

Abstract
Drug-target binding kinetics are suggested to be important parameters for the prediction of in vivo drug-efficacy. For G protein-coupled receptors (GPCRs), the binding kinetics of ligands are typically determined using association binding experiments in competition with radiolabelled probes, followed by analysis with the widely used competitive binding kinetics theory developed by Motulsky and Mahan. Despite this, the influence of the radioligand binding kinetics on the kinetic parameters derived for the ligands tested is often overlooked. To address this, binding rate constants for a series of histamine H1 receptor (H1R) antagonists were determined using radioligands with either slow (low koff) or fast (high koff) dissociation characteristics. A correlation was observed between the probe-specific datasets for the kinetic binding affinities, association rate constants and dissociation rate constants. However, the magnitude and accuracy of the binding rate constant-values was highly dependent on the used radioligand probe. Further analysis using recently developed fluorescent binding methods corroborates the finding that the Motulsky-Mahan methodology is limited by the employed assay conditions. The presented data suggest that kinetic parameters of GPCR ligands depend largely on the characteristics of the probe used and results should therefore be viewed within the experimental context and limitations of the applied methodology.
Subject terms: Biochemical assays, Receptor pharmacology, G protein-coupled receptors
Introduction
The pharmacodynamics of a drug are often related to the half-maximal modulation of target function (IC50, EC50), which typically depends on the concentration required to obtain half-maximal target binding (Ki, Kd). However, it is increasingly debated whether these pharmacological parameters provides sufficient information to predict the in vivo effectiveness of a ligand1–4. Drug-target binding kinetics have therefore received increased interest in the last decade, and the drug-target residence time has been linked to the in vivo efficacy of a number of important target classes, including the large family of membrane-bound G protein-coupled receptors (GPCRs)3,5–9. Radioligand binding is routinely used to determine ligand binding affinity and kinetics to GPCR targets10–18. To determine the binding kinetics of unlabeled ligands, the competitive effect on the association binding of a GPCR radioligand is analyzed using the theoretical model derived by Motulsky and Mahan19. Despite the wide use of this methodology in the GPCR-field, it is not known to which extent the calculated binding rate constants of unlabeled ligands depend on the binding kinetics of the radiolabeled probe used.
The histamine H1 receptor (H1R) is a prototypical Family A GPCR which is therapeutically targeted by several 2nd generation antagonists in the treatment of allergic conditions such as allergic rhinitis and urticaria20. The therapeutic success of the 2nd generation H1R antagonists is generally attributed to their reduced brain penetration compared to 1st generation H1R antagonists, which results in a decrease of on-target side effects such as sedation. Interestingly, the binding kinetics of several H1R antagonists have been investigated using the Motulsky-Mahan methodology13,21–24 and were found to have a long residence time at the H1R25. In one study the prolonged residence time of levocetirizine was linked to the presence of a carboxylic acid group, which is a frequently occurring chemical moiety for 2nd generation antihistamines13.
The success of the H1R as a drug target has resulted in a rich repertoire of antagonists that can bind the receptor, including different radiolabeled versions of commonly studied compounds20,21,25–27. Several radioligands ([3H]mepyramine, [3H]levocetirizine and [3H]olopatadine) have previously been characterized for their kinetic binding profile at the H1R. Interestingly, [3H]mepyramine and [3H]levocetirizine show similar binding affinities at the H1R, but markedly different binding kinetics21. Recently, methodologies which utilize fluorescent ligands in place of radioligands have been introduced to characterize the binding kinetics of GPCR ligands and these newer methods have advantages over radioligand binding in terms of throughput and kinetic resolution28. Both bioluminescence (BRET29) and time-resolved (HTRF30) resonance energy transfer techniques have been applied to study binding kinetics at the H1R.
Due to the wide range of radioactive and fluorescently labelled ligands available for H1R, we used this GPCR as a model system to investigate if the measured binding rate constants of unlabeled ligands are influenced by the binding kinetics of the employed labelled probe. To this end, [3H]mepyramine and [3H]levocetirizine were used to characterize the binding kinetics of a set of unlabeled H1R ligands by the Motulsky-Mahan methodology. This was followed by the determination of the binding kinetics of H1R ligands via competitive association binding using two different non-radioactive H1R binding assays (BRET-based29 or HTRF based30 approaches). The kon and Ki values, obtained from kinetic and steady-state experiments, respectively, were correlated between the various datasets employing either fluorescent ligands or radioligands as probes. However, it was found that koff-values are in part dependent on the used assay methodology. Therefore, both probe-dependent and assay-dependent factors are important contributors to the accurate determination of binding kinetics of unlabeled ligands.
Methods
Materials
The radioligand [3H]mepyramine (20 Ci/mmol) was purchased from Perkin Elmer (Waltham, MA, USA). The mepyramine based fluorescent HTRF ligand (Gmep) was purchased from Cisbio (Codolet, France). Other employed, commercially available ligands were: triprolidine hydrochloride monohydrate (Tocris Bioscience, Bristol, United Kingdom), doxepin hydrochloride (Tocris Bioscience, E/Z mixture with a ~85:15 ratio), Olopatadine hydrochloride (BOC Sciences, Shirley, NY, USA), acrivastine (BOC Sciences), levocetirizine dihydrochloride (Biotrend, Cologne, Germany), S-cetirizine dihydrochloride (TLC PharmaChem, Mississauga, Canada), Mepyramine maleate (Research Biochemicals International, Natick, MA, USA), R-fexofenadine (Sepracor Inc., Marlborough, MA, USA), S-fexofenadine (Sepracor Inc.), desloratadine (HaiHang Industry, Jinan City, China), Terfenadine (MP biomedicals, Santa Ana, CA, USA). VUF14454, VUF14544, VUF14506, VUF14493 and mianserin were synthesized at the Vrije Universiteit Amsterdam and were fully characterized with respect to purity and identity22,23. [3H]levocetirizine (25.9 Ci/mmol) was synthesized at AstraZeneca and was fully characterized with respect to purity and identity. AV082 (mepyramine-ala-ala-BY630) was synthesized at the University of Nottingham as described previously29. All other chemicals and reagents were obtained from Sigma Aldrich and Fisher, unless specified otherwise in the text.
Synthesis of [3H]olopatadine
General
The synthesis of [3H]olopatadine is schematically depicted in Fig. 1. Column chromatography was carried out using pre-packed silica gel cartridges (SiliCycle, Quebec, Canada) on an Isco Companion (Teledyne Isco, NE, USA). 1H NMR spectra were recorded on a Bruker (600 MHz or 400 MHz) using the stated solvent. Chemical shifts (δ) in ppm are quoted relative to CDCl3 (δ 7.26 ppm) and DMSO-d6 (δ 2.50 ppm). Liquid chromatography-mass spectrometry (LC-MS) data was collected using a Waters Alliance LC (Waters Corporation, MA, USA) with Waters ZQ mass detector. Analytical HPLC data was recorded using Agilent 1200 HPLC system with a β-Ram Flow Scintillation Analyser, using the following conditions: Waters Sunfire C18, 3.5 µm, 4.6 × 100 mm column at 40 °C, eluting with 5% acetonitrile/water +0.1% TFA to 95% acetonitrile/water +0.1% TFA over a 32 minute gradient. Specific activities were determined gravimetrically with a Packard TriCarb 2100CA Liquid Scintillation Analyser (Packard Instrument Company Inc., IL, USA) using Ultima GoldTM cocktail. Reactions with tritium gas were carried out on a steel manifold obtained from RC Tritec AG (Teufen, Switzerland). Specific activity was calculated by comparison of the ratio of tritium/hydrogen or carbon-14/carbon-12 for the tracer against the unlabelled reference. [3H]Methyl nosylate was obtained from Quotient Bioresearch as a solution in toluene at 3150 GBq mmol−1. Tritium gas was supplied absorbed onto a depleted uranium bed by RC Tritec AG (Teufen, Switzerland).
Figure 1.
