

Mutations in the fatty acid 2-hydroxylase gene FA2H disturb sphingolipid metabolism. In a large cohort of FA2H patients, Rattay et al. define the phenotypic spectrum, identify a unique hair phenotype (‘bristle hair’) and reveal an imaging ‘fingerprint’ of FA2H-disease captured by the acronym ‘WHAT’.
Keywords: SPG35, FAHN, FA2H, imaging biomarker, hereditary spastic paraplegia
Abstract
The endoplasmic reticulum enzyme fatty acid 2-hydroxylase (FA2H) plays a major role in the formation of 2-hydroxy glycosphingolipids, main components of myelin. FA2H deficiency in mice leads to severe central demyelination and axon loss. In humans it has been associated with phenotypes from the neurodegeneration with brain iron accumulation (fatty acid hydroxylase-associated neurodegeneration, FAHN), hereditary spastic paraplegia (HSP type SPG35) and leukodystrophy (leukodystrophy with spasticity and dystonia) spectrum. We performed an in-depth clinical and retrospective neurophysiological and imaging study in a cohort of 19 cases with biallelic FA2H mutations. FAHN/SPG35 manifests with early childhood onset predominantly lower limb spastic tetraparesis and truncal instability, dysarthria, dysphagia, cerebellar ataxia, and cognitive deficits, often accompanied by exotropia and movement disorders. The disease is rapidly progressive with loss of ambulation after a median of 7 years after disease onset and demonstrates little interindividual variability. The hair of FAHN/SPG35 patients shows a bristle-like appearance; scanning electron microscopy of patient hair shafts reveals deformities (longitudinal grooves) as well as plaque-like adhesions to the hair, likely caused by an abnormal sebum composition also described in a mouse model of FA2H deficiency. Characteristic imaging features of FAHN/SPG35 can be summarized by the ‘WHAT’ acronym: white matter changes, hypointensity of the globus pallidus, ponto-cerebellar atrophy, and thin corpus callosum. At least three of four imaging features are present in 85% of FA2H mutation carriers. Here, we report the first systematic, large cohort study in FAHN/SPG35 and determine the phenotypic spectrum, define the disease course and identify clinical and imaging biomarkers.
Introduction
Disturbances in sphingolipid metabolism have been implicated in a growing number of neurological diseases, including Parkinson’s disease, epilepsy, hereditary neuropathy, hereditary spastic paraplegia, and ataxia (reviewed in Astudillo et al., 2015). The endoplasmic reticulum enzyme fatty acid 2-hydroxylase, encoded by the FA2H gene, plays a major role in the formation of 2-hydroxy sphingolipids from ceramide (Maldonado et al., 2008). More than half of the galactolipids in myelin are 2-hydroxylated (Raghavan and Kanfer, 1972). Loss of FA2H function in mice leads to severe demyelination of optic nerves and the spinal cord (Potter et al., 2011). These changes are accompanied by pronounced axonal loss as well as altered size and distribution of Purkinje neurons in aged mice while myelination of peripheral nerves is relatively preserved. Phenotypically, Fa2h−/− mice display deficits in motor coordination, spatial memory, and learning (Potter et al., 2011).
In humans, autosomal recessive mutations in FA2H have been associated with three clinically defined disease entities with overlapping phenotypic spectra: (i) leukodystrophy with spasticity and dystonia (OMIM #612319) (Uchida et al., 2007; Edvardson et al., 2008); (ii) fatty acid hydroxylase-associated neurodegeneration (FAHN, OMIM #611026) (Kruer et al., 2010), a rare subtype of neurodegeneration with brain iron accumulation (NBIA); and (iii) hereditary spastic paraplegia (HSP) type SPG35 (OMIM #612319) (Dick et al., 2010). Taken together, 65 cases with FA2H mutations from 31 families have been published so far, carrying 40 distinct mutations. Standardized phenotypic descriptions and systematic analysis of the phenotypic spectrum in larger cohorts are lacking since prior publications contained no more than three families (Edvardson et al., 2008) or are limited to literature review of published cases (Mari et al., 2018).
