

Atmospheric oxygen levels modulate the RAS/MAPK signaling pathway in the nematode Caenorhabditis elegans through a cross-talk between the DELTA/NOTCH and hypoxia signaling pathways. These mechanisms may permit the animals to adapt their development to changes in the environment.
Abstract
Animals need to adjust many cellular functions to oxygen availability to adapt to changing environmental conditions. We have used the nematode Caenorhabditis elegans as a model to investigate how variations in oxygen concentrations affect cell fate specification during development. Here, we show that several processes controlled by the conserved RTK/RAS/MAPK pathway are sensitive to changes in the atmospheric oxygen concentration. In the vulval precursor cells (VPCs), the hypoxia-inducible factor HIF-1 activates the expression of the nuclear hormone receptor NHR-57 to counteract RAS/MAPK–induced differentiation. Furthermore, cross-talk between the NOTCH and hypoxia-response pathways modulates the capability of the VPCs to respond to RAS/MAPK signaling. Lateral NOTCH signaling positively regulates the prolyl hydroxylase EGL-9, which promotes HIF-1 degradation in uncommitted VPCs and permits RAS/MAPK–induced differentiation. By inducing DELTA family NOTCH ligands, RAS/MAPK signaling creates a positive feedback loop that represses HIF-1 and NHR-57 expression in the proximal VPCs and keeps them capable of differentiating. This regulatory network formed by the NOTCH, hypoxia, and RAS/MAPK pathways may allow the animals to adapt developmental processes to variations in oxygen concentration.
Introduction
The RAS/MAPK pathway regulates cell growth, differentiation, proliferation, apoptosis, and migration in all metazoans (Simanshu et al, 2017). Constitutively activating mutations in HRAS, NRAS, or KRAS are among the most frequent tumor-initiating mutations in human cancer. In Caenorhabditis elegans, RAS/MAPK signaling is involved in several developmental processes, such as the specification of the excretory duct cell precursor, the differentiation and maturation of meiotic germ cells or the development of the hermaphrodite vulva (Sternberg, 2005; Sundaram, 2006). During vulval development, an EGF-like ligand (LIN-3) secreted by the gonadal anchor cell (AC) activates EGFR/RAS/MAPK signaling in the adjacent vulval precursor cells (VPCs) through the EGF receptor (LET-23) pathway (Fig 1A). Strong activation of the RAS/MAPK signaling pathway in P6.p, the VPC located closest to the AC, induces the primary (1°) cell fate and up-regulates the expression of DELTA-like DSL ligands, which activate lateral NOTCH signaling in the adjacent VPCs (Chen & Greenwald, 2004). NOTCH performs two distinct functions during vulval induction (VI). First, before and after the first VPC division, NOTCH signaling maintains the VPCs competent, that is, capable of responding to the inductive AC signal (Wang & Sternberg, 1999). Second, strong NOTCH activation in P5.p and P7.p, which are the neighbors of P6.p, inhibits MAPK signaling and directly specifies the secondary (2°) fate (Berset et al, 2001; Yoo et al, 2004). The distal VPCs (P3-4.p and P8.p) that receive neither the EGFR/RAS/MAPK nor NOTCH signal adopt the tertiary (3°), uninduced fate. As a result, an invariant 3°-3°-2°-1°-2°-3° cell fate pattern is established. Activating (gain-of-function) mutations in the RAS/MAPK pathway lead to the ectopic induction of distal VPCs and a multivulva (Muv) phenotype, whereas loss-of-function mutations in RAS/MAPK pathway components cause a vulvaless (Vul) phenotype. Thus, the average number of induced VPCs per animal, termed the VI index, can be used to quantify RAS/MAPK signaling strength (e.g., VI = 3 in the wild-type, VI < 3 in Vul, and VI > 3 in Muv mutants) (Schmid et al, 2015).
Figure 1. Hypoxia represses RAS/MAPK–mediated differentiation in different tissues.

(A) Overview of vulval development showing the known interactions between the RAS/MAPK and DELTA/NOTCH pathways. During the L2 stage, LIN-3 EGF activates the RAS/MAPK cascade in P5.p-P7.p. P6.p, in which RAS/MAPK activity is highest, adopts the 1° cell fate, and expresses DSL NOTCH ligands to activate LIN-12 NOTCH signaling in the adjacent Pn.p cells, which adopt the 2° fate. The uninduced P(3-4).p and P8.p cells adopt the 3° fate and fuse with the surrounding hypodermis. (B) Vulval phenotypes of the let-60 ras G13E mutation in the N2 Bristol (left) and CB4856 Hawaii (right) background with varying oxygen concentrations. Solid lines indicate induced 1° and 2° and arrowheads uninduced 3° VPCs in L4 larvae. (C) VI of N2 Bristol and CB4856 Hawaii let-60 ras G13E mutants raised in varying oxygen concentrations. (D) Effect of hypoxia on different RTK/RAS/MAPK pathway, bar-1(ga80), lin-12(n137), and lin-12(n137n720) mutants. ∆VI indicates the change in VI of animals raised in 0.5% compared with controls grown in 21% oxygen. ∆%Muv and ∆%Vul indicate the change in the percentage of animals with VI > 3 and VI < 3, respectively. The absolute VIs at 0.5% oxygen are shown in the rightmost red column. (E) Suppression of the stacked oocyte phenotype in let-60(ga89gf) animals raised at the restrictive temperature by hypoxia. Arrowheads point at the stacked oocytes formed in the proximal gonad under normoxia. (F) Suppression of the duct cell duplication phenotype in let-60(n1046gf) mutants by hypoxia. Arrows point at the duct cell nuclei expressing LIN-48::GFP formed under normoxia (top) and hypoxia (bottom). (C, D) Error bars in (C) and (D) indicate the 95% confidence intervals, and P-values, indicated with ***P < 0.001 and **P < 0.01, were derived by bootstrapping 1,000 samples. (E, F) In (E) and (F), error bars indicate the standard error of the mean, and P-values were calculated with a Fisher’s exact test. The numbers of animals scored are indicated in brackets. The scale bars represent 5 μm.
C. elegans has evolved cellular and behavioral responses to adapt to variations in oxygen concentration (Semenza, 2001; Gray et al, 2004). At the cellular level, the hypoxia-response pathway mediates the adaptation to low oxygen conditions and a switch from aerobic to anaerobic metabolism. In ambient oxygen, the C. elegans hypoxia-inducible factor HIF-1 α is hydroxylated by the prolyl hydroxylase EGL-9 at a specific proline residue within the degradation domain (Fig 2A) (Epstein et al, 2001). Hydroxylated HIF-1 interacts with the von Hippel-Lindau E3 ubiquitin ligase VHL-1 complex and is degraded by the 26S proteasome (Bishop et al, 2004). Under low oxygen concentrations, HIF-1 is stabilized because of decreased EGL-9 activity and forms a complex with the constitutively expressed HIF-β subunit AHA-1 to promote the expression of specific target genes (Bishop et al, 2004; Shen et al, 2005).
Figure 2. The hypoxia-response pathway negatively regulates VI under normoxia.

(A) Schematic overview of the conserved hypoxia-response pathway. The C. elegans gene names are indicated. (B) Vulval phenotypes of double and triple mutants between let-60(n1046gf) and components of the hypoxia-response pathway under normoxia. Solid lines indicate induced 1° and 2° and arrowheads uninduced 3° VPCs in the L4 larvae. (C) Mutations in the hypoxia-response pathway change the VI of let-60(n1046gf) mutants. ∆VI indicates the change in VI of the genotypes relative to let-60(n1046gf) single mutant siblings obtained from the crosses. ∆%Muv indicates the change in the percentage of animals with VI > 3. The absolute VIs of the double/triple mutants are shown in the rightmost blue column. (D) Overexpression of wild-type egl-9::gfp increases the VI. ∆VI indicates the change in VI of animals carrying a wild-type (dark bars) or hydroxylase deficient (light bars) multi-copy egl-9::gfp array compared with siblings without array. Error bars indicate the 95% confidence intervals. P-values, indicated with ***P < 0.001 and **P < 0.01, were derived by bootstrapping 1,000 samples. (E–I, J) MPK-1 biosensor (ERK-nKTR) activity values measured in the VPCs of mid-L2 larvae with the indicated mutant backgrounds and (J) comparison of the MPK-1 activity levels in P6.p across the different genotypes. Relative MPK-1 activity values were measured as described under the Materials and Methods section. The numbers of animals scored are indicated in brackets. The scale bar represents 5 μm.