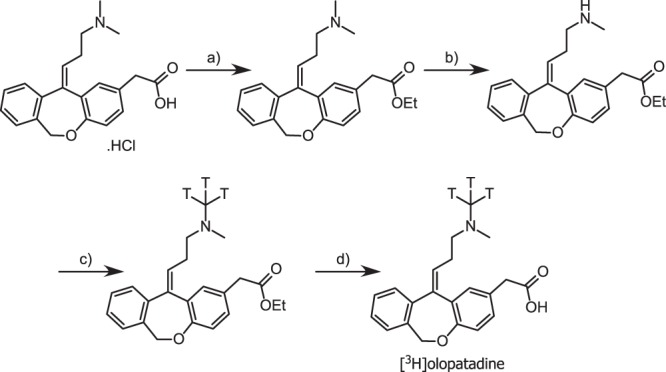
Synthesis of [3H]olopatadine. Key: (a) 4-toluenesulfonic acid, EtOH, reflux, 2 h, 89%; (b) (i), 1-chloroethyl chloroformate, DCE, reflux, 4 h; (ii) MeOH, reflux, 2 h, 10% over two steps; (c) [3H]methyl nosylate, DMF, 50 °C, 1 h; (d) NaOH, EtOH/H2O, r.t., 2 h.
Ethyl (Z)-2-(11-(3-(dimethylamino)propylidene)-6,11-dihydrodibenzo[b,e]oxepin-2-yl)acetate
(Z)-2-(11-(3-(dimethylamino)propylidene)-6,11-dihydrodibenzo[b,e]oxepin-2-yl)acetic acid hydrochloride (150 mg, 0.40 mmol), ethanol (3 mL, 0.40 mmol) and 4-toluenesulfonic acid (23 mg, 0.12 mmol) were stirred at reflux under Dean-Stark conditions for 2 h. Triethylamine (73 µL, 0.52 mmol) was added and the mixture evaporated under reduced pressure, the residue was partitioned between water (5 mL) and ethyl acetate (15 mL). The organic phase was washed with NaHCO3 (satd. aq, 5 mL), brine (5 mL), dried (MgSO4), filtered and evaporated to give ethyl (Z)-2-(11-(3-(dimethylamino)propylidene)-6,11-dihydrodibenzo[b,e]oxepin-2-yl)acetate (131 mg, 0.358 mmol, 89%) as a colourless oil. LCMS (ESI) m/z 366 [M + H]+.
Ethyl (Z)-2-(11-(3-(methylamino)propylidene)-6,11-dihydrodibenzo[b,e]oxepin-2-yl)acetate
1-Chloroethyl chloroformate (38.2 µl, 0.35 mmol) was added to a stirred solution of ethyl (Z)-2-(11-(3-(dimethylamino)propylidene)-6,11-dihydrodibenzo[b,e]oxepin-2-yl)acetate (128 mg, 0.35 mmol) in 1,2-dichloroethane (3500 µL) and the mixture heated to reflux. After 2 h additional 1-chloroethyl chloroformate (38.2 µL, 0.35 mmol) was added, and after a further 2 h the solvent was evaporated under reduced pressure. Methanol (3 mL) was added and the mixture heated to reflux for 2 h. The mixture was purified by preparative HPLC (Waters XBridge Prep C18 OBD column, 5 µ silica, 19 mm diameter, 100 mm length), using decreasingly polar mixtures of water (containing 0.1% TFA) and MeCN as eluents. Fractions containing the desired compound were combined, concentrated under vacuum, adjusted to pH 9 with NaHCO3, extracted with DCM (2 × 20 mL), then further purified by flash silica chromatography, elution gradient 0 to 6% NH3-MeOH (7 M) in DCM to afford ethyl (Z)-2-(11-(3-(methylamino)propylidene)-6,11-dihydrodibenzo[b,e]oxepin-2-yl)acetate (12 mg, 0.034 mmol, 10%) as a colourless gum. LCMS (ESI) m/z 352 [M + H]+. 1H NMR (600 MHz, DMSO-d6) 1.17 (t, J = 7.0 Hz, 3H), 2.24 (s, 3H), 2.45–2.5 (m, 2H), 2.62 (t, J = 6.8 Hz, 2H), 3.57 (s, 2H), 4.06 (q, J = 7.0 Hz, 2H), 5.16 (s, 2H), 5.70 (t, J = 7.2 Hz, 1H), 6.76 (d, J = 8.3 Hz, 1H), 7.05 (dd, J = 2.3, 8.3 Hz, 1H), 7.08 (d, J = 2.3 Hz, 1H), 7.26 (d, J = 7.6 Hz, 1H), 7.28–7.32 (m, 1H), 7.35 (t, J = 7.4 Hz, 2H).
Ethyl (Z)-2-(11-(3-(([3H]methyl)(methyl)amino)propylidene)-6,11-dihydrodibenzo[b,e]oxepin-2-yl)acetate
[3H]Methyl nosylate in toluene (2.5 mL, 1080 MBq) was concentrated under a stream of nitrogen. To this was added a solution of ethyl (Z)-2-(11-(3-(methylamino)propylidene)-6,11-dihydrodibenzo[b,e]oxepin-2-yl)acetate (0.86 mg, 2.4 µmol) in DMF (0.5 mL) and the mixture stirred at 50 °C for 1 h. After lyophilisation, a solution of di-tert-butyldicarbonate (1.1 mg, 4.9 µmol) in DCM (1 mL) was added to the residue and the mixture stirred for 1 h then purified by silica chromatography eluting with 0 to 6% NH3-MeOH (7 M) in DCM. Fractions containing product were evaporated and dissolved in ethanol (1 ml) to give ethyl (Z)-2-(11-(3-(([3H]methyl)(methyl)amino)propylidene)-6,11-dihydrodibenzo[b,e]oxepin-2-yl)acetate solution. LCMS (ESI) m/z 372 [M + H]+.
(Z)-2-(11-(3-(([3H]methyl)(methyl)amino)propylidene)-6,11-dihydrodibenzo[b,e]oxepin-2-yl)acetic acid ([3H]Olopatadine)
Sodium hydroxide (2 M aq, 200 µL) was added to the ethanol solution (1 mL) of ethyl (Z)-2-(11-(3-(([3H]methyl)(methyl)amino)propylidene)-6,11-dihydrodibenzo[b,e]oxepin-2-yl)acetate (0.11 mg, 0.28 µmol) and the mixture stirred for 2 h. The ethanol was evaporated and water (1 mL) was added. The pH was adjusted to 9 by addition of HCl (2 M) and the mixture concentrated then purified on a Waters Oasis HLB cartridge, washing with water (5 mL), drying under a flow of nitrogen and then eluting with acetonitrile (5 mL). Purification by preparative HPLC (Waters XBridge C18 column, 4.6 × 150 mm) using decreasingly polar mixtures of water (containing 0.1% NH3) and MeCN as eluents afforded [3H]olopatadine (107 MBq) which was dissolved in ethanol (2 mL) for storage as a colourless solution. Radiochemical purity >98%. LCMS (ESI) m/z 344 [M + H]+. 3H NMR (640 MHz, DMSO-d6) 2.00 (s). Specific activity by mass spectrometry: 2920 GBq mmol−1.
Cell culture
Human embryonic kidney cells transformed with large T antigen (HEK293T) and stably expressing Nluc-H1 were generated as described elsewhere29, as is the transient transfection of these HEK293T cells with the N-terminally HA-tagged H1R31. Both native and transfected HEK293T cells were maintained in Dulbecco’s Modified Eagles medium supplemented with 10% fetal calf serum at 37 °C, 5% CO2. Cell pellets of transiently transfected HEK293T cells were prepared and stored at −20 °C until used in radioligand binding experiments, as previously described31. Frozen aliquots of TagLite® cells expressing the Tb-labeled SNAP-H1R were acquired from Cisbio.
Radioligand binding assays
Radioligand binding experiments were performed as described before with minor alterations as summarized below31. Frozen cell pellets of HEK293T cells transiently expressing the H1R were thawed, resuspended in radioligand binding buffer (50 mM Na2HPO4 and 50 mM KH2PO4, pH 7.4) and homogenized with a Branson sonifier 250 (Branson Ultrasonics, Danbury, CT, USA). Homogenates (0.5–3 mg/well) were then incubated with the respective ligands at 25 °C under gentle agitation. For equilibrium saturation binding, increasing concentrations [3H]mepyramine or [3H]levocetirizine were incubated for 4 h in the absence or presence of mianserin (10 µM). Mianserin has a pKi at the H1R of 9.4 ± 0.1 (not shown) and the used concentration should therefore prevent any specific binding of the radioligands. For equilibrium competition binding, [3H]mepyramine (3 nM) was used in the presence of increasing concentrations unlabeled ligands. In radioligand association binding experiments, four concentrations [3H]mepyramine (0.2–10 nM), [3H]levocetirizine (1–60 nM) or [3H]olopatadine (9–19 nM) were used. Moreover, radioligand association binding was performed at 37 °C as well as 25 °C. For competitive association binding experiments 1–100x Ki concentrations of the respective unlabeled ligand were co-incubated with a single concentration of radioligand ranging between 1.5–12 nM for [3H]mepyramine or 5–15 nM for [3H]levocetirizine. Kinetic ligand binding was performed for the depicted incubation times.
