus, the current gold standard therapy for DMD patients remains the use of anti‐inflammatory steroids (eg, Prednisone and Deflazacort), which improve the quality of life reducing loss of muscle strength and functionality and loss of ambulation, and delaying respiratory failure.45, 90 However, corticosteroids have shown limited activity and cause several adverse effects, including gain of weight, reduction of bone mineral density, cushingoid appearance, behavioural changes, adrenal suppression, susceptibility to infection, hypertension and metabolic disorders,91, 92 so that alternative anti‐inflammatory compounds such as the NF‐κB inhibitor, VBP15, are also under investigation93 (Table 3).
5. DO PORCINE SERTOLI CELLS REPRESENT AN OPPORTUNITY FOR DUCHENNE MUSCULAR DYSTROPHY?
Having the peculiar secretory properties of SeC in mind, we treated acute and chronic dystrophic, mdx mice with a single ip injection of porcine MC‐SeC (equivalent amount, 1.0 × 106 SeC/gram of body weight) in the absence of any pharmacologic immunosuppression, and found a rapid amelioration of muscle morphology and functionality.94, 95 After 3 weeks from injection, muscles of treated mice showed ~80% reduction of the inflammatory infiltrate (as assed by evaluation of activated macrophages marker, MAC3; Figure 1), ~60%‐70% reduction of fibrous tissue deposition, and over 90% reduction of necrotic myofibres in Tibialis anterior muscle. At the same time, MC‐SeC–treated mdx mice showed increased muscle performance and resistance to exercise‐induced muscle damage. Indeed, treated mice ran similar distances in a similar time to untreated WT mice, recovering ~80% of dystrophic‐dependent deficit, and showed a ~70% reduction of damaged (Evans blue dye–positive) myofibres after treadmill exercise tests. The anti‐inflammatory effect of MC‐SeC was observed already 1 week after injection, when ~70% reduction of macrophages infiltrating muscle tissue could been observed. At the same time, these macrophages showed a tissue‐repairing (M2) phenotype, as suggested by their reduced expression of inflammatory cytokines (ie, IL‐6, IL‐12 and IFN‐γ) and up‐regulation of the anti‐inflammatory IL‐10 and the M2 markers, arginase 1, CD163 and CD206.94 Interestingly, a single ip injection of MC‐SeC conferred benefits (ie, significantly reduced necrosis, inflammatory infiltrate, and fibrotic and adipose tissues deposition) in long‐term (5 months) analysis to the diaphragm, the muscle that accumulates damage over time in the mdx animal model. No signs of immune response against injected MC‐SeC could be detected at this time.94
Figure 1.
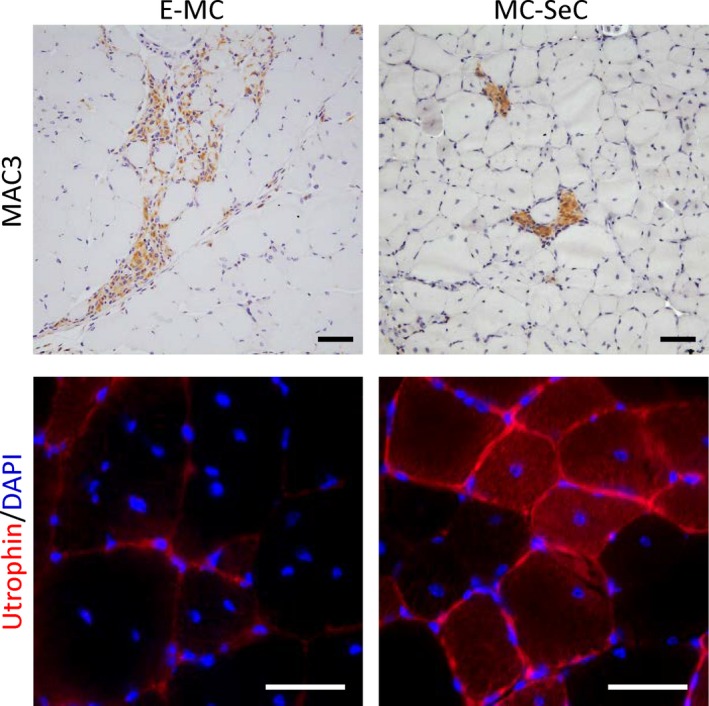
Intraperitoneal injection with microencapsulated Sertoli cells (MC‐SeC) in dystrophic mice results in reduced inflammation and re‐expression of utrophin in muscles. Tibialis anterior muscles of acute phase (4‐wk‐old) mdx mice analysed for the presence of the activated macrophage marker, MAC3, by immunohistochemistry (anti‐MAC3 antibody, clone M3/84; BD Biosciences) (upper panel) and the expression of utrophin by immunofluorescence (anti‐utrophin antibody, clone 8A4; Santa Cruz Biotechnology) (lower panel) 3 wk after ip injection with MC‐SeC or the same amounts of empty microcapsules (E‐MC). Note the significant reduction of inflammatory infiltrate (ie, MAC3‐positive areas) and the positivity for utrophin at the sarcolemma in muscles of MC‐SeC–treated mice. Original magnification, 20× (upper images) and 40× (lower images)
Another important aspect of SeC treatment is that SeC release heregulin β1, which is a major inducer of the expression of utrophin in muscle cells.96 Heregulin β1 acts by binding to erbB/HER receptor, resulting in intracellular ERK activation and subsequent binding of the ets‐related GABPα/β transcription factor complex to the utrophin‐A promoter.97 Three weeks after ip injection of MC‐SeC, a ~2.8‐fold increase in utrophin expression was observed in muscles of mdx mice,94 which is similar to that reported after 3 months of repeated ip injections each other day of the active domain of heregulin β1 (Aa 176‐246) in the same experimental model.98 In both cases, utrophin was found localized at the sarcolemma, a condition necessary for the protein to mimic the role of dystrophin (Figure 1).