We have previously used quantitative genetics to identify modifiers of the RAS/MAPK pathway by comparing two highly polymorphic C. elegans strains, N2 Bristol and CB4856 Hawaii, which had been sensitized by introgression of the G13E let-60(n1046) ras gain-of-function allele (Schmid et al, 2015). Notably, CB4856 Hawaii exhibits an overall elevated sensitivity to RAS/MAPK signaling compared with the N2 Bristol reference strain (Milloz et al, 2008; Schmid et al, 2015). Among the RAS/MAPK modifiers identified were F44F1.1, which encodes a calpain paralog (F44F1.3), and pfd-3, a prefoldin orthologous to the human VHL-binding protein 1. Both genes have independently been identified in a screen for suppressors of the egl-9 prolyl hydroxylase egg-laying–defective phenotype, suggesting that F44F1.3 and pfd-3 interact with the core hypoxia-response pathway (Gort et al, 2007). Together, these findings suggested that genetic variation in genes controlling the hypoxia-response pathway might account for the differences in RAS/MAPK signaling strength between the N2 Bristol and CB4856 Hawaii C. elegans isolates.
Here, we show that hypoxia inhibits the cellular responses to RAS/MAPK signaling in different tissues of C. elegans. Surprisingly, only part of the hypoxic inhibition of RAS/MAPK signaling depends on HIF-1 activity. We further identify the nuclear hormone receptor gene nhr-57 as a critical HIF-1 target that opposes RAS/MAPK–induced differentiation under normoxia and under hypoxia. Finally, we show that the prolyl hydroxylase EGL-9 is a NOTCH target gene. By inducing egl-9 expression, lateral NOTCH signaling between the proximal VPCs promotes HIF-1 degradation and thereby represses nhr-57 expression at the onset of vulval fate specification to keep the proximal VPCs competent to respond to the inductive AC signal. This regulatory network allows the adaptation of development to variations in oxygen availability.
Results
Hypoxia suppresses LET-60 RAS gain-of-function phenotypes in different tissues
The two polymorphic genes F44F1.1 and pfd-3 have previously been found to modify RAS/MAPK signaling during vulval development and to suppress the egg-laying–defective phenotype of egl-9(lf) mutants (Gort et al, 2007; Schmid et al, 2015). We, thus, asked if hypoxia modulates RAS/MAPK–mediated cell fate decisions and whether the differences in RAS/MAPK signaling strength observed between different C. elegans wild isolates (Milloz et al, 2008) originate from genetic variation in components of the hypoxia-response pathway. To this aim, we introduced by CRISPR/Cas9 genome editing the G13E gain-of-function mutation into the N2 Bristol (zh121gf) and CB4856 Hawaii (zh122gf) let-60 ras locus (Beitel et al, 1990; Arribere et al, 2014). We then determined the VI of N2 let-60(zh121gf) and CB4856 let-60(zh122gf) animals raised under different oxygen concentrations. Under normoxia (21% O2), CB4856 animals carrying the G13E mutation exhibited a higher VI than N2 animals carrying the same mutation, as had been observed earlier by introgressing the canonical G13E allele let-60(n1046gf) into the CB4856 background (Fig 1B and C) (Milloz et al, 2008; Schmid et al, 2015). VI was not significantly changed in either strain when the animals were raised under mild hypoxic conditions of 11%, 7%, or 3% O2 (Fig 1B and C). However, at 1% O2, the VI decreased in the CB4856 background, and at 0.5% O2, the VI was reduced to the same level in both strains. Because 0.5% is the lowest O2 concentration, under which the animals develop to adulthood, we could not test lower O2 concentrations. It, thus, appears that the same baseline level of VI is reached under severe hypoxia (0.5% O2) irrespective of the genetic background.
To further investigate the effect of oxygen on RAS/MAPK signaling and rule out allele-specific effects, we examined the effect of strong hypoxia (0.5% O2) on different RAS/MAPK pathway mutants in the N2 Bristol background. We included the Wnt pathway–mutant bar-1(ga80) and the NOTCH alleles lin-12(n137gf) and lin-12(n137n720lf) in this analysis. Even though hypoxia had no effect on the VI in the N2 Bristol wild-type strain (VI = 3, n > 50), hypoxia reduced the VI in animals carrying rf mutations in let-23 egfr, let-60 ras, or lin-45 raf or in animals expressing an activated MAPK (MPK-1) from a transgene (mpk-1(gaIs37gf)) (Fig 1D). (Note that for each genotype, ∆VI indicates the difference in the induction index between siblings grown at 0.5% and 21% O2.) By contrast, the increased VI in lin-1(lf) mutants, which encodes an Ets family transcription factor that represses VI downstream of MPK-1 (Beitel et al, 1995), was not affected by hypoxia. Moreover, the VI of bar-1 β-catenin or lin-12 notch mutants was not changed by hypoxia, indicating that hypoxia specifically affects the activity of or response to RAS/MAPK signaling (Fig 1D).
We next examined the effect of hypoxia on other tissues, in which RAS/MAPK signaling controls different cell fate decisions. The temperature-sensitive let-60(ga89gf) allele causes an accelerated exit of meiotic germ cells from the pachytene stage at the restrictive temperature and results in the stacking of smaller oocytes in the proximal gonad arms (Fig 1E) (Eisenmann & Kim, 1997). Furthermore, the excretory system of let-60(n1046gf) mutants frequently contains two duct cells expressing the lin-48::gfp marker (Fig 1F) (Berset et al, 2005). Hypoxia partially suppressed the let-60(gf) phenotypes in both tissues (Fig 1E and F).
The canonical hypoxia-response pathway opposes RAS/MAPK–induced VI
Because oxygen serves as an essential energy substrate, a reduction in the atmospheric oxygen concentration induces the adaptation of many metabolic processes (Semenza, 2001). In addition to these immediate metabolic changes, cells activate the hypoxia-response pathway via the hypoxia-inducible factor HIF-1 to change gene expression. Interestingly, VI was significantly reduced only below a threshold oxygen concentration between 1 and 0.5% oxygen rather than decreasing progressively along an O2 gradient (Fig 1C), suggesting the existence of a regulatory switch rather than a systemic metabolic effect of hypoxia on RAS/MAPK signaling. We, thus, examined the genetic interactions between mutations in the RAS/MAPK and hypoxia-response pathways under normoxic conditions (21% O2). For this purpose, we calculated the change in VI (∆VI) between the indicated double mutants and let-60(n1046gf) single mutant control siblings obtained from each cross (Fig 2B and C). The VI of let-60(n1046gf) mutants was reduced when combined with the egl-9(sa307lf) (Darby et al, 1999) or vhl-1(ok161lf) (Epstein et al, 2001) alleles, whereas the VI was increased in combination with the hif-1(ia04) allele (Jiang et al, 2001). Because the ia04 deletion in hif-1 does not affect all isoforms, we created a larger hif-1 deletion (zh111) that likely represents a null allele (Fig S1A). let-60(n1046gf); hif-1(zh111lf) double mutants exhibited an even stronger increase in VI (Figs 2C and S1B). Moreover, let-60(n1046gf); egl-9(sa307lf) hif-1(ia04lf) triple mutants exhibited the same increase in VI as let-60(n1046gf); hif-1(ia04lf) double mutants, indicating that hif-1 counteracts RAS/MAPK signaling downstream of egl-9 prolyl hydroxylase (Fig 2A–C). egl-9(sa307lf) also reduced the VI of mutants in other RAS/MAPK pathway components, such as the guanine nucleotide exchange factor sos-1 or the raf homolog lin-45 (Fig S2) (Sternberg, 2005).
Figure S1. Comparison of the zh111 and ia04 deletions in the hif-1 locus.

(A) Schematic representation of the hif-1 locus with the five isoforms shown in grey. The canonical deletion allele ia04 is depicted in light blue and the larger zh111 deletion is shown in dark blue. Note that the zh111 deletion affects all isoforms as opposed to ia04. (B) The zh111 allele has a stronger effect on the VI in the let-60(n1046gf) background than ia04. (B) Error bars in (B) indicate the 95% confidence intervals, and P-values, indicated with ***P < 0.001 and **P < 0.01, were derived by bootstrapping 1,000 samples.
Figure S2. Genetic interaction between egl-9(sa307lf) and RAS/MAPK pathway mutations.

∆VI indicates the change in VI of the indicated genotypes in the egl-9(sa307lf) background compared with single (or double) mutant siblings obtained from the crosses. ∆%Muv and ∆%Vul indicate the changes in the percentage of animals with VI > 3 and VI < 3, respectively. The absolute VIs of the double (or triple) mutants are shown in the rightmost column. Error bars indicate the 95% confidence intervals. P-values, indicated with **P < 0.01, were derived by bootstrapping 1,000 samples. The numbers of animals scored are indicated in brackets.