For dissociation experiments a single concentration of [3H]mepyramine (3–13 nM), [3H]levocetirizine (2–50 nM) or [3H]olopatadine (10–15 nM) was pre-incubated with cell homogenate for 2 h (0.5–3 mg/well), after which a saturating concentration mianserin (10 µM) was added for various incubation times (triplicate binding reactions per time point). Non-specific binding was determined by the presence of mianserin (10 µM) during the pre-incubation step. Dissociation experiments were performed at both 37 °C and 25 °C.
Binding reactions were terminated using a cell harvester (Perkin Elmer) by rapid filtration and wash steps over PEI-coated GF/C filter plates. Filter bound radioligand was then quantified by scintillation counting using Microscint-O and a Wallac Microbeta counter (Perkin Elmer).
HTRF binding assays
HTRF based binding assays were performed as described before30 with minor changes as summarized below. The mepyramine based fluorescent ligand (Gmep, Cisbio; time-resolved fluorescence resonance energy transfer (TR-FRET) acceptor) and unlabeled ligands were predispensed in 384-well plates. Binding reactions were started upon addition of TagLite® cells expressing the Tb-labeled SNAP-H1R (Cisbio) (1:8 predilution and 1:2.5 dilution in well; TR-FRET donor). TR-FRET signals arising from Gmep binding were measured at room temperature with an excitation wavelength of 337 nm and emission wavelengths of 490 ± 10 and 520 ± 10 nm, using a PHERAstar FS plate reader (BMG Labtech) with syringes for sample injection. The ratio values (520 nm/490 nm * 10000) were calculated as defined in the instrument software.
The steady-state affinity of the probe was determined by saturation binding experiments. Increasing concentrations of Gmep (2 fold serial dilution; 3.66 × 10−10 M–3 × 10−6 M; and 0 M) were incubated for 2.5 h in the absence and presence of doxepin hydrochloride (1 µM). Kinetic rate constants of Gmep binding were obtained with “association then dissociation” experiments32. Briefly, H1R expressing cells were added to increasing concentrations of Gmep in the absence and presence of doxepin hydrochloride (1 µM) and association was measured for 25.6 min with kinetic intervals of 26 s. Dissociation of Gmep was immediately initiated by addition of doxepin hydrochloride (1.1 µM) and detected for a further 40 min with kinetic intervals of 100 s. Competitive binding experiments were performed to quantify the affinities and kinetic rate constants of ligand binding: Gmep (100 nM) was co-incubated with increasing concentrations unlabeled ligands for 3 h as an endpoint measurement (11-point 3.5 fold serial dilutions of ligand, and 0 nM) or with a kinetic interval of 60 s for competitive association experiments (kPCA; 4-point 10 fold serial dilutions of ligand).
NanoBRET binding assays
For NanoBRET assays, HEK293Tcells stably expressing Nluc-H1 were seeded 24 h before experimentation in white Thermo Scientific 96-well microplates in normal growth medium. For saturation and competition experiments, the medium was removed and replaced with HEPES-buffered saline solution (HBSS; 25 mM HEPES, 10 mM glucose, 146 mM NaCl, 5 mM KCl, 1 mM MgSO4, 2 mM sodium pyruvate, 1.3 mM CaCl2) with the required concentration of AV082 and competing ligand. Cells were then incubated for 1 h at 37 °C (no CO2). The Nluc substrate furimazine (Promega) was then added to each well at a final concentration of 10 µM and allowed to equilibrate for 5 min prior to measurement of fluorescence and luminescence. For association kinetic and competitive association kinetic experiments, medium was replaced by HBSS containing furimazine (10 µM) and incubated at room temperature in the dark for 15 min to allow the luminescence signal to reach equilibrium. For association kinetic experiments, the required concentration of AV082 in the presence and absence of doxepin (10 µM) was then added simultaneously. Immediately after, all wells of the microplate were read once per minute for 60 min. For competitive association experiments, AV082 (10 nM) was added simultaneously with the required concentration of unlabeled ligand or doxepin (10 µM) and read once per minute for 60 min. For all experiments fluorescence and luminescence was read sequentially using the PHERAstar FS plate reader (BMG Labtech) at room temperature. Filtered light emissions were measured at 460 nm (80-nm bandpass) and at >610 nm (longpass) and the raw BRET ratio was calculated by dividing the >610-nm emission by the 460-nm emission.
Data analysis
Analysis of saturation binding experiments, competition binding experiments and association binding experiments are fully described elsewhere29–31. The kinetic experiments were analyzed by GraphPad Prism (GraphPad Software, Inc., La Jolla, CA, USA) using non-linear regression of the data to pharmacological models that assume a one-step binding of the ligand to the receptor.
Competitive association – Motulsky-Mahan model
| 1 |
| 2 |
The baseline signal was subtracted from the total signal obtained in competitive association experiments. In the case of radioligand binding experiments and NanoBRET experiments equation (1) was employed to fit the data. In the case of HTRF experiments the adapted equation (2) is used to account for the observed signal drift (using kdrift as a fitting constant). RL* is the baseline corrected signal that corresponds to the level of receptor binding by the labeled ligands. Bmax is the theoretical RL* in the case that all receptors would be occupied by the labeled ligand. [L*] and [I] stand for the concentrations labeled ligand and unlabeled ligand, respectively. Association rate constants are denoted by k1 or k3 and the dissociation rate constants by k2 or k4 for labeled ligand or cold ligand respectively. For the kinetics of competitive binding model, binding rate constants of the labeled ligands are required to fit the binding rate constants of the unlabeled ligand. The relative error was calculated for k3 and k4 values by dividing the reported error of the non-linear regression (SE) by the fitted mean value.
Dissociation experiments
Non-specific binding was subtracted from the total bound radioligand and the resulting specific radioligand binding over time was analyzed with a one-phase dissociation model (Graphpad prism: ‘Dissociation – One phase exponential decay’). When the radioligand was not fully dissociated within the timespan of the experiment, the final steady-state radioligand binding was constrained to baseline during analysis.
Results
Characterization of radioligand probes
To explore how the binding kinetics of a radioligand affects the Motulsky-Mahan analysis of radioligand association in competition with unlabeled GPCR ligands, three different radioligands and two fluorescence-based probes for the H1R were investigated in this study. In addition to the widely used and commercially available [3H]mepyramine (fast off rate), radiolabeled versions of the 2nd generation antihistamine radioligands olopatadine and levocetirizine (slow off rate)21,33 were synthesized as described in the method section and structures are depicted in Fig. 2. Equilibrium binding of increasing concentrations of [3H]mepyramine (Fig. 3a), [3H]levocetirizine (Fig. 3b) or [3H]olopatadine (Fig. 3c) to H1R-expressing HEK293T cell homogenates, revealed that all radioligands saturably bind to the H1R with high affinities, resulting in pKd values of 8.6 ± 0.1, 8.1 ± 0.1 and 8.7 ± 0.1, respectively (Table 1). Moreover, saturation binding experiments revealed similar Bmax values (25–31 pmol receptor per mg protein) for all three radioligands, indicating that these three radioligands interact with the same H1R population. To determine the binding rate constants of the radioligands at the H1R, four different concentrations of either [3H]mepyramine (Fig. 3d), [3H]levocetirizine (Fig. 3e) or [3H]olopatadine (Fig. 3f) were incubated with cell homogenate for increasing incubation times and the data was fitted using a one-step binding model. The rate constants (kon and koff) of [3H]levocetirizine binding to the H1R are 30- to 100-fold lower than the binding rate constants of [3H]mepyramine binding (Table 1), as was previously described21. The binding rate constants of the 2nd generation antihistamines [3H]levocetirizine and [3H]olopatadine were comparable (<2 fold differences). Moreover, equilibrium dissociation constants calculated from the binding rate constants (pKd,kin = koff/kon) were in good agreement with equilibrium dissociation constants determined by saturation binding experiments (pKd) (Table 1).