Microcapsules containing SeC–based protocol resulted efficacious also in pre‐symptomatic (2‐week‐old) and in chronic (12‐month‐old) mdx mice.95 In the diaphragms of these latter mice, a significant reduction of adipose and fibrous tissue deposition (~43% and ~58% reduction, respectively), macrophage infiltrate (~70% reduction of MAC3‐positive areas) and damaged myofibres (more than 80% reduction of EBD‐positive myofibres) were observed 3 weeks after ip injection of MC‐SeC.95
The above reported results about the use of MC‐SeC have opened an additional avenue in the scenario of DMD treatment. Intraperitoneally injected MC‐SeC act as a micro‐biofactory that from the peritoneal cavity of dystrophic animals release factors into the bloodstream thus being able to reach every muscle where they exert a double effect: (a) an anti‐inflammatory effect (as expected) due to immunomodulatory factors; and (b) the induction of utrophin expression due to the SeC‐secreted heregulin β1.94 These two effects are independent from each other but cooperate to ameliorate muscle morphology activating a positive loop that leads to reduction of necrosis, fibrosis and adipose tissue deposition, finally culminating in rescue of muscle architecture and performance. Indeed, the use of an anti‐heregulin β1 antibody nullified the SeC‐dependent induction of utrophin in mdx muscles. However, anti‐heregulin β1 antibody had no significant effects on the reduction of the inflammatory infiltrate, suggesting that this latter effect is under the control of anti‐inflammatory factors. On the converse, only a partial loss of anti‐necrotic effects could be observed on myofibres of mdx mice treated with MC‐SeC in the presence of anti‐heregulin β1 antibody, due to a balance between increased damage extent of (dystrophin‐negative/utrophin‐negative) myofibres and the effects of anti‐inflammatory factors.94
Several considerations give particular relevance to the porcine MC‐SeC–based therapeutic approach (Table 4). On the side of the biomaterial used, (a) highly biocompatible, clinical grade alginate (endotoxin content less than 0.5 EU/mL, as required for human transplants) was used for the production of microcapsules15, 16, 17, 18, 19, 20, 21, 22, 94, 95, 101, 102, 103, 105; (b) alginate‐based microcapsules have shown long‐term survival and activity of entrapped cells22, 94, 99, 100 with porcine IGF‐1 being detected in the serum of mice treated with porcine‐derived SeC up to 1 year after injection22; and (c) alginate‐based microcapsules containing human pancreatic islets have been employed in a phase I clinical trial in which they were transplanted ip in non‐immunosuppressed type 1 diabetic patients with no undesired effects reported.101, 102, 103 On the side of SeC, (a) SeC were purified from testis of SPF (specific pathogen free) piglets, that is, animals suitable for engraftment in humans; 104 (b) MC‐SeC were injected ip in spontaneous type 2 diabetes non‐human primates (rhesus macaques) resulting in reduction of plasma glucose and B lymphocytes, and absence of adverse effects105; (c) neonatal porcine SeC were inserted together with pancreatic islets subcutaneously in a porous chamber in the abdominal wall of young diabetic patients, in the absence of immunosuppressive treatment, and half patients significantly diminished their insulin doses with no complications reported in a 7‐year follow‐up.106, 107
Table 4.
Advantages and disadvantages of the MC‐SeC approach to DMD
| Approach | Advantages | Disadvantages | References |
|---|---|---|---|
| Intraperitoneal injection of MC‐SeC | Independent from DMD gene mutation | Need for xenogeneic source of SeC | 94, 95 |
| All muscles interested thanks to the systemic release of SeC‐derived factors | Caution for PERV presence in pig‐derived SeC, especially in immunosuppressed patients | [108, 109, 110, 111, 112, 113] | |
| Combinatorial approach (ie, anti‐inflammatory effect, induction of utrophin expression and release of trophic factors) | ‐ | 94, 95 | |
| No need for immunosuppression | ‐ | 15, 18, 19, 20, 21, 22, 94, 95, 106 | |
| Single ip injection not requiring incision of the abdominal wall | ‐ | 15, 18, 19, 20, 21, 22, 94, 95, 106 | |
| No undesired effects reported in several pre‐clinical settings (including non‐human primates) | ‐ | [15, 18, 19, 20, 21, 22, 94, 95, 102] | |
| SeC (non‐encapsulated) already used in clinical trials; no undesired effects reported | ‐ | [106, 107] | |
| Alginate‐based microcapsules (containing cells other than SeC) already used in clinical trials; no undesired effects reported | [99, 100, 101] |
MC‐SeC, microencapsulated Sertoli cells; PERVs, porcine endogenous retroviruses; SeC, Sertoli cells.
The use of pig cells, tissues and organs meets the general need to satisfy the increasing request for transplantation by humans who do not find sufficient availability among members of their species. Pigs represent suitable animals for human xenotransplantation because they share a similar organ physiology and size, for the relatively low costs of breeding and for the possibility to be genetically modified. One concern in xenotransplantation using pigs as a donor species is represented by porcine endogenous retroviruses (PERVs). This is because PERVs are present in almost all strains of pigs and cannot be removed even if pigs are being raised in sterile conditions. PERVs are inactive and harmless in pigs; however, transplantation into humans could activate the viruses and lead to human diseases with the risk of spreading to the entire community.108 However, although PERV can infect human cells in vitro, (a) transmission of PERV was not observed in animals (including non‐human primates) inoculated with PERV preparations or in pre‐clinical xenotransplantations (reviewed in 109); (b) patients xenotransplanted with porcine islets and SeC showed no PERV infection in their white blood cell DNA in long‐term clinical follow‐up110; and (c) studies of around 200 people worldwide who had been transplanted with pig tissue or had their blood pass through pig cells have shown no evidence of infection of porcine origin, and neither antibodies against PERV nor provirus integration in patients’ blood cells was observed.111 The reason why PERVs are not transmitted is that they probably are not released from the transplants or they are neutralized by the host cellular defence and immune system.109
However, porcine pancreatic islets were demonstrated to produce PERV and infect NOD/SCID (non‐obese diabetic, severe combined immunodeficiency) mice after transplantation, suggesting that PERV infection is a risk to take into account when pig xenotransplantation involves immunocompromised subjects.112 In this regard, it is noteworthy that PERV‐inactivated pigs have been recently generated via somatic cell nuclear transfer using a cell line in which PERVs were inactivated by CRISPR/Cas9 technology.113
Among the factors secreted by SeC are mitogenic factors that could potentially sustain cell growth and induce the formation of tumour masses in the recipient. However, this event has not been reported in any study involving the use of SeC, including the above‐mentioned clinical study.110 Probably, this is because SeC secrete a cocktail of molecules whose global effect results from the combination of all factors rather than being the sum of each single factor activity.
Another issue about the use of SeC is related to their immunosuppressive effect, representing a potential problem in case of lifelong clinical applications. However, results from several pre‐clinical and clinical studies suggest that SeC have an immunomodulatory rather than immunosuppressive effect.2, 5 Interestingly, SeC responded to viruses and bacteria eliciting an inflammatory response via the recruitment of Toll‐like receptors expressed on SeC surface and subsequent release of proinflammatory cytokines and chemokines.114
Although data obtained in macaques 105 and data from the experimentation in106 support the safety of the use of SeC in humans, absence of SeC‐induced tumour formation and the immunomodulatory vs immunosuppressive role of SeC should be addressed definitively in animal models.