Because HIF-1 is efficiently degraded under normoxia, the increased VI caused by hif-1(lf) mutations under normoxia was somewhat surprising. It, thus, appears that low residual levels of HIF-1 present under normoxia are sufficient to oppose RAS/MAPK signaling. We, therefore, investigated the effect of wild-type and hydroxylase deficient egl-9::gfp transgenes on VI (Shao et al, 2009). A wild-type egl-9::gfp transgene efficiently rescued the reduced VI of let-60(n1046gf); egl-9(sa307lf) and further increased the VI of let-60(n1046gf) single mutants (Fig 2D). By contrast, the hydroxylase-deficient egl-9::gfp transgene had no effect on the VI in either background. We conclude that EGL-9 enhances RAS/MAPK–induced VPC differentiation by inhibiting HIF-1 protein stability, rather than by acting through a prolyl hydroxylase-independent pathway (Shao et al, 2009; Park et al, 2012). Moreover, EGL-9 enzymatic activity appears to be a rate-limiting step in the inhibition of VI by HIF-1, as the introduction of additional transgenic copies of egl-9 into an egl-9(wt) background caused a further increase in the VI. Thus, low levels of HIF-1 that persist under normoxia are sufficient to repress VI.
The hypoxia pathway modifies the response to rather than the activity of the RAS/MAPK pathway
Even though mutations in the hypoxia pathway mutants significantly affected VI in sensitized genetic backgrounds, egl-9(sa307lf),hif-1(ia04lf), or hif-1(zh111lf) single mutants raised at normoxia or hypoxia exhibited normal vulval development (VI = 3, n > 50 for each condition and genotype). Because VI is very robust even under varying environmental conditions (Félix & Barkoulas, 2012), small alterations in RAS/MAPK pathway activity caused by hypoxia or mutations in the hypoxia-response pathway may not manifest as a vulval fate change, unless the RAS/MAPK pathway is simultaneously compromised. Alternatively, the hypoxia pathway may not directly modify RAS/MAPK pathway activity but rather inhibit the response of the VPCs to RAS/MAPK signaling. To distinguish between these two scenarios, we directly measured MPK-1 activity in the VPCs of hypoxia pathway mutants at the mid-L2 stage that were grown under normoxia. For this purpose, we used a recently developed ERK-nKTR activity biosensor (de la Cova et al, 2017) (for a detailed description of the biosensor quantification, see the Materials and Methods section). All mutant backgrounds exhibited the highest MPK-1 activity levels in P6.p, as observed in wild-type controls (Fig 2E–I). Furthermore, we found no significant decrease in MPK-1 activity in P6.p of egl-9(sa307) and no increase in hif-1(ia04lf) or hif-1(zh111lf) animals relative to the wild-type controls (Fig 2J). (All P-values in a one-way ANOVA test with Dunnett’s multiple comparison correction were above 0.05.) Thus, we did not detect substantial changes in MPK-1 activity within the dynamic range of the ERK-nKTR biosensor. We conclude that the hypoxia-response pathway attenuates the response of the VPCs to MAPK signaling, rather than directly inhibiting RAS/MAPK pathway activity.
HIF-1 inhibits VI cell autonomously in the VPCs
Because HIF-1 exerts both cell autonomous and cell nonautonomous functions (Shao et al, 2010; Sendoel et al, 2010), we investigated in which tissue HIF-1 functions to inhibit VI. First, we analyzed the expression pattern of a translational hif-1::gfp reporter (Sendoel et al, 2010). The hif-1::gfp reporter was undetectable under normoxia, but visible in many somatic cells, including the VPCs and the intestinal cells in egl-9(sa307lf) or vhl-1(ok161) mutants (Fig 3A). Because we had previously described a cell nonautonomous suppression of VI via the amx-2 monoamine oxidase acting in intestinal cells (Schmid et al, 2015) and because expression of egl-9::gfp was prominent in intestinal cells, we tested a possible function of HIF-1 in the intestine and VPCs. To this aim, we performed Pn.p cell–specific and intestine-specific RNA interference using rde-1(ne219lf); let-60(n1046gf) RNAi-resistant mutants expressing rde-1(wt) from the Pn.p cell–specific lin-31 and the intestine-specific elt-2 promoters (Schmid et al, 2015). Whereas the knockdown of hif-1 in the intestinal cells did not change the VI, Pn.p cell–specific hif-1 RNAi significantly increased the VI compared with empty vector control animals (Fig 3B). It is noteworthy that, for unknown reasons, the genetic background used to establish the tissue-specific RNAi strains also affected the VI of the let-60(n1046gf) mutation, as reported previously (Schmid et al, 2015). Nevertheless, these results indicate that HIF-1 acts autonomously in the VPCs to oppose RAS/MAPK signaling during VI.
Figure 3. egl-9 is a NOTCH target that promotes HIF-1 degradation in the induced VPCs.

(A) HIF-1::GFP expression in wild-type, egl-9(sa307lf), and vhl-1(ok161lf) larvae before VI. Arrowheads point at the nuclei of the VPCs. (B) Pn.p cell– and gut-specific hif-1 RNAi (see main text and the Material and Methods section). ∆VI indicates the change in VI in hif-1 RNAi compared with empty vector–treated control animals. Error bars indicate the 95% confidence interval, and P-values were derived by bootstrapping 1,000 samples. (C) EGL-9::GFP expression in a wild-type mid-L2 larva (top panel) at the onset of VI, a late-L2/early-L3 larva before the proximal and after the distal VPCs had divided (second panel) and a mid-L3 larva after all VPCs had divided once (third panel, Pn.px stage). The arrowheads point at the VPC nuclei and the small arrows at the nuclei of ventral nerve cord neurons expressing EGL-9::GFP. Expression in let-60 ras(n2021rf), lin-12 notch(n137n720lf), and lin-12 notch(n137gf) mutants is shown in mid-L3 larvae, after the first round of VPC divisions had been completed (Pn.px stage). Induced VPCs are underlined. The scale bar represents 5 μm. (D) Schematic representation of the wild-type EGL-9::GFP pattern during vulval development. (E–G) Quantification of EGL-9::GFP expression in the four genotypes shown in (C) after VI (early to mid L3 and at the Pn.pxx stage in (G)). Error bars indicate the standard error of the mean, and P-values, indicated with ***P < 0.001 and *P < 0.05, were calculated in a two-tailed t test. The numbers of animals scored are indicated in brackets.
EGL-9 is a NOTCH target gene
Whereas hif-1::gfp reporter expression was not detectable in wild-type animals raised under normoxia (Fig 3A), the expression of the functional egl-9::gfp reporter could be observed in the VPCs and their descendants throughout vulval development (Fig 3C and D). At the onset of VI in mid L2 larvae, the egl-9::gfp reporter was expressed in all VPCs, although expression levels were slightly higher in the three proximal VPCs P5-7.p than the three distal VPCs P3.p, P4.p, and P8.p (Fig 3C, top panel). In late-L2/early-L3 stage animals, EGL-9::GFP expression remained prominent in the proximal VPCs but decreased in the distal VPCs as they started dividing (Fig 3C, second panel). After completion of the first round of VPC divisions in mid-L3 larvae, EGL-9::GFP expression faded in the 1° P6.p descendants and increased in the 2° P5.p and P7.p descendants (Fig 3C, third panel).
This expression pattern suggested that either RAS/MAPK signaling inhibits or lateral DELTA/NOTCH signaling induces EGL-9::GFP expression in the VPCs. We distinguished between these two scenarios by introducing the egl-9::gfp reporter into let-60(n2021rf), lin-12(n137n720lf), and lin-12 (n137gf) mutants and quantifying the EGL-9::GFP signal intensities. EGL-9::GFP expression was strongly reduced in let-60(n2021rf); egl-9(sa307lf) mutants (Fig 3C and E; note that for unknown reasons we only obtained viable progeny from let-60(n2021rf) mutants carrying the egl-9::gfp transgene in the egl-9(sa307lf) background). Furthermore, EGL-9::GFP expression was absent in the VPCs of lin-12(n137n720lf) mutants, whereas lin-12(n137gf) mutants showed strong and uniform expression in all VPCs (Fig 3C, F, and G). Note that, because no AC is formed in lin-12(n137gf) mutants, the RAS/MAPK is not activated in lin-12(n137gf) mutants. Furthermore, we identified eight predicted CSL (LAG-1)-binding sites (G/ATGGGAA) within a 6-kb egl-9 genomic region (Fig S3), which is a significant enrichment in CSL-binding site frequency (one site per 8 kb corresponds to a random distribution) and comparable with the known LIN-12 NOTCH target genes lip-1, lin-12, lag-1, or glp-1 (Christensen et al, 1996; Berset et al, 2001; Yoo & Greenwald, 2005). Finally, we tested if egl-9 modulates lateral NOTCH signaling strength by determining the VI of lin-12; egl-9(sa307lf) double mutants. The egl-9(sa307lf) allele neither affected the VI of lin-12(n137n720lf) nor of lin-12(n137gf) mutants (Fig S4).