Figure 2.
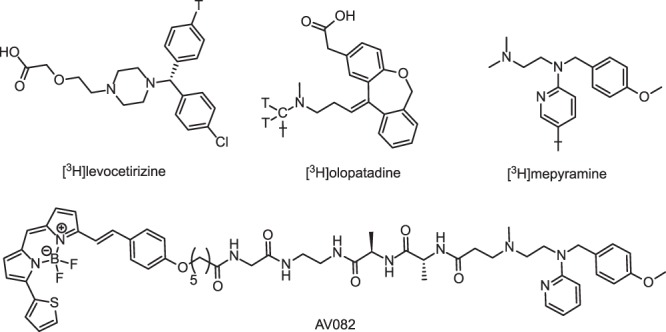
Structures of the used probes. T indicates a tritium atom.
Figure 3.

Binding of [3H]mepyramine, [3H]levocetirizine and [3H]olopatadine to the H1R. Increasing concentrations of [3H]mepyramine (a), [3H]levocetirizine (b) or [3H]olopatadine (c) were incubated with H1R-expressing cell homogenates for 4 h at 25 °C. Indicated concentrations of [3H]mepyramine (d), [3H]levocetirizine (e) or [3H]olopatadine (f) were incubated with cell homogenate for several incubation times at 25 °C. Representative graphs are shown of ≥3 experiments and the depicted data points represent the mean ± SEM of triplicate values (a–c) or depict individual measurements with duplicate values per time point (d–f). Extracted binding constants and statistical information are shown in Table 1 and Supplementary Table 2.
Table 1.
H1R binding parameters of radioligand and fluorescent probes at 25 °C.
| [3H]mepyramine | [3H]levocetirizine | [3H]olopatadine | Gmepa | AV082b | ||
|---|---|---|---|---|---|---|
| pKd | 8.6 ± 0.1 | 8.1 ± 0.1 | 8.7 ± 0.1 | 7.3, 7.3 | 8.1 ± 0.1 | |
| pKd,kin | (k off /k on ) | 8.7 ± 0.0 | 8.7 ± 0.2 | 9.3 ± 0.2 | 7.0, 6.8 | 8.3 ± 0.0 |
| kon | 106 min−1 M−1 | 112 ± 5 | 1.1 ± 0.1 | 2.0 ± 0.1 | 1.58, 0.86 | 46 ± 6 |
| koff | min−1 | 0.22 ± 0.01 | 0.0023 ± 0.0006 | 0.0016 ± 0.0003 | 0.17, 0.13 | 0.21 ± 0.03 |
| RT | min | 4.7 ± 0.3 | 600 ± 200 | 600 ± 100 | 5.9, 7.9 | 5.0 ± 0.8 |
| Bmax | (pmol/mg) | 26 ± 4 | 31 ± 4 | 25 ± 4 | NA | NA |
The koff values of the radioligand probes were verified by radioligand displacement experiments, in which pre-bound radioligand is forced to dissociate by a high concentration of the unlabeled competitor mianserin (Supplementary Fig. 1). The koff values of [3H]mepyramine and [3H]levocetirizine deviated less than 1.5-fold between the association and dissociation experiments (Supplementary Table 2). However, there was very little dissociation of [3H]olopatadine within the 6 hour time frame measured (Supplementary Fig. 1c). To accelerate the association and dissociation of the radioligands and, thereby obtain a more robust quantification of the binding kinetics for the three radioligands, experiments were also performed at 37 °C (Supplementary Fig. 1)21. As expected, both the kon and the koff values of [3H]mepyramine and [3H]levocetirizine increased by 3–10 fold (Supplementary Table 2). However, even at 37 °C there was still limited dissociation of [3H]olopatadine within the 6 h time span. Interestingly, at 37 °C the association of [3H]olopatadine was not described well by a mono-exponential increase in binding as expected for a one-step binding mechanism. Consequently, [3H]olopatadine was therefore excluded as probe, as the Motulsky-Mahan model that is used to describe competitive association ligand binding is based a one-step binding mechanism.
Quantifying the binding characteristics of unlabeled H1R antagonist
A chemically diverse set of unlabeled H1R ligands (structures are depicted in Supplementary Fig. 2), including reference molecules with known differences in their H1R binding kinetics, was selected for characterization of their H1R binding kinetics using either [3H]mepyramine or [3H]levocetirizine13,23,31. To guide the design of competitive association experiments, binding affinities (Ki) of the unlabeled ligands were first determined by equilibrium competition binding. Cell homogenates were therefore co-incubated with [3H]mepyramine and increasing concentrations of the unlabeled ligands (Supplementary Fig. 3). Binding affinities (Ki) for H1R were calculated from the determined IC50 values using the Cheng-Prusoff equation34 and are depicted in Supplementary Table 3.
The binding rate constants of the unlabeled ligands at the H1R were determined by competitive association binding experiments, in which H1R binding of the radioligand probes is quantified over time in the absence or presence of unlabeled ligands at three different concentrations. Concentrations of unlabeled ligand were varied ten-fold between the lowest and highest used concentration which was within an equipotent range of 1–100 times the respective Ki of the ligands at the H1R. From the resulting radioligand association binding curves, the binding rate constants of unlabeled ligands can be determined by Motulsky-Mahan analysis (Fig. 4, Table 2)19. As each time point requires a new binding reaction, the kinetic resolution for quantifying radioligand binding is limited and dependent on the number of parallel incubations. Therefore, incubation times were adjusted for the individual radioligands to best capture their kinetic profile. For the rapidly binding radioligand [3H]mepyramine a relatively short 80 min incubation time was chosen (Fig. 4a–c), whereas a 360 min incubation time was employed for the slowly binding probe [3H]levocetirizine (Fig. 4d–f). The association of the radiolabeled probes to the H1R in the presence and absence of three competing unlabeled ligands with (from left to right) fast, intermediate, and slow binding kinetics, is depicted in Fig. 4 and covers the diversity in binding kinetics observed within the full set of unlabeled ligands. Binding of [3H]mepyramine in the presence of unlabeled mepyramine leads to a gradual increase in radioligand binding until binding equilibrium has been established after approximately 10 min (Fig. 4a). In the presence of doxepin and levocetirizine there is first a transient overshoot in the binding of [3H]mepyramine which results from the relatively slow dissociation (lower koff value) of both unlabeled ligands compared to the rapid binding of [3H]mepyramine (Fig. 4b,c). Conversely, since [3H]levocetirizine binds much slower than [3H]mepyramine (Fig. 3d,e), no overshoot pattern is observed for [3H]levocetirizine binding to the H1R in the presence of the same three unlabeled ligands (Fig. 4d–f). The selected time points and length of incubation depended on the employed radioligand (Fig. 4), which might also affect the resulting binding rate constants in competitive association experiments.
Figure 4.

Association binding of [3H]mepyramine and [3H]levocetirizine in the presence of competing unlabeled ligands at the H1R at 25 °C. The kinetic binding of [3H]mepyramine to H1R-expressing cell homogenates was measured with various concentrations of either mepyramine (a) doxepin (b) or levocetirizine (c). Similarly, the kinetic binding of [3H]levocetirizine to H1R-expressing cell homogenates was measured in competition with various concentrations of either mepyramine (d) doxepin (e) or levocetirizine (f). Representative graphs are shown of ≥3 experiments and each condition was measured in duplicate. Extracted binding constants and statistical information are shown in Table 2 and Supplementary Table 2.
Table 2.