6. CONCLUSIONS
Duchenne muscular dystrophy is a lethal muscular dystrophy affecting 1 in 3600‐5000 male live births worldwide.115 The progressive muscle degeneration subsequent to lack of dystrophin creates a condition of chronic inflammation that culminates in the progressive substitution of myofibres with fibrous and adipose tissues, impaired locomotion and premature death. Several intrinsic properties of the DMD pathology have nullified the huge effort to find a cure so far. In addition, many suggested therapeutic treatments have immunosuppression as a necessary co‐treatment, which means adding problem to problem, especially in a lifelong perspective. Therefore, investigation is still particularly active on DMD, and combinatorial therapeutic approaches might be envisaged.
Intraperitoneal injection of MC‐SeC translates into the release into the bloodstream of a cocktail of factors of which at least two components (ie, anti‐inflammatory factors and heregulin β1) are independently active on dystrophic muscles,94 representing in this sense a combinatorial approach per se (Figure 2). Interestingly, both mechanisms involved in MC‐SeC treatment are known to be very efficacious in DMD patients: the extinction of the inflammatory response and the induction of expression of the dystrophin paralogue, utrophin, at the sarcolemma. Based on the biology of SeC, it cannot be excluded that other factors secreted by this cell type (eg, IGF‐1) may additionally concur to the amelioration of dystrophic muscle morphology (Figure 2), which deserves further investigation.
Figure 2.
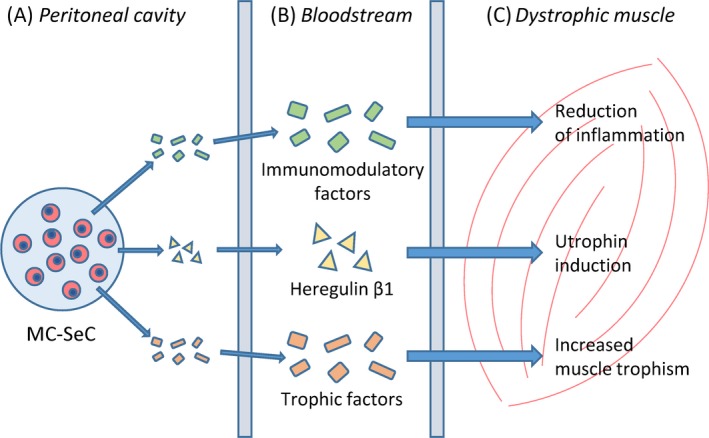
Schematics of the microencapsulated Sertoli cells (MC‐SeC) therapeutic approach. Once injected into the peritoneal cavity of dystrophic mice, MC‐SeC release a cocktail of factors including immunomodulatory factors, heregulin β1 and trophic factors (A). From the peritoneum, SeC‐released factors enter the systemic circulation (B) through which they can reach all skeletal muscle compartments (including cardiac muscle). At muscle tissue level, SeC‐released factors reduce the inflammatory response, induce utrophin expression and favour muscle trophism (C), thus recovering muscle morphology and functionality. It is noteworthy that thanks to the immunomodulatory properties of SeC, the procedure does not require pharmacological immunosuppression
Encapsulation represents an additional point of force of the proposed approach as encapsulation encloses the cells in a defined space, avoiding cell migration throughout the host body, and potentially allowing the recovery of injected cells if the injection is performed in a confined region of the body, such as the peritoneal cavity.
It is noteworthy that treatment with MC‐SeC has the advantage to be a universal approach to treat DMD since it is potentially applicable to the entire cohort of DMD patients regardless of the kind of mutation. Moreover, it is potentially applicable in general to myopathies characterized by chronic inflammation or immune dysregulation, such as autoimmune myositis.
Elucidation of several aspects in the near future, including the biological status of SeC inside the microcapsules over time, the dose‐response of MC‐SeC, potential direct effects of SeC on muscle precursor cells, the analysis of the mechanism(s) underpinning the anti‐inflammatory effect of SeC, and the demonstration of the safety of MC‐SeC in long‐term treatments will accelerate the application of this novel therapeutic approach to DMD patients.
CONFLICT OF INTEREST
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses or interpretation of data; in the writing of the manuscript; and in the decision to publish the results.
ACKNOWLEDGEMENTS
L.S. was recipient of a fellowship by Parent Project Onlus, Italy. G.L. and R.C. were supported by a grant from Mr. Gary Harlem (Altucell, NY, USA). R.D. was supported by the AFM (Project 15679). G.S. is supported by Parent Project Onlus, Italy.
Chiappalupi S, Salvadori L, Luca G, et al. Do porcine Sertoli cells represent an opportunity for Duchenne muscular dystrophy? Cell Prolif. 2019;52:e12599 10.1111/cpr.12599
Sara Chiappalupi and Laura Salvadori contributed equally to the present work.