Figure S3. Enrichment of predicted LAG-1–binding sites in the egl-9 locus.

(A) Predicted CSL-binding sites defined by the RTGGGAA motif in the egl-9 genomic locus. (B) The location of each CSL site is indicated relative to the transcriptional start site and the consensus sequence is present either in the direction of transcription (“+”) or against (“−”).
Figure S4. EGL-9 does not modulate LIN-12 NOTCH signaling.
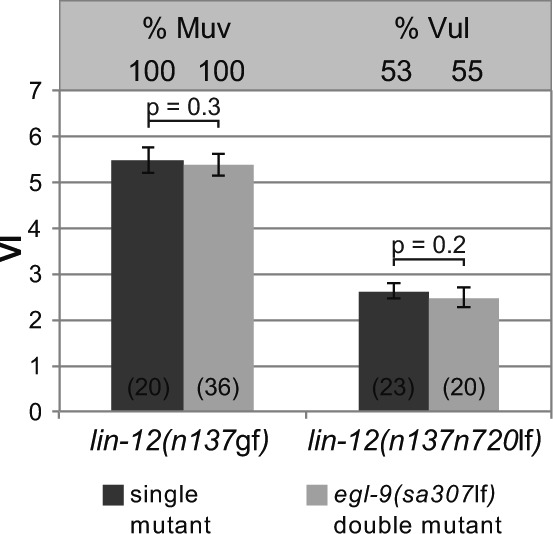
The absolute VIs of lin-12(n137gf) and lin-12(n137n720lf) mutants in the egl-9(sa307lf) background compared with egl-9(+) single mutant siblings obtained from the crosses are shown. %Muv and %Vul indicate the percentage of animals with VI > 3 and VI < 3, respectively. Error bars indicate the 95% confidence intervals. P-values were derived by bootstrapping 1,000 samples. The numbers of animals scored are indicated in brackets.
Taken together, we conclude that EGL-9 does not regulate LIN-12 NOTCH signaling but rather acts downstream of LIN-12 NOTCH to down-regulate HIF-1. LIN-12 up-regulates EGL-9::GFP expression independently of RAS/MAPK signaling, and the enrichment of CSL-binding sites in the egl-9 locus suggests that egl-9 may be a direct LIN-12 NOTCH target gene. Because RAS/MAPK signaling induces the expression of the DSL NOTCH ligands (Chen & Greenwald, 2004) (Fig 1A), inhibiting RAS/MAPK signaling most likely reduces EGL-9::GFP expression indirectly via the loss of NOTCH activation.
The HIF-1 target gene nhr-57 inhibits VI under normoxia
HIF-1 up-regulates the expression of numerous target genes under hypoxia and induces multiple cellular responses (Semenza, 2001). Previous studies have identified a large number of HIF-1 regulated genes in C. elegans (Bishop et al, 2004; Shen et al, 2005). We used the available expression data to screen for genes that act downstream of HIF-1 and negatively regulate VI. We selected a total of 80 predicted HIF-1 target genes that exhibited an at least twofold induction by HIF-1 and performed RNAi in let-60(n1046gf); egl-9(sa307lf) and let-60(n1046gf); hif-1(zh111lf) double mutants grown under normoxia (Tables S1 and S2). We used these two double-mutant backgrounds to distinguish between RAS/MAPK pathway modifiers that are partially or fully HIF-1 dependent. We hypothesized that the knockdown of a gene that inhibits VI and depends on HIF-1 for its expression should increase the VI of let-60(n1046gf); egl-9(sa307lf) mutants in which HIF-1 is stabilized. However, knockdown of the same gene in let-60(n1046gf); hif-1(zh111lf) mutants should not increase the VI because this gene would not be expressed in the absence of HIF-1.
Based on these criteria, we identified a single HIF-1 target gene, nhr-57, as a negative regulator of RAS/MAPK-induced VPC differentiation (Fig 4A and Table S1). nhr-57 encodes a nuclear hormone receptor similar to the Drosophila estrogen-related receptor ERR2 and the vertebrate glucocorticoid hormone receptor NR3C1 (Shaye & Greenwald, 2011). We next used the nhr-57(tm4533lf) deletion allele to confirm that NHR-57 acts as an inhibitor of VPC differentiation. The VI of let-60(n1046gf); hif-1(zh111lf); nhr-57(tm4533lf) triple mutants was not significantly different from let-60(n1046gf); hif-1(zh111lf); nhr-57(+) control siblings (Fig 4B). However, the nhr-57(tm4533lf) allele increased the VI of let-60(n1046gf); egl-9(sa307lf) double and of let-60(n1046gf) single mutants compared with the corresponding nhr-57(+) control siblings.
Figure 4. The HIF-1 target NHR-57 inhibits RAS/MAPK signaling under normoxia and hypoxia.

(A) Knock-down of nhr-57 increases the VI of let-60(n1046gf) in an egl-9(sa307lf) but not in a hif-1(zh111lf) background. ∆VI and ∆%Muv indicate the changes in VI and percentage of animals with VI > 3 after nhr-57 RNAi compared with empty vector–treated control animals. (B) Deletion of the nhr-57(tm4533lf) allele increases the VI of let-60(n1046gf) single and let-60(n1046gf); egl-9(sa307lf) double mutants but has no effect in a let-60(n1046gf); hif-1(zh111lf) background. ∆VI and ∆%Muv indicate the changes in VI and the percentage of animals with VI > 3 compared with nhr-57(+) control siblings. (C) Hypoxic treatment decreases the VI in the hif-1(ia04lf), hif-1(zh111lf), and egl-9(sa307lf) hif-1(ia04lf) but not the nhr-57(tm4533lf) background. ∆VI and ∆%Muv indicate the changes in VI and percentage of animals with VI > 3 raised in hypoxia compared with normoxia. Error bars indicate 95% confidence intervals, and P-values, indicated with ***P < 0.001 and **P < 0.01, were derived by bootstrapping 1,000 samples. The numbers of animals scored are indicated in brackets. (D) NHR-57::GFP expression pattern in the wild-type (left panels) and egl-9(sa307lf) (right panels) background in late-L2/early-L3 larvae at the Pn.p stage (top panels), in mid L3 larvae (Pn.px stage) after induction (middle panels), and at the end of vulval differentiation (bottom panels). Arrowheads and solid lines point at the nuclei of the induced or uninduced VPCs and their descendants, respectively. The scale bar represents 5 μm. (E) Schematic representation of the NHR-57::GFP pattern during vulval development in the wild-type (left) and in an egl-9(sa307lf) background (right).
We next assessed the expression pattern of NHR-57 by constructing a translational nhr-57::gfp reporter transgene. The reporter includes the complete nhr-57 promoter/enhancer region and contains the predicted HIF-1–binding sites (Miyabayashi et al, 1999; Shen et al, 2006). In animals raised under normoxia, the nhr-57::gfp reporter was weakly expressed in several somatic cells including the intestinal cells, but not in the VPCs before VI. After the first round of VPC divisions in mid-L3 larvae, nhr-57::gfp expression became visible in the descendants of the uninduced distal VPCs but remained undetectable in the descendants of the induced proximal VPCs until they had completed their last round of division and terminally differentiated (Fig 4D and E). Consistent with its function as an HIF-1 target, NHR-57::GFP expression was up-regulated in the VPCs of egl-9(sa307lf) mutants with the signal being most prominent in the descendants of uninduced VPCs (Fig 4D and E). Taken together, we propose that LIN-12 NOTCH signaling induces EGL-9 expression in the proximal VPCs to counteract the inhibitory effect of HIF-1 and its target NHR-57 on VPC fate specification.
NHR-57 mediates the HIF-1–dependent and HIF-1–independent hypoxic inhibition of VI
Our results so far have suggested that under normoxic conditions, EGL-9 induces HIF-1 degradation to prevent the HIF-1 target NHR-57 from inhibiting vulval differentiation (Fig 4A and B). However, HIF-1–independent factors also play a role in the transcriptional response to hypoxia (Shen et al, 2005; Padmanabha et al, 2015). We, thus, investigated if the inhibitory effect of hypoxia on vulval development depends entirely on HIF-1 activity. Surprisingly, the VI of let-60(n1046gf); hif-1(lf) double and let-60(n1046gf); egl-9(sa307lf) hif-1(ia04lf) triple mutants was significantly decreased under hypoxia (Fig 4C). Therefore, the reduction in VI under hypoxia only partially depends on HIF-1.