Binding rate constants and calculated pKd and RT of unlabeled ligands at the H1R. Values represent the mean ± SEM of n experiments as reported in Supplementary Table 1. Competitive association experiments were performed with [3H]mepyramine or [3H]levocetirizine as the probe, with representative graphs shown for a subset unlabeled ligands in Fig. 4.
| [3H]mepyramine | [3H]levocetirizine | |||||||
|---|---|---|---|---|---|---|---|---|
| pKd,kina | konb | koffb | RTb | pKd,kina | kon | koff | RTb | |
| 106 min−1 M−1 | min−1 | min | 106 min−1 M−1 | min−1 | min | |||
| olopatadine | 8.5 ± 0.0 | 1.8 ± 0.1 | 0.0061 ± 0.0004 | 170 ± 10 | 9.3 ± 0.3 | 1.6 ± 0.5 | 0.0007 ± 0.0002 | 1600 ± 400 |
| levocetirizine | 8.2 ± 0.1 | 1.2 ± 0.2 | 0.008 ± 0.001 | 140 ± 20 | 8.5 ± 0.0 | 1.4 ± 0.1 | 0.0049 ± 0.0008 | 230 ± 30 |
| desloratadine | 9.5 ± 0.0 | 30 ± 10 | 0.008 ± 0.003 | 160 ± 50 | 9.3 ± 0.1 | 9 ± 2 | 0.0044 ± 0.0006 | 240 ± 30 |
| (S)fexofenadine | 7.4 ± 0.1 | 0.23 ± 0.03 | 0.011 ± 0.003 | 110 ± 30 | 7.9 ± 0.0 | 0.19 ± 0.01 | 0.0022 ± 0.0002 | 450 ± 40 |
| (R)fexofenadine | 7.3 ± 0.1 | 0.24 ± 0.03 | 0.013 ± 0.003 | 90 ± 20 | 7.7 ± 0.1 | 0.18 ± 0.01 | 0.0040 ± 0.0006 | 260 ± 40 |
| doxepin | 9.1 ± 0.1 | 70 ± 10 | 0.06 ± 0.02 | 22 ± 7 | 9.7 ± 0.1 | 80 ± 30 | 0.014 ± 0.003 | 80 ± 20 |
| (S)cetirizine | 6.4 ± 0.1 | 0.21 ± 0.01 | 0.09 ± 0.01 | 11 ± 1 | 6.8 ± 0.1 | 0.4 ± 0.2 | 0.05 ± 0.02 | 27 ± 8 |
| triprolidine | 8.1 ± 0.1 | 36 ± 5 | 0.30 ± 0.03 | 3.5 ± 0.4 | 8.7 ± 0.0 | 30 ± 10 | 0.05 ± 0.02 | 30 ± 9 |
| mepyramine | 8.8 ± 0.1 | 200 ± 50 | 0.28 ± 0.05 | 3.9 ± 0.6 | 8.8 ± 0.1 | 40 ± 10 | 0.07 ± 0.02 | 20 ± 8 |
| VUF14454 | 8.6 ± 0.1 | 250 ± 90 | 0.6 ± 0.1 | 1.8 ± 0.3 | 8.7 ± 0.0 | 11 ± 2 | 0.026 ± 0.005 | 41 ± 7 |
| VUF14493 | 8.4 ± 0.1 | 300 ± 100 | 0.9 ± 0.2 | 1.1 ± 0.2 | 8.6 ± 0.0 | 80 ± 70 | 0.2 ± 0.2 | 20 ± 10 |
| VUF14544 | 7.8 ± 0.1 | 100 ± 40 | 1.3 ± 0.3 | 0.9 ± 0.2 | 7.9 ± 0.0 | 2.4 ± 0.4 | 0.032 ± 0.004 | 33 ± 5 |
apKd,kin = koff/kon.
bValues were reported before except for (S)fexofenadine, (R)fexofenadine and (S)cetirizine23.
cRT = residence time = 1/koff.
Probe dependent differences in binding characteristics
The observed association binding data of both radioligands (Fig. 4a–f) agreed well with the fitted non-linear regression lines based on the Motulsky-Mahan model from which binding rate constants (kon and koff) of the unlabeled ligands could be calculated (Table 2). Thus, two datasets were obtained with the binding rate constants of unlabeled ligands that were determined by using either [3H]mepyramine or [3H]levocetirizine as competitive probe. The measured binding rate constants correlated well between datasets as is depicted in Fig. 5a (kon values: R2 = 0.80, P < 0.001) and in Fig. 5b (koff values: R2 = 0.77, P = 0.002). However, the regression lines (solid lines) deviate from unity (dashed line) and some unlabeled ligands showed larger differences in binding kinetics between the two datasets than others. For example, more than 10-fold differences in the kon and koff values were observed for VUF14454 and VUF14544 between both datasets (with the Kd,kin, calculated as the koff/kon, deviating less than 2-fold). The differences in the kon values between datasets were largest for ligands with a relatively high koff value (Table 2). Additionally, a probe-dependent difference for the range in koff values was observed, with [3H]mepyramine discriminating unlabeled ligands over a range with higher koff-values (Fig. 5b, logkoff −2.2 and 0.1) and [3H]levocetirizine discriminating unlabeled ligands over a range with lower koff-values (Fig. 5b, logkoff −3.2 and −0.7). These data suggest that the [3H]mepyramine-based assay better distinguishes fast dissociating unlabeled ligands(high koff values), whereas the [3H]levocetirizine-based assay better distinguishes slow dissociating unlabeled ligands (low koff values). From the determined binding rate constants, the binding affinity (pKd,kin = koff/kon) and the residence time (RT = 1/koff), a proposed metric to relate binding kinetics to in vivo drug efficacy3,5–9, were calculated (Table 2). The pKd,kin values correspond well with the respective pKi values (Fig. 5c), with a good correlation for both the [3H]mepyramine-dataset (R2 = 0.93, P < 0.0001) and [3H]levocetirizine-dataset (R2 = 0.87, P < 0.0001). Furthermore, the pKd,kin values correlate nicely between the probe specific datasets as well (R2 = 0.87, P < 0.0001, data not shown). Since the Kd,kin value is directly derived from the binding rate constants (koff/kon), these correlations highlight the reciprocal changes in kon- and koff values in both binding assays and suggests that the ratio between the binding rate constants (e.g., reflected by the steady state level of radioligand binding, Fig. 4) is more robustly determined by the Motulsky-Mahan model than the binding rate constants themselves.
Figure 5.

Comparison of the binding properties of unlabeled ligands as measured by using two different probes at 25 °C. Binding rate constants of unlabeled ligands were determined in radioligand binding studies. A correlation plot is depicted for the logkon (a) and logkoff (b) as determined from competitive association experiments using either [3H]mepyramine (x-axis) or [3H]levocetirizine (y-axis) as competitive probe. (c) The correlation plot between the affinity calculated from the kinetic binding rate constants (pKd,kin) and the affinity from competition binding experiments (pKi) is depicted. Errors represent SEM values. Dashed lines represent a perfect correlation respective to the X-axis values and solid lines represent the linear regression lines.
To investigate the accuracy of the obtained kon and koff values, the relative errors of the fitted binding rate constants were calculated for each individual experiment for both data sets. The relative errors of the kon and the koff values were plotted against the corresponding mean koff value for each individual competitive association experiment, as depicted in Fig. 6. It is shown that the relative error on the determined binding rate constants depends on the fitted mean koff values of unlabeled ligands at the H1R (Fig. 6). For both binding-rate-constants a decrease in accuracy, i.e. an increase in the relative error, is observed for unlabeled ligands with high mean koff-values at the H1R (Fig. 6a,b) as is apparent for, e.g., −logkoff values > −1. Additionally, an increase in the relative error on the koff-values (Fig. 6b), but not on the kon-values (Fig. 6a), is observed for unlabeled ligands with low mean koff-values as is apparent for, e.g., −logkoff values < −2. Interestingly, Fig. 6 clearly shows a probe-dependent accuracy for the determination of the kon and the koff- values of unlabeled ligands. The kon value for H1R binding is generally more accurate when determined in a [3H]mepyramine binding experiment (blue curve, Fig. 6a), whereas the koff value is more accurate for the [3H]levocetirizine dataset in the case of unlabeled ligands with a logkoff < −2 and less accurate for ligands with a logkoff > −2 (red curve, Fig. 6b).
Figure 6.

Accuracy of the measured binding rate constants depend on the fitted mean koff of unlabeled ligands at the H1R at 25 °C. The accuracy in which the Motulsky-Mahan model fitted the kon (a) and koff (b) by non-linear regression was examined for the different experimental conditions that were employed in this study. To compare the accuracy of the fitted mean kon and koff values over a broad range, the relative magnitude of the error (SE), as derived from non-linear regression, was calculated for each individual replicate experiment and pooled for all ligands. The relative error was calculated by normalizing the SE by the mean (relative error = SE/mean). Subsequently, the relative error for the kon and koff were plotted against the corresponding mean koff determined from the same competitive association curve. Data points derived from competitive association experiments that employed [3H]levocetirizine are depicted in red. The arrows depict the koff of the used probes with [3H]levocetirizine in red and [3H]mepyramine in blue as reported in Table 1. Dashed lines represent a relative error of 1 (mean = SE).