REFERENCES
- 1. Russell LD, Griswold MD. The Sertoli Cell. Clearwater, FL: Cache River Press; 1993. [Google Scholar]
- 2. Mital P, Kaur G, Dufour JM. Immunoprotective Sertoli cells: making allogeneic and xenogeneic transplantation feasible. Reproduction. 2010;139:495‐504. [DOI] [PubMed] [Google Scholar]
- 3. Meinhardt A, Hedger MP. Immunological, paracrine and endocrine aspects of testicular immune privilege. Mol Cell Endocrinol. 2011;335:60‐68. [DOI] [PubMed] [Google Scholar]
- 4. Kaur G, Thompson LA, Dufour JM. Sertoli cells‐immunological sentinels of spermatogenesis. Semin Cell Dev Biol. 2014;30:36‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Luca G, Arato I, Sorci G, et al. Sertoli cells for cell transplantation: preclinical studies and future perspectives. Andrology. 2018;6:385‐395. [DOI] [PubMed] [Google Scholar]
- 6. Qvist G. John Hunter 1728–1793. London, UK: William Heinemann Medical Books Ltd; 1981. [Google Scholar]
- 7. Selawry HP, Cameron DF. Sertoli cell‐enriched fractions in successful islet cell transplantation. Cell Transplant. 1993;2:123‐129. [DOI] [PubMed] [Google Scholar]
- 8. Dufour JM, Rajotte RV, Kin T, Korbutt GS. Immunoprotection of rat islet xenografts by cotransplantation with Sertoli cells and a single injection of antilymphocyte serum. Transplantation. 2003;75:1594‐1596. [DOI] [PubMed] [Google Scholar]
- 9. Sanberg PR, Borlongan CV, Saporta S, Cameron DF. Testis‐derived Sertoli cells survive and provide localized immunoprotection for xenografts in rat brain. Nat Biotechnol. 1996;14:1692‐1695. [DOI] [PubMed] [Google Scholar]
- 10. Willing AE, Sudberry JJ, Othberg AI, et al. Sertoli cells decrease microglial response and increase engraftment of human hNT neurons in the hemiparkinsonian rat striatum. Brain Res Bull. 1999;48:441‐444. [DOI] [PubMed] [Google Scholar]
- 11. Shamekh R, El‐Badri NS, Saporta S, Pascual C, Sanberg PR, Cameron DF. Sertoli cells induce systemic donor‐specific tolerance in xenogenic transplantation model. Cell Transplant. 2006;15:45‐53. [DOI] [PubMed] [Google Scholar]
- 12. Lim HG, Lee HM, Oh BC, Lee JR. Cell‐mediated immunomodulation of chemokine receptor 7‐expressing porcine sertoli cells in murine heterotopic heart transplantation. J Heart Lung Transplant. 2008;28:72‐78. [DOI] [PubMed] [Google Scholar]
- 13. Rodriguez AI, Willing AE, Saporta S, et al. Effects of Sertoli cells transplants in a 3 nitropropionic acid model of early Huntington’s disease: a preliminary study. Neurotox Res. 2003;5:443‐450. [DOI] [PubMed] [Google Scholar]
- 14. Hemendinger R, Wang J, Malik S, et al. Sertoli cells improve survival of motor neurons in SOD1 transgenic mice, a model of amyotrophic lateral sclerosis. Exp Neurol. 2005;196:235‐243. [DOI] [PubMed] [Google Scholar]
- 15. Fallarino F, Luca G, Calvitti M, et al. Therapy of experimental type 1 diabetes by isolated Sertoli cell xenografts alone. J Exp Med. 2009;206:2511‐2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Luca G, Calafiore R, Basta G, et al. Improved function of rat islets upon co‐microencapsulation with Sertoli's cells in alginate/poly‐L‐ornithine. AAPS Pharm Sci Tech. 2001;2:E15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rahman TM, Diakanov I, Selden C, Hodgson H. Cotransplantation of encapsulated HepG2 and rat Sertoli cells improves outcome in a thioacetamide induced rat model of acute hepatic failure. Transpl Int. 2005;18:1001‐1009. [DOI] [PubMed] [Google Scholar]
- 18. Luca G, Fallarino F, Calvitti M, et al. Xenograft of microencapsulated sertoli cells reverses T1DM in NOD mice by inducing neogenesis of beta‐cells. Transplantation. 2010;90:1352‐1357. [DOI] [PubMed] [Google Scholar]
- 19. Bistoni G, Calvitti M, Mancuso F, et al. Prolongation of skin allograft survival in rats by the transplantation of microencapsulated xenogeneic neonatal porcine Sertoli cells. Biomaterials. 2012;33:5333‐5340. [DOI] [PubMed] [Google Scholar]
- 20. Luca G, Bellezza I, Arato I, et al. Terapeutic potential of microencapsulated Sertoli cells in Huntington disease. CNS Neurosci Ther. 2016;22:686‐690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luca G, Arato I, Mancuso F, et al. Xenograft of microencapsulated Sertoli cells restores glucose homeostasis in db/db mice with spontaneous diabetes mellitus. Xenotransplantation. 2016;23:429‐439. [DOI] [PubMed] [Google Scholar]
- 22. Luca G, Calvitti M, Mancuso F, et al. Reversal of experimental Laron syndrome by xenotransplantation of microencapsulated porcine Sertoli cells. J Control Release. 2013;165:75‐81. [DOI] [PubMed] [Google Scholar]
- 23. Selawry HP, Kotb M, Herrod HG, Lu ZN. Production of a factor, or factors, suppressing IL‐2 production and T cell proliferation by Sertoli cell‐enriched preparations. A potential role for islet transplantation in an immunologically privileged site. Transplantation. 1991;52:846‐850. [DOI] [PubMed] [Google Scholar]
- 24. De Cesaris PD, Filippini A, Cervelli C, et al. Immunosuppressive molecules produced by Sertoli cells cultured in vitro: biological effects on lymphocytes. Biochem Biophys Res Commun. 1992;186:1639‐1646. [DOI] [PubMed] [Google Scholar]
- 25. Takeda Y, Gotoh M, Dono K, et al. Protection of isllografts transplanted together with Fas ligand expressing testicular allografts. Diabetologia. 1998;41:315‐321. [DOI] [PubMed] [Google Scholar]
- 26. Dufour JM, Hamilton M, Rajotte RV, Korbutt GS. Neonatal porcine Sertoli cells inhibit human natural antibody‐mediated lysis. Biol Reprod. 2005;72:1224‐1231. [DOI] [PubMed] [Google Scholar]
- 27. Sipione S, Simmen KC, Lord SJ, et al. Identification of a novel human granzyme B inhibitor secreted by cultured sertoli cells. J Immunol. 2006;177:5051‐5058. [DOI] [PubMed] [Google Scholar]
- 28. Winnall WR, Muir JA, Hedger MP. Rat resident testicular macrophages have an alternatively activated phenotype and constitutively produce interleukin‐10 in vitro. J Leukoc Biol. 2011;90:133‐143. [DOI] [PubMed] [Google Scholar]
- 29. Campese AF, Grazioli P, de Cesaris P, et al. Mouse Sertoli cells sustain de novo generation of regulatory T cells by triggering the notch pathway through soluble JAGGED1. Biol Reprod. 2014;90:53. [DOI] [PubMed] [Google Scholar]