NHR-57 appeared to be a good candidate for a factor that mediates the HIF-1–independent effect of hypoxia for a number of reasons. First, the translational nhr-57::gfp reporter was expressed under normoxia in cells that also expressed the egl-9::gfp reporter. Second, nhr-57::gfp expression in the VPCs of egl-9(sa307lf) mutants that uniformly express hif-1::gfp (Fig 3A) was not uniform (Fig 4D), pointing at a regulation of nhr-57 expression by additional factors besides HIF-1. Third, the transcription of nhr-57 was significantly enhanced under hypoxia even in the absence of HIF-1 activity (Bishop et al, 2004). Together, these findings suggested that NHR-57 integrates multiple inputs together with the HIF-1–mediated hypoxic response. In contrast to let-60(n1046gf); hif-1(lf) mutants, the VI of let-60(n1046gf); nhr-57(tm4533lf) double mutants did not change under hypoxia (Fig 4C). Thus, NHR-57 is necessary for both the HIF-1–dependent and HIF-1–independent repression of VI by hypoxia.
Discussion
We have found that different tissues in C. elegans can modulate their cellular responses to RAS/MAPK signaling according to the availability of oxygen. Under limiting oxygen concentrations, energetically demanding processes that are dispensable for viability, such as oogenesis or VI, may be suppressed to preserve essential cellular functions. By connecting the cellular hypoxia response to an essential developmental signaling pathway, the animals can adapt their development to fluctuations in oxygen concentration. In wild-type animals raised under the optimal laboratory growth conditions, the effect of hypoxia does not manifest as a change in cell fate specification in the tissues we examined. The effect of hypoxia only becomes apparent if the RAS/MAPK pathway has been sensitized. However, the hypoxic effect may become relevant when animals in the wild encounter stressful conditions, in which developmental robustness is challenged. The capability to adapt to varying oxygen concentrations could increase developmental robustness.
The CB4856 Hawaii wild isolate exhibits overall higher levels of VI than the N2 Bristol reference laboratory strain when grown under normoxia (Milloz et al, 2008; Schmid et al, 2015) as well as under mild hypoxic conditions (>3% O2) (this study). Strong hypoxia (<1% O2) not only reduces vulval differentiation to a baseline level but also eliminates the difference between the two strains. Thus, differences in their hypoxia response may account for some of the differences in VI between the Bristol N2 and Hawaii CB4856 strains. Most wild C. elegans isolates, including CB4856, are adapted to lower oxygen levels and aggregate when grown under normoxia (Hodgkin & Doniach, 1997; Rogers et al, 2006). Aggregation is often followed by burrowing, which may further decrease the oxygen concentrations the animals experience. By contrast, the laboratory strain N2 Bristol does not aggregate because it has become habituated to atmospheric oxygen concentrations during the prolonged laboratory cultivation (de Bono & Bargmann, 1998). Thus, in wild strains, the cellular sensitivity to RAS/MAPK signaling may be elevated to allow development under lower oxygen levels.
We next focused on the process of vulval fate specification to dissect how the hypoxia-response and RAS/MAPK signaling pathways interact at the molecular level. Because the activity of the prolyl hydroxylase EGL-9 is a rate-limiting step in the HIF-1 degradation pathway (Fig 2D), the hypoxia-response pathway exerts its inhibitory effect even under normoxia. This observation allowed us to investigate the cross-talk between the hypoxia and RAS/MAPK pathways under standard growth conditions. By screening a selection of candidate genes regulated by HIF-1, we have identified the nuclear hormone receptor NHR-57 as a key downstream integrator of both the HIF-1–dependent and HIF-1–independent pathways (Fig 5). Because we did not detect significant changes in MAPK biosensor activity in hif-1(lf) or egl-9(lf) mutants, the hypoxia-response pathway and NHR-57 must act downstream of or in parallel with the RAS/MAPK pathway. NHR-57 may interact with the ETS family transcription factor LIN-1, which is an MAPK substrate that represses VPC differentiation when the RAS/MAPK pathway is inactive (Beitel et al, 1995; Jacobs et al, 1998; Tan et al, 1998). Consistent with this hypothesis, multiple genetic and physical interactions between ETS family transcription factors and steroid hormone receptors have been reported in other systems (Mullick et al, 2001; Geng & Vedeckis, 2005; Kalet et al, 2013; Cao et al, 2015). Among the steroid hormone receptors, the glucocorticoid receptor NR3C1 exhibits closest homology to NHR-57 (Shaye & Greenwald, 2011). Also, NR3C1 accumulates under hypoxia to promote erythropoiesis (Bauer et al, 1999; Mense, 2006). Although NHR-57 is a critical HIF-1 target that counteracts RAS/MAPK signaling in the VPCs, NHR-57 expression appears to be regulated by additional factors besides HIF-1 (Bishop et al, 2004; Padmanabha et al, 2015). Similarly, the expression of the mammalian glucocorticoid receptor NR3C1 is not only regulated by hypoxia but also by serotonin (5-HT) (Mitchell et al, 1990). It thus seems likely that nhr-57 expression is controlled by several additional cues besides the HIF-1–induced up-regulation under hypoxia.
Figure 5. EGL-9 inhibits the HIF-1 target NHR-57 from reducing RAS/MAPK signaling through a DELTA/NOTCH-induced negative feedback loop.

(A) Model illustrating the cross-talk of the hypoxia-response, DELTA/NOTCH, and RAS/MAPK pathways at normoxia. At the onset of VI, before a distinction between 1° and 2° VPCs is made, lateral DELTA/NOTCH signaling between the proximal VPCs induces EGL-9 expression to maintain low HIF-1 and NHR-57 levels, thereby keeping the VPCs competent to respond to RAS/MAPK signaling. RAS/MAPK signaling, in turn, promotes the expression of the DELTA family NOTCH ligands. Distal VPCs lose their competence because of higher NHR-57 levels and adopt the 3° fate. Thereafter, NOTCH signaling directly induces the 2° fate and inhibits RAS/MAPK signaling in P5.p and P7.p. (B) Under hypoxia, the inhibition of HIF-1 and NHR-57 by EGL-9 is reduced because of the lack of oxygen and vulval fate acquisition is compromised. An as-of-yet unidentified factor activates NHR-57 during hypoxia independently of HIF-1.
Interestingly, the regulation of egl-9 expression by NOTCH signaling provides a link between the VPC fate specification and the hypoxia-response pathways. This link could be used to modulate the sensitivity of the VPCs to the inductive signal according to oxygen concentrations. At the beginning of VI, EGFR/RAS/MAPK signaling is predominantly activated in the proximal VPCs (P5.p, P6.p, and P7.p) by the LIN-3 EGF signal secreted from the AC. EGFR/RAS/MAPK signaling then up-regulates the expression of multiple DELTA family ligands in P6.p to activate lateral NOTCH signaling in the neighboring VPCs P5.p and P7.p and focus the inductive signal on P6.p, the future 1° VPC (Fig 1A) (Berset et al, 2001; Yoo et al, 2004; Sternberg, 2005). However, in early L3 larvae, both the NOTCH ligand LAG-2 and its receptor LIN-12 are expressed in all VPCs (Chen & Greenwald, 2004; Levitan & Greenwald, 1998). Only after VI before the VPCs divide at the mid-L3 stage, LAG-2 expression becomes restricted to P6.p, whereas LIN-12 levels decrease in P6.p. Thus, reciprocal LIN-12 NOTCH signaling between the VPCs during an early phase of vulval fate specification may induce EGL-9 expression to reduce HIF-1 and NHR-57 levels and keep the proximal VPCs competent to differentiate (Fig 5A). In line with this model, NOTCH signaling has previously been shown to maintain the VPCs competent to respond to the inductive LIN-3 EGF signal even after the VPCs have started dividing (Wang & Sternberg, 1999; Nusser-Stein et al, 2012). The lack of inductive EGFR/RAS/MAPK and lateral NOTCH signaling in the distal uninduced VPCs and their descendants results in higher NHR-57 levels, which reduce their capability to differentiate. However, under hypoxic conditions, the elevated NHR-57 levels may render all VPCs less responsive to the inductive LIN-3 EGF signal (Fig 5B).
The activity of the NOTCH signaling pathway and short-term hypoxic periods involving activation of the hypoxia-response pathway are important during normal animal development (Patterson & Zhang, 2010; Siebel & Lendahl, 2017). To our knowledge, prolyl hydroxylases have so far not been implicated in RAS/MAPK or DELTA/NOTCH–mediated cell fate decisions in other animals. However, a dependence of mammalian EGLN3 hypoxia-inducible factor prolyl hydroxylase transcription downstream of NOTCH has been reported (Ohashi et al, 2011; Li et al, 2012). It remains to be investigated whether these two signaling pathways are interconnected in other organisms in a similar manner as in C. elegans to couple cell fate decision with oxygen availability.