Cross-comparison between fluorescent-ligand and radioligand binding experiments
Recently, promising advances using resonance energy transfer techniques have been made in the use of fluorescent ligands as probes to characterize the binding kinetics of unlabeled ligands to GPCRs such as the H1R29,30. Since the binding of a fluorescent ligand can be measured continuously using a HTRF or NanoBRET-based approach, the kinetic resolution and throughput of such assays are much higher than conventional radioligand binding kinetics experiments. The availability of these assays for H1R, allows them to be compared to traditional radioligand binding assays for measuring the binding kinetics of unlabeled ligands. We initially sought to characterize the binding kinetics of the fluorescent probes in both the NanoBRET and HTRF assays. For the NanoBRET binding assay a BODIPY630/650-labeled mepyramine analog which emits in the red range (AV082; formally described as compound 10 in Stoddart et al.29 and depicted in Fig. 2) is used as fluorescent probe and is used with HEK293T cells expressing H1R tagged on the N-terminus with NanoLuc which are grown in a mono-layer. For the HTRF binding assay, a commercially available fluorescent analog of mepyramine (structure unknown) which emits light in the green range (Gmep) was employed alongside TagLite® cells expressing an N-terminally SNAP-tagged H1R and labelled with terbium cryptate. Characterization of both fluorescent probes was as previously described (Table 1)29,30. Although the relative large size of the attached fluorophore is likely to affect the binding properties of the unlabeled ligand29, the koff value for the binding of mepyramine-analogs AV082 and Gmep to the H1R resembled those of [3H]mepyramine (<2-fold difference), albeit with differences in their kon values (2–100 fold) (Table 1).
Both non-radioactive assays were used to characterize the H1R binding properties of the set of unlabeled ligands depicted in Supplementary Fig. 2. Equilibrium competition binding experiments were performed to obtain pKi values of the unlabeled ligands as described before29,30 (Supplementary Fig. 3, Supplementary Table 3) and ligands were further characterized in kinetic competition association experiments (Fig. 7, Supplementary Table 4). For the binding of AV082 measured by NanoBRET, in line with the competitive association experiments using [3H]mepyramine (Fig. 7a–c), an overshoot was apparent when co-incubated with doxepin and levocetirizine but not with mepyramine. Kinetic binding rate constants were determined by fitting the NanoBRET signal over time to the Motulsky-Mahan model.
Figure 7.

Association binding of AV082 and Gmep in the presence of competing unlabeled ligands at the H1R at 25 °C. The kinetic binding of AV082 to the H1R, stably expressed on adherent cells, was measured in the presence of various concentrations mepyramine (a) doxepin (b) or levocetirizine (c). AV082 binding was measured continuously by NanoBRET for 60 min. The kinetic binding of Gmep to the H1R, stably expressed on freshly thawed cells in suspension, was measured in the presence of various concentrations mepyramine (d) doxepin (e) or levocetirizine (f). Gmep binding was measured continuously by HTRF for 180 min. Representative graphs are shown of ≥3 experiments (a–f).
For the competitive association experiments with Gmep (Fig. 7d–f), a signal drift was observed in the absence of unlabeled ligand. To allow robust fitting of the HTRF signal (including the signal drift), an additional one-phase decay function was incorporated into the Motulsky-Mahan model, as described before30. Despite the signal drift, an overshoot in Gmep binding is apparent in competition association experiments with unlabeled ligands with slow binding kinetics. After an initial rapid increase of Gmep binding, the HTRF-signal decreases much faster when co-incubated with 25 nM or 250 nM levocetirizine than in the absence of any competitor (Fig. 7f). In contrast, in the presence of mepyramine (Fig. 7d), the HTRF-signal never decreased faster than was observed for Gmep in the absence of unlabeled ligand.
A comparison of the binding constants that were obtained with [3H]mepyramine binding (x-axis) and the two fluorescent binding assays (y-axis) are depicted in Fig. 8a–c and Supplementary Table 3. Interestingly, a good correlation was observed between assays for the relative binding affinities (pKi) (Fig. 8a; (AV082: R2 = 0.88, P < 0.0001; Gmep: R2 = 0.94, P < 0.0001) and logkon values (Fig. 8b; AV082: R2 = 0.84, P < 0.0001; Gmep: R2 = 0.92, P < 0.0001). Although, the use of AV082 in probe-displacement experiments resulted additionally in a log-unit lower pKi-values compared to values obtained in the orthogonal assays (Fig. 8a). Moreover, the logkoff values determined with Gmep as probe (HTRF assay) also correlated with those determined using [3H]mepyramine as probe (Fig. 8c; R2 = 0.96, P < 0.0001). However, when employing AV082 (NanoBRET assay) in competitive association experiments, the relative differences in the koff values between the unlabeled ligands differ from those observed in the orthogonal assays (Fig. 8c). Since both the Ki-values and kon values correlate between orthogonal assays, the koff can be calculated (Ki × kon) for the NanoBRET assay in order to estimate the relative differences in the koff between unlabeled ligands. As expected, these calculated values correlate better with the observed values in the orthogonal assays (Supplementary Fig. 4).
Figure 8.

Comparison of the binding properties of unlabeled ligands as measured by using orthogonal assays at 25 °C. The binding rate constants of unlabeled ligands that were determined using the fluorescent probes AV082 and Gmep were compared with the binding constants obtained with [3H]mepyramine. A comparison of the pKi values (a) (from equilibrium competition binding experiments) is depicted as well as the comparison of kon (b) and koff (c) values (from kinetic competition association experiments). Dashes lines represent a perfect correlation respective to the X-axis values and solid lines represent the linear regression lines.
Discussion
In GPCR drug discovery, drug-receptor binding kinetics are often quantified using competition association experiments with a radioligand probe. Despite the increased use of this methodology, it is unclear whether the kinetic properties of the probe influence the obtained kinetic binding parameters of unlabeled ligands. Therefore, in this study we employed two radioligand probes, that differ in their H1R binding kinetics ([3H]mepyramine and [3H]levocetirizine), to measure the binding rate constants of a diverse set of unlabeled antagonists. The analysis shows that the kon and koff values obtained with each probe correlate between both probe-specific datasets (Fig. 5a,b). However, large differences in the binding rate constants are observed for some compounds, e.g., VUF14454 and VUF14544. Moreover, although more than 10-fold differences in the koff are observed among (S)-cetirizine, triprolidine, mepyramine, VUF14454, VUF14493 and VUF14544 when using [3H]mepyramine as probe, no difference is observed when the koff are measured for the same set of unlabeled ligands with [3H]levocetirizine as probe. The comparison of these two datasets therefore suggests a probe-dependent limit to discriminate binding rate constants of unlabeled ligands. A related probe-dependent effect is apparent in the relative errors of the determined kon (Fig. 6a) and koff values (Fig. 6b) of the unlabeled ligands. In our competitive binding experiments, only the binding of the probe can be directly observed. When unlabeled ligands reach a binding equilibrium rapidly, before noticeable binding of the probe, binding kinetics of these unlabeled ligands is masked by the slow onset of probe binding at each time point. Since the onset of a receptor-binding equilibrium is faster with increasing koff of the respective ligands19, it seems logical that at some point, when the unlabeled ligands have an increasingly high koff compared to that of the probe, kinetic binding of the unlabeled ligands can no longer be distinguished. This is in line with the observation that the relative error in the fitted binding rate constants increases when the corresponding mean koff value (i.e. from the same competitive association curve) increases (Fig. 6). Moreover, a pronounced increase in the relative error is observed when the unlabeled ligands dissociate faster than the respective radioligand (koff unlabeled > koff radioligand, see arrows Fig. 6a,b). This implies that [3H]mepyramine, which has a 100-fold higher koff than [3H]levocetirizine, is better suited to discriminate the binding kinetics of fast dissociating (high koff) unlabeled ligands at the H1R. Moreover, in our dataset the kon (Fig. 7a) and koff (Fig. 7b) are fitted with a higher accuracy using [3H]mepyramine as probe for unlabeled ligands with a residence time less than 100 min (log koff > −2).