- 30. Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin related proteins in muscle. Physiol Rev. 2002;82:291‐329. [DOI] [PubMed] [Google Scholar]
- 31. Dalkilic I, Kunkel LM. Muscular dystrophies: genes to pathogenesis. Curr Opin Genet Dev. 2003;13:231‐238. [DOI] [PubMed] [Google Scholar]
- 32. Pichavant C, Aartsma‐Rus A, Clemens PR, et al. Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol Ther. 2011;19:830‐840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bladen CL, Salgado D, Monges S, et al. The TREAT‐NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36:395‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol. 2006;10:762‐773. [DOI] [PubMed] [Google Scholar]
- 35. Evans NP, Misyak SA, Robertson JL, Bassaganya‐Riera J, Grange RW. Immune mediated mechanisms potentially regulate the disease time course of Duchenne muscular dystrophy and provide targets for therapeutic intervention. Am J Phys Med Rehabil. 2009;1:755‐768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harper SQ, Hauser MA, DelloRusso C, et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002;8:253‐261. [DOI] [PubMed] [Google Scholar]
- 37. Ramos J, Chamberlain JS. Gene therapy for Duchenne muscular dystrophy. Expert Opin Orphan Drugs. 2015;3:1255‐1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Naso MF, Tomkowicz B, Perry WL 3rd, Strohl WR. Adeno‐associated virus (AAV) as a vector for gene therapy. BioDrugs. 2017;31:317‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shimizu‐Motohashi Y, Komaki H, Motohashi N, et al. Restoring dystrophin expression in Duchenne muscular dystrophy: current status of therapeutic approaches. J Pers Med. 2019;9(1):pii:E1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chamberlain JR, Chamberlain JS. Progress toward gene therapy for Duchenne muscular dystrophy. Mol Ther. 2017;25:1125‐1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang B, Li J, Xiao X. Adeno‐associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci U S A. 2000;97:13714‐13719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gregorevic P, Allen JM, Minami E, et al. rAAV6‐microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med. 2006;12:787‐789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yue Y, Pan X, Hakim CH, et al. Safe and body wide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno‐associated virus. Hum Mol Genet. 2015;24:5880‐5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bowles DE, McPhee S, Li C, et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol Ther. 2012;20:443‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Falzarano MS, Scotton C, Passarelli C, Ferlini A. Duchenne muscular dystrophy: from diagnosis to therapy. Molecules. 2015;20:18168‐18184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aartsma‐Rus A, Fokkema I, Verschuuren J, et al. Theoretic applicability of antisense‐mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. 2009;30:293‐299. [DOI] [PubMed] [Google Scholar]
- 47. Niks EH, Aartsma‐Rus A. Exon skipping: a first in class strategy for Duchenne muscular dystrophy. Expert Opin Biol Ther. 2017;17:225‐236. [DOI] [PubMed] [Google Scholar]
- 48. Aartsma‐Rus A, Krieg AM. FDA approves Eteplirsen for Duchenne muscular dystrophy: the next chapter in the Eteplirsen saga. Nucleic Acid Ther. 2017;27:1‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shimizu‐Motohashi Y, Murakami T, Kimura En, Komaki H, Watanabe N. Exon skipping for Duchenne muscular dystrophy: a systematic review and meta‐analysis. Orphanet J Rare Dis. 2018;13:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lu QL, Cirak S, Partridge T. What can we learn from clinical trials of exon skipping for DMD? Mol Ther Nucleic Acids. 2014;3:e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Partridge TA, Morgan JE, Coulton GR, Hoffman EP, Kunkel LM. Conversion of mdx myofibres from dystrophin‐negative to positive by injection of normal myoblasts. Nature. 1989;337:176‐179. [DOI] [PubMed] [Google Scholar]
- 52. Sampaolesi M, Blot S, D’Antona G, et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature. 2006;444:574‐579. [DOI] [PubMed] [Google Scholar]
- 53. Darabi R, Arpke R, Irion S, et al. Human ES‐ and iPS‐derived myogenic progenitors restore dystrophin and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell. 2012;10:610‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bonfanti C, Rossi G, Tedesco FS, et al. PW1/Peg3 expression regulates key properties that determine mesoangioblast stem cell competence. Nat Commun. 2015;6:6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Quenneville SP, Tremblay JP. Ex vivo modification of cells to induce a muscle‐based expression. Curr Gene Ther. 2006;6:625‐632. [DOI] [PubMed] [Google Scholar]
- 56. Skuk D, Goulet M, Tremblay JP. Transplanted myoblasts can migrate several millimeters to fuse with damaged myofibers in nonhuman primate skeletal muscle. J Neuropathol Exp Neurol. 2011;70:770‐778. [DOI] [PubMed] [Google Scholar]
- 57. Price FD, Kuroda K, Rudnicki MA. Stem cell based therapies to treat muscular dystrophy. Biochim Biophys Acta. 2007;1772:272‐283. [DOI] [PubMed] [Google Scholar]
- 58. Skuk D, Goulet M, Roy B, et al. Dystrophin expression in muscles of duchenne muscular dystrophy patients after high‐density injections of normal myogenic cells. J Neuropathol Exp Neurol. 2006;65:371‐386. [DOI] [PubMed] [Google Scholar]
- 59. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663‐676. [DOI] [PubMed] [Google Scholar]
- 60. Danisovic L, Culenova M, Csobonyeiova M. Induced pluripotent stem cells for Duchenne muscular dystrophy modeling and therapy. Cells. 2018;7:pii:E253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cossu G, Previtali Sc, Napolitano S, et al. Intra‐arterial transplantation of HLA‐matched donor mesoangioblasts in Duchenne muscular dystrophy. EMBO Mol Med. 2015;7:1513‐1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Konieczny P, Swiderski K, Chamberlain JS. Gene and cell‐mediated therapies for muscular dystrophy. Muscle Nerve. 2013;47:649‐663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve. 2000;23:1456‐1471. [DOI] [PubMed] [Google Scholar]