Materials and Methods
C. elegans alleles
C. elegans strains were maintained at 20°C using standard procedures (Brenner, 1974) except N2 strains carrying the let-60(ga89) allele that were maintained at 20°C and shifted to 25°C for mutant analysis. C. elegans Bristol refers to the wild-type N2 strain and Hawaii to CB4856 (Erik C Andersen, 2015). The transgenic lines were generated as described in (Mello et al, 1991). The following mutations and transgenes were used in this study: LGII: let-23(sy1); LG III: unc-32(e189) lin-12(n137n720), dpy-19(e1259) lin-12(n137), hT2[bli-4(e937) let-?(q782) qIs48] (I;III). LG IV: let-60(n1046), let-60(zh121) (this study), let-60(zh122) (this study), let-60(ga89), let-60(n2021), lin-1(n304), lin-3(e1417), lin-45(sy96). LG V: egl-9(sa307), hif-1(ia04), hif-1(zh111) (this study), nhr-57(tm4533) (Mitani lab). LG X: vhl-1(ok161), rde-1(ne219), bar-1(ga80). Transgenes: gaIs37[lin-31::mpk-1(gf); lin-31::D-mek(gf)] (Lackner & Kim, 1998), iaIs38[egl-9p::egl-9::tag, unc-119(+)] (Shao et al, 2009), iaEx101[egl-9p::egl-9(H487A)::tag, unc-119(+)] (Shao et al, 2009), duIs?[elt-2p::rde-1(+); pRF4], zhEx418[lin-31p::rde-1;myo-2::mCherry], saIs14[lin-48p::gfp], opIs206[hif-1p::hif-1::gfp::hif-1 3′UTR, unc-119(+)] (Sendoel et al, 2010), zhEx605[nhr-57p::nhr-57::gfp:: nhr-57 3′UTR, unc-119(+)] (this study), arTi85[lin-31p::ERK-KTR(NLS3)-mClover-T2A-mCherry-H2B::unc-54 3′UTR, rps-27p::NeoR::unc-54 3′UTR] (de la Cova et al, 2017).
Detailed information on the alleles used can be found under www.wormbase.org.
A list of all strains used can be found in Table S3.
Table S3 List of strains used. (63.6KB, pdf)
RNAi experiments
RNAi was performed as described in (Kamath et al, 2003). Larvae were synchronized by hypochloride treatment of gravid adults and raised on bacteria expressing dsRNA. The empty vector plasmid L4440 was used as a negative control and dsRNA against rpn-6.1 as a positive control. VI was scored in the F1 generation. Animals were anesthetized in in a drop of 5 mM tetramisole and analyzed using differential interference contrast and fluorescence microscopy.
Staging of the animals used for microscopy analysis
Synchronized population of L1 larvae were obtained by hypochloride treatment of gravid adults and letting the embryos hatch in S-basal buffer without OP50 bacteria for 16–20 h. Arrested L1 larvae were then fed with OP50 bacteria until they reached the stage to be analyzed. The developmental stages were further distinguished using Nomarski image stacks taken along with the fluorescent images by scoring VPC divisions and the dorsal turning of the distal tip cell (DTC) and by measuring the gonad length as described (Kimble & Hirsh, 1979). Developmental stages were assigned as follows: mid-L2 stage (gonad: 35–75 μm, VPCs undivided, and DTC not turned), late-L2 stage (gonad: 75–110 μm and DTC not turned), early-L3 stage (gonad: >110 μm and DTC begins to turn), and mid-L3 stage (gonad: >110 μm, all VPCs divided once, and DTC turned).
VI counts
VI was scored by examining worms at the L4 stage under Nomarski optics as described in (Sternberg & Horvitz, 1986). The number of VPCs that had adopted a 1° or 2° vulval cell fate was counted for each animal, and the VI index was calculated by dividing the total number of induced cells by the number of animals scored. Animals with a VI > 3 were scored as hyperinduced and animals with VI < 3 as hypoinduced. The different allele combinations compared were generated from progeny obtained in the same crosses. The raw data used to compile the induction graphs shown in the figures can be found in Table S4.
Table S4 Complete dataset of VI counts. (148.1KB, xlsx)
ERK-nKTR biosensor quantification
MPK-1 activity in the VPCs was measured in mid-L2 larvae (Pn.p stage) using the recently established ERK-nKTR biosensor (arTi85), which is based on the MPK-1 activity-dependent nuclear export of the biosensor (de la Cova et al, 2017). Images were recorded on a Leica wide-field microscope equipped with a piezo focus drive, a beam splitter, and two scientific Complementary metal–oxide–semiconductor cameras to simultaneously record the mCherry and mClover signals. We used custom-made ImageJ and CellProfiler scripts provided in the Supplemental Information to process and quantify the images taken in the indicated mutant backgrounds under standardized illumination conditions. Flat field illumination and background corrections were done using blank and dark-field images obtained for every experiment. The nuclear RED/GREEN (mCherry::H2B/nKTR::mClover) average intensity ratios were measured in each VPC (except for P3.p) in summed z-projections of the five central slices (z-spacing of 0.2 μm) relative to the focus of the nuclear mCherry::H2B signal, using a modified Cell Profiler script as described by de la Cova et al (2017). For each VPC, the RED/GREEN nuclear ratio was normalized to the mean of the RED/GREEN ratios in all VPCs in the same animal, and these values are shown in the graphs of Fig 2. Statistical analysis was carried out with one-way ANOVA followed by Dunnett’s multiple comparison correction.
LSA-2018-00255_Supplemental_Data_1.txt (4.3KB, txt)
LSA-2018-00255_Supplemental_Data_2.txt (10.6KB, txt)
Hypoxic treatment
Hypoxic chambers were adapted from Fawcett et al (2012) as follows. Briefly, a glass bowl with flange served as container for small Nematode Growth Medium plates. A customized lid was made, which consists of an acrylic glass plate surrounded by a metal ring equipped with an indentation to fit in an O-ring. Two holes in the acrylic glass served as the gas in- and output apertures. A third larger hole accommodates an oxygen measuring device (GOX 100). A hypoxic environment was generated via oxygen replacement by N2 until the desired O2 concentration was obtained. Nematode Growth Medium plates containing mixed-stage C. elegans were incubated for 3 to 5 d and analyzed at the microscope.
Generation of the hif-1(zh111) null allele
CRISPR/CAS9 was performed as described in Friedland et al (2013), Arribere et al (2014). 25 ng/μl dpy-10 sgRNA pJA58 was co-injected into N2 Bristol animals with 25 ng/μl of a sgRNA targeting hif-1 (GATAGAAAAGTGAGTCCTAA), 500 nM of dpy-10(cn64) repair template AF-ZF-827, and 50 ng/μl of pDD162. Candidate worms were selected from plates harboring a large number of animals showing a roller phenotype progeny and analyzed by PCR. The zh111 deletion spans 2,464 bp and removes three to five exons (region upstream: ATTTCAAAAAATTTTTGACA; region downstream: CTAAGTTAAAAAAACAACAG).
Generation of let-60(zh121) and let-60(zh122)
The sgRNA sequence AATGACGGAGTACAAGCTTG was inserted into plasmid #46169 (PU6::unc-119_sgRNA) via site-directed mutagenesis. The let-60 genomic locus was partially amplified using the primers AAGGAGCAAATCGAACAGAC and ATCCATTTTATTAGGCACGCAC and cloned into p-GEM-T easy. The silently mutated PAM and the G13E mutation were obtained using the primers TGAGTGCTGATTTACCAACTCCTCCATCTCCAACTACCACAAGCTTGTACTCCGTC and AAGCTTGTGGTAGTTGGAGATGGAGGAGTTGGTAAATCAGCACTCACCATTCAACTCATC. N2 Bristol or CB4856 Hawaii animals were injected with 50 ng/μl sgRNA plasmid, 50 ng/μl pDD162, 50 ng/μl repair template, and 5 ng/μl pCFJ104. The progeny was screened for the multivulva phenotype, and candidate animals were verified by PCR and sequencing.