In contrast to the determined kon-values, which show a growing inaccuracy upon increases of the linked koff-value (Fig. 6a), the koff-values are increasingly inaccurate at the lower end of the spectrum as well, i.e. both with low koff as well as high koff values (Fig. 6b). Interestingly, a probe dependent difference in inaccuracy in these determinations is again apparent. However, for slowly binding unlabeled ligands (residence time of more than 100 min; fitted log koff < −2) a moderately better accuracy is obtained when using [3H]levocetirizine (and not [3H]mepyramine) as probe in competition association experiments (Fig. 6b).
Taken together, analysis of the competitive association radioligand binding data show probe-dependent differences in the measured binding rate constants of unlabeled ligands, which result (at least to some extent) from the accuracy with which these binding rate constants can be fitted to the competitive association curves of the radioligand probes. Considering that the accuracy of the measured binding rate constants decreased most extensively for unlabeled ligands that bind the receptor faster than the probe (koff unlabeled > koff radioligand), it is recommended to use a fast binding probe for the GPCR of interest (like [3H]mepyramine for the H1R).
To avoid that the kinetics of the probe will mask the binding kinetics of the unlabeled ligands, the koff of the probe should ideally be higher than that of the unlabeled ligands. In radioligand binding experiments, the bound radioligand should not dissociate during the wash steps. A minimum residence time is therefore required and the koff should probably not go beyond 1 min−1 (at room temperature). For probes in fluorescent binding experiments, which do not require wash steps, probes could be designed to have a very high koff. There are not yet sufficient structure-kinetics-relationships available that allow clear cut optimization of the koff. However, reducing the binding affinity sufficiently will in most cases increase the koff35. Introducing subtle steric clashes in the binding pocket22 might therefore be a way to fine-tune the koff of fluorescent probes.
Comparing the binding kinetics of unlabeled H1R antagonists determined in experiments using the fluorescently labelled H1R probes AV082 and Gmep with those obtained using [3H]mepyramine, again reveals that the determined pKi values and the binding rate constants are highly correlated (Fig. 8). This might be explained (partially) by the fact that all three H1R probes have quite similar koff values (Table 1). In fact, the correlations for the measured binding rate constants were stronger when comparing the different assays (Fig. 8a,b) than when comparing the datasets obtained with the different radioligands (Fig. 6a,b). The notable exception were the koff-values determined with AV082 competition association binding experiments (measured by NanoBRET), which deviated only slightly among unlabeled ligands, suggesting again a probe-dependent limitation for discriminating the binding kinetics of unlabeled ligands. However, this effect is most likely not explained by the binding kinetics of the H1R probe as the binding rate constants of AV082 and [3H]mepyramine differed only <3-fold. Assay-dependent differences might therefore also underlie the observed disconnect between koff values. It has been described that the measurement of drug-target binding kinetics can be additionally convoluted in pharmacological assays by rebinding36,37, the mechanism of ligand binding38,39, and the differences in local ligand concentration40, all of which cannot be accounted for during this analysis. Interestingly, in silico docking suggests that the hydrophobic fluorophore of large fluorescent ligands, like AV082, can protrude out of the GPCR 7TM pocket and might incorporate in the membrane29, which is distinct from the binding mode of [3H]mepyramine, which is deeply buried within the transmembrane region of the H1R41–43. One can speculate that the simple one-step binding mechanism that is the conceptual basis of the Motulsky-Mahan model for probe binding to the H1R, is not a valid approximation in the case of AV082. Besides the probe, the extracellular N-terminal tag of the employed H1R was different between the NanoBRET-based detection of AV082 binding and the HTRF-based detection of Gmep binding to the H1R protein. Moreover, since AV082 was the only probe that was employed in binding experiments on adherent living cells, the extracellular environment might shape a unique exosite that promotes ligand-rebinding. For example, the epithelial layer of human lungs in organ bath perfusion experiments proved crucial for the insurmountable antagonism of the H1R imposed by azelastine44. Since the insurmountable antagonism depends on the length of the receptor-occupancy by azelastine23,44,45, the extracellular environment may in some cases contribute to the observed ligand binding kinetics.
In conclusion, the Motulsky-Mahan approach is a useful approach to quantify the binding rate constants of unlabeled GPCR ligands, especially with the high throughput and kinetic resolution that can be obtained with fluorescent ligand binding experiments. However, it should be taken into account that probe-dependent and assay-dependent factors can have an impact on the measured binding kinetics of the unlabeled ligands. It is recommended to use orthogonal approaches to confirm the binding kinetics of a set of reference compounds, for example, by studying the kinetics by which ligands functionally modulate GPCR activity. Previously, we found the ligand binding kinetics at the H1R of a set of H1R antagonists to correlate well with their kinetic effects on functional H1R responses23,31. Furthermore, the use of benchmark ligands will also allow comparison between different methodologies and will allow the selection of the best method to reach highest confidence for discriminating the target-binding kinetics of unlabeled ligands by the Motulsky-Mahan method. Considering that the Motulsky-Mahan model is by far the most frequently used way to derive the residence time of GPCR ligands, this study provides important considerations for the study of drug-target binding kinetics at GPCRs.
Supplementary information
Acknowledgements
This research was financially supported by the EU/EFPIA Innovative Medicines Initiative (IMI) Joint Undertaking, K4DD (Grant No. 115366).
Author Contributions
R.B., L.I., H.V. and R.L. contributed the data and analysis for the “radioligand binding assays” methodology. L.S., M.B., S.B. and S.H. contributed the data and analysis for the “NanoBRET binding assays” methodology. V.G. and A.F. contributed the data and analysis for the “HTRF binding assays” methodology. Nick Bushby contributed to the synthetic procedures. All authors participated in writing of the manuscript.
Data Availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-44025-5.
References
- 1.Swinney DC. Biochemical mechanisms of drug action: what does it take for success? Nat. Rev. Drug Discov. 2004;3:801–8. doi: 10.1038/nrd1500. [DOI] [PubMed] [Google Scholar]
- 2.Swinney DC. Biochemical mechanisms of New Molecular Entities (NMEs) approved by United States FDA during 2001–2004: mechanisms leading to optimal efficacy and safety. Curr Top Med Chem. 2006;6:461–78. doi: 10.2174/156802606776743093. [DOI] [PubMed] [Google Scholar]
- 3.Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov. 2006;5:730–9. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- 4.Cumming JG, et al. Potential strategies for increasing drug-discovery productivity. Future Med. Chem. 2014;6:515–27. doi: 10.4155/fmc.14.7. [DOI] [PubMed] [Google Scholar]
- 5.Hoffmann C, et al. Ligand Residence Time at G-protein-Coupled Receptors–Why We Should Take Our Time To Study It. Mol. Pharmacol. 2015;88:552–60. doi: 10.1124/mol.115.099671. [DOI] [PubMed] [Google Scholar]
- 6.Swinney DC. Applications of Binding Kinetics to Drug Discovery. Pharmaceut. Med. 2008;22:23–34. [Google Scholar]
- 7.Copeland RA. The drug–target residence time model: a 10-year retrospective. Nat. Rev. Drug Discov. 2015;15:1–9. doi: 10.1038/nrc3883. [DOI] [PubMed] [Google Scholar]
- 8.Lu H, Tonge PJ. Drug-target residence time: critical information for lead optimization. Curr. Opin. Chem. Biol. 2010;14:467–74. doi: 10.1016/j.cbpa.2010.06.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo D, Hillger JM, IJzerman AP, Heitman LH. Drug-Target Residence Time-A Case for G Protein-Coupled Receptors. Med. Res. Rev. 2014;34:856–92. doi: 10.1002/med.21307. [DOI] [PubMed] [Google Scholar]
- 10.Unett, D., Gatlin, J. & Anthony, T. Kinetics of 5-HT2B receptor signaling: Profound agonist-dependent effects on signaling onset and duration. Pharmacol. 645–659 (2013). [DOI] [PubMed]
- 11.Guo D, et al. Dual-point competition association assay: a fast and high-throughput kinetic screening method for assessing ligand-receptor binding kinetics. J. Biomol. Screen. 2013;18:309–20. doi: 10.1177/1087057112464776. [DOI] [PubMed] [Google Scholar]
- 12.Ramsey SJ, Attkins NJ, Fish R, van der Graaf PH. Quantitative pharmacological analysis of antagonist binding kinetics at CRF1 receptors in vitro and in vivo. Br. J. Pharmacol. 2011;164:992–1007. doi: 10.1111/j.1476-5381.2011.01390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gillard M, Van Der Perren C, Moguilevsky N, Massingham R, Chatelain P. Binding characteristics of cetirizine and levocetirizine to human H(1) histamine receptors: contribution of Lys(191) and Thr(194) Mol. Pharmacol. 2002;61:391–9. doi: 10.1124/mol.61.2.391. [DOI] [PubMed] [Google Scholar]
- 14.Dowling MR, Charlton SJ. Quantifying the association and dissociation rates of unlabelled antagonists at the muscarinic M3 receptor. Br. J. Pharmacol. 2006;148:927–37. doi: 10.1038/sj.bjp.0706819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sullivan SK, et al. Kinetics of nonpeptide antagonist binding to the human gonadotropin-releasing hormone receptor: Implications for structure-activity relationships and insurmountable antagonism. Biochem. Pharmacol. 2006;72:838–49. doi: 10.1016/j.bcp.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 16.Cassel JA, Daubert JD, DeHaven RN. [(3)H]Alvimopan binding to the micro opioid receptor: comparative binding kinetics of opioid antagonists. Eur. J. Pharmacol. 2005;520:29–36. doi: 10.1016/j.ejphar.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 17.Hoare SR, Usdin TB. Tuberoinfundibular peptide (7-39) [TIP(7-39)], a novel, selective, high-affinity antagonist for the parathyroid hormone-1 receptor with no detectable agonist activity. J. Pharmacol. Exp. Ther. 2000;295:761–70. [PubMed] [Google Scholar]
- 18.Bosma, R., Mocking, T. A. M., Leurs, R. & Vischer, H. F. In Histamine Receptors as Drug Targets (eds Tiligada, E. & Ennis, M.) 115–55, 10.1007/978-1-4939-6843-5_5 (Springer New York, 2017).