- 64. Khurana TS, Watkins SC, Chafey P, et al. Immunolocalization and developmental expression of dystrophin related protein in skeletal muscle. Neuromuscul Disord. 1991;1:185‐194. [DOI] [PubMed] [Google Scholar]
- 65. Nguyen TM, Ellis JM, Love DR, et al. Localization of the DMDL gene‐encoded dystrophin‐related protein using a panel of nineteen monoclonal antibodies: presence at neuromuscular junctions, in the sarcolemma of dystrophic skeletal muscle, in vascular and other smooth muscles, and in proliferating brain cell lines. J Cell Biol. 1991;115:1695‐1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Clerk A, Morris GE, Dubowitz V, Davies KE, Sewry CA. Dystrophin‐related protein, utrophin, in normal and dystrophic human fetal skeletal muscle. Histochem J. 1993;25:554‐561. [PubMed] [Google Scholar]
- 67. Rafael JA, Tinsley JM, Potter AC, Deconinck AE, Davies KE. Skeletal muscle‐specific expression of a utrophin transgene rescues utrophin‐dystrophin deficient mice. Nat Genet. 1998;19:79‐82. [DOI] [PubMed] [Google Scholar]
- 68. Tinsley J, Deconinck N, Fisher R, et al. Expression of full‐length utrophin prevents muscular dystrophy in mdx mice. Nat Med. 1998;4:1441‐1444. [DOI] [PubMed] [Google Scholar]
- 69. Guiraud S, Roblin D, Kay DE. The potential of utrophin modulators for the treatment of Duchenne muscular dystrophy. Expert Opin Orphan Drugs. 2018;6:179‐192. [Google Scholar]
- 70. Ricotti V, Spinty S, Roper H, et al. Safety, tolerability, and pharmacokinetics of SMT C1100, a 2‐arylbenzoxazole utrophin modulator, following single‐ and multiple‐dose administration to pediatric patients with Duchenne muscular dystrophy. PLoS ONE. 2016;11:e0152840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Colussi C, Mozzetta C, Gurtner A, et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc Natl Acad Sci U S A. 2009;105:19183‐19187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Consalvi S, Mozzetta C, Bettica P, et al. Preclinical studies in the mdx mouse model of duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol Med. 2013;19:79‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bettica P, Petrini S, D'Oria V, et al. Histological effects of givinostat in boys with Duchenne muscular dystrophy. Neuromuscul Disord. 2016;26:643‐649. [DOI] [PubMed] [Google Scholar]
- 74. Percival JM, Whitehead NP, Adams ME, Adamo CM, Beavo JA, Froehner SC. Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J Pathol. 2012;228:77‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nelson Md, Rader F, Tang X, et al. PDE5 inhibition alleviates functional muscle ischemia in boys with Duchenne muscular dystrophy. Neurology. 2014;82:2085‐2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Victor RG, Sweeney HL, Finkel R, et al. A phase 3 randomized placebo‐controlled trial of tadalafil for Duchenne muscular dystrophy. Neurology. 2017;89:1811‐1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Buyse GM, Goemans N, van den Hauwe M, Meier T. Effects of glucocorticoids and idebenone on respiratory function in patients with duchenne muscular dystrophy. Pediatr Pulmonol. 2013;48:912‐920. [DOI] [PubMed] [Google Scholar]
- 78. Buyse GM, Voit T, Schara U, et al. Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): a double‐blind randomised placebo‐controlled phase 3 trial. Lancet. 2015;385:1748‐1757. [DOI] [PubMed] [Google Scholar]
- 79. McDonald CM, Meier T, Voit T, et al. Idebenone reduces respiratory complications in patients with Duchenne muscular dystrophy. Neuromuscul Disord. 2016;26:473‐480. [DOI] [PubMed] [Google Scholar]
- 80. Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014;50:477‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. McDonald CM, Campbell C, Torricelli RE, et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACTDMD): a multicentre, randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2017;390:1489‐1498. [DOI] [PubMed] [Google Scholar]
- 82. Bodanovsky A, Guttman N, Barzilai‐Tutsch H, et al. Halofuginone improves muscle‐cell survival in muscular dystrophies. Biochim Biophys Acta. 2014;1843:1339‐1347. [DOI] [PubMed] [Google Scholar]
- 83. Singh P, Rong H, Gordi T, Bosley J, Bhattacharya I. Translational pharmacokinetic/pharmacodynamic analysis of MYO‐029 antibody for muscular dystrophy. Clin Transl Sci. 2016;9:302‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bhattacharya I, Manukyan Z, Chan P, Heatherington A, Harnisch L. Application of quantitative pharmacology approaches in bridging pharmacokinetics and pharmacodynamics of Domagrozumab from adult healthy subjects to pediatric patients with Duchenne muscular disease. J Clin Pharmacol. 2017;58:314‐326. [DOI] [PubMed] [Google Scholar]
- 85. Nelson CE, Hakim CH, Ousterout DG, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tabebordbar M, Zhu K, Cheng J, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR‐Cas9 system. Nat Protoc. 2013;8:2281‐2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gee P, Xu H, Hotta A. Cellular reprogramming, genome editing, and alternative CRISPR Cas9 technologies for precise gene therapy of Duchenne muscular dystrophy. Stem Cells Int. 2017;2017:8765154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lim K, Yoon C, Yokota T. Applications of CRISPR/Cas9 for the treatment of Duchenne muscular dystrophy. J Pers Med. 2018;8(4):pii:E38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Muntoni F, Fisher I, Morgan JE, Abraham D. Steroids in Duchenne muscular dystrophy: from clinical trials to genomic research. Neuromuscul Disord. 2002;12:S162‐S165. [DOI] [PubMed] [Google Scholar]
- 91. Fenichel GM, Florence JM, Pestronk A, et al. Long‐term benefit from prednisone therapy in Duchenne muscular dystrophy. Neurol. 1991;41:1874‐1877. [DOI] [PubMed] [Google Scholar]
- 92. Angelini C, Peterle E. Old and new therapeutic developments in steroid treatment in Duchenne muscular dystrophy. Acta Myol. 2012;31:9‐15. [PMC free article] [PubMed] [Google Scholar]