Translational nhr-57 reporter
A 1,206-bp fragment upstream of the nhr-57 transcriptional start site was amplified together with the coding genomic region (without the stop codon) using the primers ctaacaacttggaaatgaaatCACCAACACCTTCTACACAGCTGC and gttcttctcctttactcatTTGTCCATCAA-TGATTTTATAGATTTTGTCG. The gfp sequence was amplified from pPD95.75 using the primers ATGAGTAAAGGAGAAGAAC and gagatctggttcaaatagCTATTTGTATAGTTCATCCATGC. 459 bp of the nhr-57 3′ UTR were amplified using the primers GCTATTTGAACCAGATCTCTTC and cagtacggccgactagtagGAATAAATCATCCCAAAGCCGTTTTTG. A vector backbone containing a CB-unc-119(+) rescue as well as the AmpR gene was amplified using the primers ATTTCATTTCCAAGTTGTTAGCG and CTACTAGTCGGCCGTACTGAGGTGTTGTCGCTT-TTATTGGG. The four fragments were combined into a 10.170-kb plasmid (pSMa32) using Gibson Assembly. N2 animals were injected (because of mutation in the CB-unc-119(+) gene) with 5 ng/μl pSMa32, 2.5 ng/μl pCFJ104 and 100 ng/μl pBS. Transformants were isolated based on the presence of the co-injection marker.
Supplementary Material
Acknowledgements
We wish to thank the members of the Hajnal laboratory, Jan Kammenga, Beatrice Beck-Schimmer, and Konrad Basler for critical discussion and comments on the manuscript, and R Maier and D Schnarwiler for their help in designing and manufacturing the hypoxic chambers. We are also grateful to the C. elegans Genetics Center CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440), and the Mitani lab (National Bioresource Project) for providing some strains, Andrew Fire for GFP vectors, and J Ahringer for RNAi clones. This work was supported by a grant from the Swiss National Science Foundation to A.H. no. 31003A-166580 and the Kanton of Zürich.
Author Contributions
S Maxeiner: conceptualization, data curation, formal analysis, supervision, investigation, and writing—original draft.
J Grolleman: validation and investigation.
T Schmid: conceptualization, resources, data curation, and investigation.
J Kammenga: funding acquisition and project administration.
A Hajnal: conceptualization, data curation, formal analysis, supervision, funding acquisition, investigation, project administration, and writing—review and editing.
Conflict of Interest Statement
The authors declare that they have no conflict of interest.
References
- Arribere JA, Bell RT, Fu BX, Artiles KL, Hartman PS, Fire AZ (2014) Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics 198: 837–846. 10.1534/genetics.114.169730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen EC, Shimko TC, Crissman JR, Ghosh R, Bloom JS, Seidel HS, Gerke JP, Kruglyak L (2015) A powerful new quantitative genetics platform, combining Caenorhabditis elegans high-throughput fitness assays with a large collection of recombinant strains. G3 (Bethesda) 5: 911–920. 10.1534/g3.115.017178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer A, Tronche F, Wessely O, Kellendonk C, Reichardt HM, Steinlein P, Schütz G, Beug H (1999) The glucocorticoid receptor is required for stress erythropoiesis. Genes Dev 13: 2996–3002. 10.1101/gad.13.22.2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beitel GJ, Clark SG, Horvitz HR (1990) Caenorhabditis elegans ras gene let-60 acts as a switch in the pathway of vulval induction. Nature 348: 503–509. 10.1038/348503a0 [DOI] [PubMed] [Google Scholar]
- Beitel GJ, Tuck S, Greenwald I, Horvitz HR (1995) The Caenorhabditis elegans gene lin-1 encodes an ETS-domain protein and defines a branch of the vulval induction pathway. Genes Dev 9: 3149–3162. 10.1101/gad.9.24.3149 [DOI] [PubMed] [Google Scholar]
- Berset T, Hoier EF, Battu G, Canevascini S, Hajnal A (2001) Notch inhibition of RAS signaling through MAP kinase phosphatase LIP-1 during C. elegans vulval development. Science 291: 1055–1058. 10.1126/science.1055642 [DOI] [PubMed] [Google Scholar]
- Berset TA, Hoier EF, Hajnal A (2005) The C. elegans homolog of the mammalian tumor suppressor Dep-1/Scc1 inhibits EGFR signaling to regulate binary cell fate decisions. Genes Dev 19: 1328–1340. 10.1101/gad.333505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop T, Lau KW, Epstein ACR, Kim SK, Jiang M, O’Rourke D, Pugh CW, Gleadle JM, Taylor MS, Hodgkin J, et al. (2004) Genetic analysis of pathways regulated by the von Hippel-Lindau tumor suppressor in Caenorhabditis elegans. PLoS Biol 2: e289 10.1371/journal.pbio.0020289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. (1974) The genetics of Caenorhabditis elegans. Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P, Feng F, Dong G, Yu C, Feng S, Song E, Shi G, Liang Y, Liang G (2015) Estrogen receptor α enhances the transcriptional activity of ETS-1 and promotes the proliferation, migration and invasion of neuroblastoma cell in a ligand dependent manner. BMC Cancer 15: 491 10.1186/s12885-015-1495-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Greenwald I (2004) The lateral signal for LIN-12/Notch in C. elegans vulval development comprises redundant secreted and transmembrane DSL proteins. Dev Cell 6: 183–192. 10.1016/s1534-5807(04)00021-8 [DOI] [PubMed] [Google Scholar]
- Christensen S, Kodoyianni V, Bosenberg M, Friedman L, Kimble J (1996) lag-1, a gene required for lin-12 and glp-1 signaling in Caenorhabditis elegans, is homologous to human CBF1 and Drosophila Su(H). Development 122: 1373–1383. [DOI] [PubMed] [Google Scholar]
- Darby C, Cosma CL, Thomas JH, Manoil C (1999) Lethal paralysis of Caenorhabditis elegans by Pseudomonas aeruginosa. Proc Natl Acad Sci USA 96: 15202–15207. 10.1073/pnas.96.26.15202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono M, Bargmann CI (1998) Natural variation in a neuropeptide Y receptor homolog modifies social behavior and food response in C. elegans. Cell 94: 679–689. 10.1016/s0092-8674(00)81609-8 [DOI] [PubMed] [Google Scholar]
- de la Cova de C, Townley R, Regot S, Greenwald I (2017) A real-time biosensor for ERK activity reveals signaling dynamics during C. elegans cell fate specification. Dev Cell 42: 542–553.e4. 10.1016/j.devcel.2017.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenmann DM, Kim SK (1997) Mechanism of activation of the Caenorhabditis elegans ras homologue let-60 by a novel, temperature-sensitive, gain-of-function mutation. Genetics 146: 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein ACR, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, et al. (2001) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107: 43–54. 10.1016/s0092-8674(01)00507-4 [DOI] [PubMed] [Google Scholar]
- Fawcett EM, Horsman JW, Miller DL (2012) Creating defined gaseous environments to study the effects of hypoxia on C. elegans. J Vis Exp: e4088 10.3791/4088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félix M-A, Barkoulas M (2012) Robustness and flexibility in nematode vulva development. Trends Genet 28: 185–195. 10.1016/j.tig.2012.01.002 [DOI] [PubMed] [Google Scholar]
- Friedland AE, Tzur YB, Esvelt KM, Colaiácovo MP, Church GM, Calarco JA (2013) Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods 10: 741–743. 10.1038/nmeth.2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng CD, Vedeckis WV (2005) c-Myb and members of the c-Ets family of transcription factors act as molecular switches to mediate opposite steroid regulation of the human glucocorticoid receptor 1A promoter. J Biol Chem 280: 43264–43271. 10.1074/jbc.m508245200 [DOI] [PubMed] [Google Scholar]
- Gort EH, van Haaften G, Verlaan I, Groot AJ, Plasterk RHA, Shvarts A, Suijkerbuijk KPM, van Laar T, van der Wall E, Raman V, et al. (2007) The TWIST1 oncogene is a direct target of hypoxia-inducible factor-2α. Oncogene 27: 1501–1510. 10.1038/sj.onc.1210795 [DOI] [PubMed] [Google Scholar]
- Gray JM, Karow DS, Lu H, Chang AJ, Chang JS, Ellis RE, Marletta MA, Bargmann CI (2004) Oxygen sensation and social feeding mediated by a C. elegans guanylate cyclase homologue. Nature 430: 317–322. 10.1038/nature02714 [DOI] [PubMed] [Google Scholar]