- 19.Motulsky H, Mahan L. The kinetics of competitive radioligand binding predicted by the law of mass action. Mol. Pharmacol. 1984;25:1–9. [PubMed] [Google Scholar]
- 20.Simons FER, Simons KJ. Histamine and H1-antihistamines: celebrating a century of progress. J. Allergy Clin. Immunol. 2011;128:1139–50. doi: 10.1016/j.jaci.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 21.Gillard M, Chatelain P. Changes in pH differently affect the binding properties of histamine H1 receptor antagonists. Eur. J. Pharmacol. 2006;530:205–14. doi: 10.1016/j.ejphar.2005.11.051. [DOI] [PubMed] [Google Scholar]
- 22.Kuhne S, et al. Identification of Ligand Binding Hot Spots of the Histamine H1 Receptor following Structure-Based Fragment Optimization. J. Med. Chem. 2016;59:9047–61. doi: 10.1021/acs.jmedchem.6b00981. [DOI] [PubMed] [Google Scholar]
- 23.Bosma R, et al. The target residence time of antihistamines determines their antagonism of the G protein-coupled histamine H1 receptor. Front. Pharmacol. 2017;8:667–667. doi: 10.3389/fphar.2017.00667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wittmann H-J, Seifert R, Strasser A. Influence of the N-terminus and the E2-loop onto the binding kinetics of the antagonist mepyramine and the partial agonist phenoprodifen to H(1)R. Biochem. Pharmacol. 2011;82:1910–8. doi: 10.1016/j.bcp.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 25.Panula P, et al. International Union of Basic and Clinical Pharmacology. XCVIII. Histamine Receptors. Pharmacol. Rev. 2015;67:601–55. doi: 10.1124/pr.114.010249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hill SJ, Emson PC, Young JM. The binding of [3H]mepyramine to histamine H1 receptors in guinea-pig brain. J. Neurochem. 1978;31:997–1004. doi: 10.1111/j.1471-4159.1978.tb00139.x. [DOI] [PubMed] [Google Scholar]
- 27.Kanba S, Richelson E. Histamine H1 receptors in human brain labelled with [3H]doxepin. Brain Res. 1984;304:1–7. doi: 10.1016/0006-8993(84)90856-4. [DOI] [PubMed] [Google Scholar]
- 28.Schuetz DA, et al. Kinetics for Drug Discovery: an industry-driven effort to target drug residence time. Drug Discov. Today. 2017;22:896–911. doi: 10.1016/j.drudis.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Stoddart LA, et al. Development of novel fluorescent histamine H1-receptor antagonists to study ligand-binding kinetics in living cells. Sci. Rep. 2018;8:1572. doi: 10.1038/s41598-018-19714-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schiele F, Ayaz P, Fernández-Montalván A. A universal homogeneous assay for high-throughput determination of binding kinetics. Anal. Biochem. 2015;468:42–49. doi: 10.1016/j.ab.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 31.Bosma R, Moritani R, Leurs R, Vischer HF. BRET-based β-arrestin2 recruitment to the histamine H1 receptor for investigating antihistamine binding kinetics. Pharmacol. Res. 2016;111:679–87. doi: 10.1016/j.phrs.2016.07.034. [DOI] [PubMed] [Google Scholar]
- 32.Nederpelt I, et al. Characterization of 12 GnRH peptide agonists - A kinetic perspective. Br. J. Pharmacol. 2016;173:128–41. doi: 10.1111/bph.13342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nonaka H, et al. Unique binding pocket for KW-4679 in the histamine H1 receptor. Eur. J. Pharmacol. 1998;345:111–7. doi: 10.1016/S0014-2999(97)01620-8. [DOI] [PubMed] [Google Scholar]
- 34.Cheng Y, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 35.Pan AC, Borhani DW, Dror RO, Shaw DE. Molecular determinants of drug-receptor binding kinetics. Drug Discov Today. 2013;18:667–673. doi: 10.1016/j.drudis.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 36.Vauquelin G, Charlton SJ. Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br. J. Pharmacol. 2010;161:488–508. doi: 10.1111/j.1476-5381.2010.00936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vauquelin G, Van Liefde I. Radioligand dissociation measurements: potential interference of rebinding and allosteric mechanisms and physiological relevance of the biological model systems. Expert Opin. Drug Discov. 2012;7:583–95. doi: 10.1517/17460441.2012.687720. [DOI] [PubMed] [Google Scholar]
- 38.Swinney DC, et al. A study of the molecular mechanism of binding kinetics and long residence times of human CCR5 receptor small molecule allosteric ligands. Br. J. Pharmacol. 2014;171:3364–75. doi: 10.1111/bph.12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vauquelin G, Van Liefde I, Swinney DC. On the different experimental manifestations of two-state ‘induced-fit’ binding of drugs to their cellular targets. Br. J. Pharmacol. 2016;173:1268–85. doi: 10.1111/bph.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sykes DA, et al. Observed drug-receptor association rates are governed by membrane affinity: the importance of establishing ‘micro-pharmacokinetic/pharmacodynamic relationships’ at the β2-adrenoceptor. Mol. Pharmacol. 2014;85:608–17. doi: 10.1124/mol.113.090209. [DOI] [PubMed] [Google Scholar]
- 41.Blandina Patrizio, Passani Maria Beatrice., editors. Histamine Receptors. Cham: Springer International Publishing; 2016. [Google Scholar]
- 42.Shimamura T, et al. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kooistra AJ, Kuhne S, de Esch IJP, Leurs R, de Graaf C. A structural chemogenomics analysis of aminergic GPCRs: lessons for histamine receptor ligand design. Br. J. Pharmacol. 2013;170:101–26. doi: 10.1111/bph.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slack RJ, Hart AD, Luttmann MA, Clark KL, Begg M. In vitro characterisation of the duration of action of the histamine-1 receptor antagonist azelastine. Eur. J. Pharmacol. 2011;670:586–92. doi: 10.1016/j.ejphar.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 45.Kenakin T, Jenkinson S, Watson C. Determining the potency and molecular mechanism of action of insurmountable antagonists. J. Pharmacol. Exp. Ther. 2006;319:710–723. doi: 10.1124/jpet.106.107375. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.