- 93. Heier CR, Damsker JM, Yu Q, et al. VBP15, a novel anti‐inflammatory and membrane‐stabilizer, improves muscular dystrophy without side effects. EMBO Mol Med. 2013;5:1569‐1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chiappalupi S, Luca G, Mancuso F, et al. Intraperitoneal injection of microencapsulated Sertoli cells restores muscle morphology and performance in dystrophic mice. Biomaterials. 2016;75:313‐326. [DOI] [PubMed] [Google Scholar]
- 95. Chiappalupi S, Luca G, Mancuso F, et al. Effects of intraperitoneal injection of microencapsulated Sertoli cells on chronic and presymptomatic dystrophic mice. Data Brief. 2015;15:1015‐1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Gramolini Ao, Angus Lm, Schaeffer L, et al. Induction of utrophin gene expression by heregulin in skeletal muscle cells: role of the N‐box motif and GA binding protein. Proc Natl Acad Sci U S A. 1999;96:3223‐3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Basu U, Gyrd‐Hansen M, Baby SM, et al. Heregulin‐induced epigenetic regulation of the utrophin‐A promoter. FEBS Lett. 2007;581:4153‐4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Krag T, Bogdanovich S, Jensen CJ, et al. Heregulin ameliorates the dystrophic phenotype in mdx mice. Proc Natl Acad Sci U S A. 2004;101:13856‐13860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Calafiore R, Basta G, Luca G, et al. Microencapsulated pancreatic isllografts into nonimmunosuppressed patients with type 1 diabetes: first two cases. Diabetes Care. 2006;29:137‐138. [DOI] [PubMed] [Google Scholar]
- 100. Calafiore R, Basta G, Luca G, et al. Standard technical procedures for microencapsulation of human islets for graft into nonimmunosuppressed patients with type 1 diabetes mellitus. Transplant Proc. 2006;38:1156‐1157. [DOI] [PubMed] [Google Scholar]
- 101. Soon‐Shiong P, Heintz RE, Merideth N, et al. Insulin independence in a type 1 diabetic patient after encapsulated islet transplantation. Lancet. 1994;343:950‐951. [DOI] [PubMed] [Google Scholar]
- 102. Luca G, Cameron DF, Arato I, et al. Xenograft of microencapsulated Sertoli cells for the cell therapy of type 2 diabetes mellitus in spontaneously diabetic non human primates: preliminary data. Transplant Proc. 2014;46:1999‐2000. [DOI] [PubMed] [Google Scholar]
- 103. Kobayashi T, Aomatsu Y, Iwata H, et al. Survival of microencapsulated islets at 400 days posttransplantation in the omental pouch of NOD mice. Cell Transplant. 2006;15:359‐365. [DOI] [PubMed] [Google Scholar]
- 104. Luca G, Mancuso F, Calvitti M. Long-term stability, functional competence, and safety of microencapsulated specific pathogen-free neonatal porcine Sertoli cells: a potential product for cell transplant therapy. Xenotransplantation. 2015;22:273–283. [DOI] [PubMed] [Google Scholar]
- 105. Basta G, Montanucci P, Luca G, et al. Long‐term metabolic and immunological follow‐up of nonimmunosuppressed patients with type 1 diabetes treated with microencapsulated isllografts: four cases. Diabetes Care. 2011;34:2406‐2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Valdés‐González RA, White D, Dorantes LM, et al. Three‐yr follow‐up of a type 1 diabetes mellitus patient with an islet xenotransplant. Clin Transplant. 2007;21:352‐357. [DOI] [PubMed] [Google Scholar]
- 107. Esquivel‐Pérez R, Rodriguez‐Ventura AL, Dorantes LM, Ramírez‐González B, López‐Santos MG, Valdes‐Gonzalez R. Correlation between insulin requirements and anti‐galactose antibodies in patients with type 1 diabetes transplanted with neonatal pig islets. Clin Exp Immunol. 2011;165:104‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wilson CA. Porcine endogenous retroviruses and xenotransplantation. Cell Mol Life Sci. 2008;65:3399‐3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Valdes‐Gonzalez R, Dorantes LM, Bracho‐Blanchet E, RodrÃguez‐Ventura A, Djg W. No evidence of porcine endogenous retrovirus in patients with type 1 diabetes after long‐term porcine islet xenotransplantation. J Med Virol. 2010;82:331‐334. [DOI] [PubMed] [Google Scholar]
- 110. Denner J, Tönjes RR. Infection barriers to successful xenotransplantation focusing on porcine endogenous retroviruses. Clin Microbiol Rev. 2012;25:318‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Denner J. Why was PERV not transmitted during preclinical and clinical xenotransplantation trials and after inoculation of animals? Retrovirology. 2018;15:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. van der Laan L, Lockey C, Griffeth BC, et al. Infection by porcine endogenous retrovirus after islet xenotransplantation in SCID mice. Nature. 2000;407:90‐94. [DOI] [PubMed] [Google Scholar]
- 113. Niu D, Wei H‐J, Lin L, et al. Inactivation of porcine endogenous retrovirus in pigs using CRISPR‐Cas9. Science. 2017;357:1303‐1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Riccioli A, Starace D, Galli R, et al. Sertoli cells initiate testicular innate immune responses through TLR activation. J Immunol. 2006;177:7122‐7130. [DOI] [PubMed] [Google Scholar]
- 115. Emery A. Duchenne Muscular Dystrophy, 2nd edn Oxford, UK: Oxford University Press; 1993. [Google Scholar]
- 116. Meehan T, Schlatt S, O'Bryan MK, de Kretser DM, Loveland KL. Regulation of germ cell and Sertoli cell development by activin, follistatin, and FSH. Dev Biol. 2000;220:225‐237. [DOI] [PubMed] [Google Scholar]
- 117. Bitgood MJ, Shen L, McMahon AP. Sertoli cell signaling by Desert hedgehog regulates the male germline. Curr Biol. 1996;6:298‐304. [DOI] [PubMed] [Google Scholar]
- 118. Bouraïma‐Lelong H, Vanneste M, Delalande C, Zanatta L, Wolczynski S, Carreau S. Aromatase gene expression in immature rat Sertoli cells: age‐related changes in the FSH signalling pathway. Reprod Fertil Dev. 2010;22:508‐515. [DOI] [PubMed] [Google Scholar]
- 119. Vincent S, Segretain D, Nishikawa S, et al. Stage‐specific expression of the Kit receptor and its ligand (KL) during male gametogenesis in the mouse: a Kit‐KL interaction critical for meiosis. Development. 1998;125:4585‐4593. [DOI] [PubMed] [Google Scholar]
- 120. Hakovirta H, Yan W, Kaleva M, et al. Function of stem cell factor as a survival factor of spermatogonia and localization of messenger ribonucleic acid in the rat seminiferous epithelium. Endocrinology. 1999;140:1492‐1498. [DOI] [PubMed] [Google Scholar]