- Hodgkin J, Doniach T (1997) Natural variation and copulatory plug formation in Caenorhabditis elegans. Genetics 146: 149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Guo R, Powell-Coffman JA (2001) The Caenorhabditis elegans hif-1 gene encodes a bHLH-PAS protein that is required for adaptation to hypoxia. Proc Natl Acad Sci USA 98: 7916–7921. 10.1073/pnas.141234698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs D, Beitel GJ, Clark SG, Horvitz HR, Kornfeld K (1998) Gain-of-function mutations in the Caenorhabditis elegans lin-1 ETS gene identify a C-terminal regulatory domain phosphorylated by ERK MAP kinase. Genetics 149: 1809–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalet BT, Anglin SR, Handschy A, O’Donoghue LE, Halsey C, Chubb L, Korch C, Duval DL (2013) Transcription factor Ets1 cooperates with estrogen receptor α to stimulate estradiol-dependent growth in breast cancer cells and tumors. PLoS One 8: e68815 10.1371/journal.pone.0068815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. (2003) Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421: 231–237. 10.1038/nature01278 [DOI] [PubMed] [Google Scholar]
- Kimble J, Hirsh D (1979) The postembryonic cell lineages of the hermaphrodite and male gonads in Caenorhabditis elegans. Dev Biol 70: 396–417. 10.1016/0012-1606(79)90035-6 [DOI] [PubMed] [Google Scholar]
- Lackner M, Kim S (1998) Genetic analysis of the Caenorhabditis elegans MAP kinase gene mpk-1. Genetics 150: 103–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan D, Greenwald I (1998) LIN-12 protein expression and localization during vulval development in C. elegans. Development 125: 3101–3109. [DOI] [PubMed] [Google Scholar]
- Li Y, Hibbs MA, Gard AL, Shylo NA, Yun K (2012) Genome-wide analysis of N1ICD/RBPJ targets in vivo reveals direct transcriptional regulation of Wnt, SHH, and hippo pathway effectors by Notch1. Stem Cells 30: 741–752. 10.1002/stem.1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V (1991) Efficient gene transfer in C.elegans: Extrachromosomal maintenance and integration of transforming sequences. EMBO J 10: 3959–3970. 10.1002/j.1460-2075.1991.tb04966.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mense SM. (2006) Gene expression profiling reveals the profound upregulation of hypoxia-responsive genes in primary human astrocytes. Physiol Genomics 25: 435–449. 10.1152/physiolgenomics.00315.2005 [DOI] [PubMed] [Google Scholar]
- Milloz J, Duveau F, Nuez I, Félix MA (2008) Intraspecific evolution of the intercellular signaling network underlying a robust developmental system. Genes Dev 22: 3064–3075. 10.1101/gad.495308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JB, Iny LJ, Meaney MJ (1990) The role of serotonin in the development and environmental regulation of type II corticosteroid receptor binding in rat hippocampus. Brain Res Dev Brain Res 55: 231–235. 10.1016/0165-3806(90)90204-c [DOI] [PubMed] [Google Scholar]
- Miyabayashi T, Palfreyman MT, Sluder AE, Slack F, Sengupta P (1999) Expression and function of members of a divergent nuclear receptor family in Caenorhabditis elegans. Dev Biol 215: 314–331. 10.1006/dbio.1999.9470 [DOI] [PubMed] [Google Scholar]
- Mullick J, Anandatheerthavarada HK, Amuthan G, Bhagwat SV, Biswas G, Camasamudram V, Bhat NK, Reddy SEP, Rao V, Avadhani NG (2001) Physical interaction and functional synergy between glucocorticoid receptor and Ets2 proteins for transcription activation of the rat cytochrome P-450c27 promoter. J Biol Chem 276: 18007–18017. 10.1074/jbc.m100671200 [DOI] [PubMed] [Google Scholar]
- Nusser-Stein S, Beyer A, Rimann I, Adamczyk M, Piterman N, Hajnal A, Fisher J (2012) Cell-cycle regulation of NOTCH signaling during C. elegans vulval development. Mol Syst Biol 8: 618 10.1038/msb.2012.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi S, Natsuizaka M, Naganuma S, Kagawa S, Kimura S, Itoh H, Kalman RA, Nakagawa M, Darling DS, Basu D, et al. (2011) A NOTCH3-mediated squamous cell differentiation program limits expansion of EMT-competent cells that express the ZEB transcription factors. Cancer Res 71: 6836–6847. 10.1158/0008-5472.can-11-0846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabha D, Padilla PA, You YJ, Baker KD (2015) A HIF-independent mediator of transcriptional responses to oxygen deprivation in Caenorhabditis elegans. Genetics 199: 739–748. 10.1534/genetics.114.173989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EC, Ghose P, Shao Z, Ye Q, Kang L, Xu XZ, Powell-Coffman JA, Rongo C (2012) Hypoxia regulates glutamate receptor trafficking through an HIF-independent mechanism. EMBO J 31: 1379–1393. 10.1038/emboj.2011.499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson AJ, Zhang L (2010) Hypoxia and fetal heart development. Current Mol Med 10: 653 10.2174/156652410792630643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers C, Persson A, Cheung B, De Bono M (2006) Behavioral motifs and neural pathways coordinating O2 responses and aggregation in C. elegans. Curr Biol 16: 649–659. 10.1016/j.cub.2006.03.023 [DOI] [PubMed] [Google Scholar]
- Schmid T, Snoek LB, Fröhli E, van der Bent ML, Kammenga J, Hajnal A (2015) Systemic regulation of RAS/MAPK signaling by the serotonin metabolite 5-HIAA. PLoS Genet 11: e1005236 10.1371/journal.pgen.1005236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. (2001) Hypoxia-inducible factor 1: Oxygen homeostasis and disease pathophysiology. Trends Mol Med 7: 345–350. 10.1016/s1471-4914(01)02090-1 [DOI] [PubMed] [Google Scholar]
- Sendoel A, Kohler I, Fellmann C, Lowe SW, Hengartner MO (2010) HIF-1 antagonizes p53-mediated apoptosis through a secreted neuronal tyrosinase. Nature 465: 577–583. 10.1038/nature09141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Zhang Y, Powell-Coffman JA (2009) Two distinct roles for EGL-9 in the regulation of HIF-1-mediated gene expression in Caenorhabditis elegans. Genetics 183: 821–829. 10.1534/genetics.109.107284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Zhang Y, Ye Q, Saldanha JN, Powell-Coffman JA (2010) C. elegans SWAN-1 binds to EGL-9 and regulates HIF-1-mediated resistance to the bacterial pathogen Pseudomonas aeruginosa PAO1. PLOS Pathog 6: e1001075 10.1371/journal.ppat.1001075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaye DD, Greenwald I (2011) OrthoList: A compendium of C. elegans genes with human orthologs. PLoS One 6: e20085 10.1371/journal.pone.0020085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Nettleton D, Jiang M, Kim SK, Powell-Coffman JA (2005) Roles of the HIF-1 hypoxia-inducible factor during hypoxia response in Caenorhabditis elegans. J Biol Chem 280: 20580–20588. 10.1074/jbc.m501894200 [DOI] [PubMed] [Google Scholar]
- Shen C, Shao Z, Powell-Coffman JA (2006) The Caenorhabditis elegans rhy-1 gene inhibits HIF-1 hypoxia-inducible factor activity in a negative feedback loop that does not include vhl-1. Genetics 174: 1205–1214. 10.1534/genetics.106.063594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebel C, Lendahl U (2017) Notch signaling in development, tissue homeostasis, and disease. Physiol Rev 97: 1235–1294. 10.1152/physrev.00005.2017 [DOI] [PubMed] [Google Scholar]
- Simanshu DK, Nissley DV, McCormick F (2017) RAS proteins and their regulators in human disease. Cell 170: 17–33. 10.1016/j.cell.2017.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg PW. (2005) Vulval development. WormBook: 1–28. 10.1895/wormbook.1.6.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg PW, Horvitz HR (1986) Pattern formation during vulval development in C. elegans. Cell 44: 761–772. 10.1016/0092-8674(86)90842-1 [DOI] [PubMed] [Google Scholar]
- Sundaram MV. (2006) RTK/Ras/MAPK signaling. WormBook: 1–19. 10.1895/wormbook.1.80.1 [DOI] [Google Scholar]
- Tan PB, Lackner MR, Kim SK (1998) MAP kinase signaling specificity mediated by the LIN-1 Ets/LIN-31 WH transcription factor complex during C. elegans vulval induction. Cell 93: 569–580. 10.1016/s0092-8674(00)81186-1 [DOI] [PubMed] [Google Scholar]
- Wang M, Sternberg PW (1999) Competence and commitment of Caenorhabditis elegans vulval precursor cells. Dev Biol 212: 12–24. 10.1006/dbio.1999.9357 [DOI] [PubMed] [Google Scholar]
- Yoo AS, Greenwald I (2005) LIN-12/Notch activation leads to microRNA-mediated down-regulation of Vav in C. elegans. Science 310: 1330–1333. 10.1126/science.1119481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo AS, Bais C, Greenwald I (2004) Crosstalk between the EGFR and LIN-12/Notch pathways in C. elegans vulval development. Science 303: 663–666. 10.1126/science.1091639 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3 List of strains used. (63.6KB, pdf)
Table S4 Complete dataset of VI counts. (148.1KB, xlsx)
LSA-2018-00255_Supplemental_Data_1.txt (4.3KB, txt)
LSA-2018-00255_Supplemental_Data_2.txt (10.6KB, txt)