- 121. Munsterberg A, Lovell‐Badge R. Expression of the mouse anti‐Müllerian hormone gene suggests a role in both male and female sexual differentiation. Development. 1991;113:613‐624. [DOI] [PubMed] [Google Scholar]
- 122. Park C, Choi WS, Kwon H, Kwon YK. Temporal and spatial expression of neurotrophins and their receptors during male germ cell development. Mol Cells. 2001;12:360‐367. [PubMed] [Google Scholar]
- 123. Mullaney BP, Skinner MK. Basic fibroblast growth factor (bFGF) gene expression and protein production during pubertal development of the seminiferous tubule: follicle‐stimulating hormone‐induced Sertoli cell bFGF expression. Endocrinology. 1992;131:2928‐2934. [DOI] [PubMed] [Google Scholar]
- 124. Pellegrini M, Grimaldi P, Rossi P, et al. Developmental expression of BMP4/ALK3/SMAD5 signaling pathway in the mouse testis: a potential role of BMP4 in spermatogonia differentiation. J Cell Sci. 2003;116:3363‐3372. [DOI] [PubMed] [Google Scholar]
- 125. Sun Y‐P, Yan Y‐C, Zhang S‐Q, Zhao F. Immunohistochemical localization of epidermal growth factor in rat and mouse testis. Chin J Histochem Cytochem. 1997;6:19‐22. [Google Scholar]
- 126. Viglietto G, Dolci S, Bruni P, et al. Glial cell line‐derived neutrotrophic factor and neurturin can act as paracrine growth factors stimulating DNA synthesis of Ret‐expressing spermatogonia. Int J Oncol. 2000;16:689‐694. [DOI] [PubMed] [Google Scholar]
- 127. Hedger MP, Meinhardt A. Cytokines and the immune‐testicular axis. J Reprod Immunol. 2003;58:1‐26. [DOI] [PubMed] [Google Scholar]
- 128. Chatelain PG, Naville D, Saez JM. Somatomedin‐C/insulin‐like growth factor 1‐like material secreted by porcine Sertoli cells in vitro: characterization and regulation. Biochem Biophys Res Commun. 1987;146:1009‐1017. [DOI] [PubMed] [Google Scholar]
- 129. Naville D, Chatelain PG, Avallet O, Saez JM. Control of production of insulin‐like growth factor I by pig Leydig and Sertoli cells cultured alone or together. Cell‐cell interactions. Mol Cell Endocrinol. 1990;70:217‐224. [DOI] [PubMed] [Google Scholar]
- 130. Tsuruta JK, O'Brien DA. Sertoli cell‐spermatogenic cell interaction: the insulin‐like growth factor‐II/cation‐independent mannose 6‐phosphate receptor mediates changes in spermatogenic cell gene expression in mice. Biol Reprod. 1995;53:1454‐1464. [DOI] [PubMed] [Google Scholar]
- 131. Jonsson CK, Zetterström RH, Holst M, et al. Constitutive expression of interleukin‐1alpha messenger ribonucleic acid in rat Sertoli cells is dependent upon interaction with germ cells. Endocrinology. 1999;140:3755‐3761. [DOI] [PubMed] [Google Scholar]
- 132. Söder O, Sultana T, Jonsson C, et al. The interleukin‐1 system in the testis. Andrologia. 2000;32:52‐55. [PubMed] [Google Scholar]
- 133. Buch JP, Lamb DJ, Lipshultz LI, Smith RG. Partial characterization of a unique growth factor secreted by human Sertoli cells. Fertil Steril. 1988;49:658‐665. [PubMed] [Google Scholar]
- 134. Cupp AS, Kim GH, Skinner MK. Expression and action of neurotropin‐3 and nerve growth factor in embryonic and early postnatal rat testis development. Biol Reprod. 2000;63:1617‐1628. [DOI] [PubMed] [Google Scholar]
- 135. Loveland KL, Zlatic K, Stein‐Oakley A, Risbridger G, deKretser DM. Platelet‐derived growth factor ligand and receptor subunit mRNA in the Sertoli and Leydig cells of the rat testis. Mol Cell Endocrinol. 1995;108:155‐159. [DOI] [PubMed] [Google Scholar]
- 136. Morales CR, el‐Alfy M, Zhao Q, Igdoura S. Molecular role of sulfated glycoprotein‐1 (SGP‐1/prosaposin) in Sertoli cells. Histol Histopathol. 1995;10:1023‐1034. [PubMed] [Google Scholar]
- 137. Stallard BJ, Griswold MD. Germ cell regulation of Sertoli cell transferrin mRNA levels. Mol Endocrinol. 1990;4:393‐401. [DOI] [PubMed] [Google Scholar]
- 138. Petersen C, Boitani C, Fröysa B, Söder O. Transforming growth factor‐alpha stimulates proliferation of rat Sertoli cells. Mol Cell Endocrinol. 2001;181:221‐227. [DOI] [PubMed] [Google Scholar]
- 139. Avallet O, Vigier M, Leduque P, Dubois PM, Saez JM. Expression and regulation of transforming growth factor‐beta 1 messenger ribonucleic acid and protein in cultured porcine Leydig and Sertoli cells. Endocrinology. 1994;134:2079‐2087. [DOI] [PubMed] [Google Scholar]
- 140. Ergün S, Kiliç N, Fiedler W, Mukhopadhyay AK. Vascular endothelial growth factor and its receptors in normal human testicular tissue. Mol Cell Endocrinol. 1997;131:9‐20. [DOI] [PubMed] [Google Scholar]
- 141. Yan W, Samson M, Jégou B, Toppari J. Bcl‐w forms complexes with Bax and Bak, and elevated ratios of Bax/Bcl‐w and Bak/Bcl‐w correspond to spermatogonial and spermatocyte apoptosis in the testis. Mol Endocrinol. 2000;14:682‐699. [DOI] [PubMed] [Google Scholar]
- 142. Grima J, Pineau C, Bardin CW, Cheng CY. Rat Sertoli cell clusterin, alpha 2‐macroglobulin, and testins: biosynthesis and differential regulation by germ cells. Mol Cell Endocrinol. 1992;89:127‐140. [DOI] [PubMed] [Google Scholar]
- 143. Lee J, Richburg JH, Shipp EB, et al. The Fas system, a regulator of testicular germ cell apoptosis, is differentially up‐regulated in Sertoli cell versus germ cell injury of the testis. Endocrinology. 1999;140:852‐858. [DOI] [PubMed] [Google Scholar]
- 144. Xu JP, Li X, Mori E, et al. Expression of Fas‐Fas ligand in murine testis. Am J Reprod Immunol. 1999;42:381‐388. [DOI] [PubMed] [Google Scholar]
- 145. Huleihel M, Abofoul‐Azab M, Abarbanel Y, Einav I, Levitas E, Lunenfeld E. Production of macrophage inhibitory factor (MIF) by primary sertoli cells; its possible involvement in migration of spermatogonial cells. J Cell Physiol. 2017;232:2869‐2877. [DOI] [PubMed] [Google Scholar]
- 146. Huleihel M, Zeyse D, Lunenfeld E, Zeyse M, Mazor M. Induction of transferrin secretion in murine Sertoli cells by FSH and IL‐1: the possibility of different mechanism(s) of regulation. Am J Reprod Immunol. 2002;47:112‐117. [DOI] [PubMed] [Google Scholar]
- 147. Suire S, Fontaine I, Guillou F. Transferrin gene expression and secretion in rat Sertoli cells. Mol Reprod Dev. 1997;48:168‐175. [DOI] [PubMed] [Google Scholar]