Here, we report the first systematic, large cohort study in FAHN/SPG35 to determine the phenotypic spectrum, define the disease course and identify clinical biomarkers as well as confirm and specify imaging characteristics.
Materials and methods
Clinical characterization
All cases received a detailed clinical examination by a movement disorder specialist including standardized rating of disease severity using the spastic paraplegia rating scale (SPRS) (Schule et al., 2006). The majority of cases were re-evaluated clinically after their genetic diagnosis had been made. Neurophysiological characterization included motor evoked potentials, sensory evoked potentials, and nerve conduction studies; hereby, retrospective data were used.
Unless stated otherwise (Supplementary Table 1), no formal neurophysiological testing was available for the majority of cases. Cognitive deficits were evaluated clinically as well as based on clinical history. Cognitive decline was noted when loss of cognitive abilities was reported over time.
All subjects included in the study gave their written informed consent according to protocols approved by the respective institutional human ethics review boards.
MRI
Retrospective routine MRI data from 13 patients were collected from eight different sites and was re-evaluated by an experienced neuroradiologist (T.L.). As MRIs were obtained at different time points and clinical centres and not within a study setting, no harmonized MRI protocol was used and field strengths varied between scanners. To assess midbrain and pons atrophy, the area of the midbrain and pons was measured on mid-sagittal MRI; an area smaller than 88 mm2 or 417 mm2 [average −2 standard deviations as described in Oba et al. (2005)] was regarded as midbrain or pons atrophy, respectively.
Genetic analyses
FA2H mutations were detected by different genetic approaches including Sanger sequencing, a next generation sequencing (NGS)-based HSP gene panel and whole exome sequencing (see ‘Results’ section for more details). All sequence abnormalities were confirmed by Sanger sequencing. Mutated positions were annotated in reference to transcript ENST00000219368.
We used PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.bii.a-star.edu.sg) and MutationTaster (http://www.mutationtaster.org/) to predict the impact of missense mutations on the structure and function of the protein (Table 1). The ExAC Browser and Exome Variant Server (EVS) data on minor allele frequency (MAF) were retrieved on 4 September 2018. We recently published genetic information, focusing on an unusual inheritance pattern of uniparental disomy, about Families F1, F8, F10, and F13 (Soehn et al., 2016).
Table 1.
Overview over FA2H mutations identified in this study
| cDNA | Protein | MAF ExAC (All), % | GERP | Phastcon | Functional domain | PolyPhen-2 (HumDiv) | SIFT | Mutation taster | Classification [ACMG] | Family/ reference |
|---|---|---|---|---|---|---|---|---|---|---|
| c.21delC | p.A8Pfs*91 | - | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | Pathogenic | F12, F14 |
| c.131C>A | p.P44Q | - | 4.1 | 1.0 | cB5 | 1.000 | D | D | Likely pathogenic | F8, Soehn et al., 2016 |
| c.133G>T | p.G45W | - | 4.1 | 1.0 | cB5 | 1.000 | D | D | Likely pathogenic | F10, Soehn et al., 2016 |
| c.160_169del GCGGGCCAGG | p.A54Tfs*42 | - | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | Pathogenic | F4, F11 |
| c.205C>T | p.H69Y | - | 4.1 | 0.991 | cB5 | 0.338 | D | D | Likely pathogenic | F15 |
| c.232G>A | p.E78K | 0.035 | 3.91 | 0.999 | cB5 | 0.889 | T | D | Unknown significance | F9, F14 |
| c.262G>T | p.E88* | - | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | Pathogenic | F16 |
| c.443C>T | p.P148L | - | 5.45 | 0.982 | no | 1.000 | D | D | Likely pathogenic | F3 |
| c.503_506del TCTG | p.V168Gfs*72 | - | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | Pathogenic | F11 |
| c.460C>T | p.P154C | 0.002 | 5.32 | 1.0 | no | 1.00 | D | D | Pathogenic | F16, Kruer et al., 2010; Kara et al., 2016 |
| c.510_511delCA | p.Y170* | - | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | Pathogenic | F5; c.509_510delAC, p.Y170* in Kruer et al., 2010; Donkervoort et al., 2014 |
| c.527G>A | p.W176* | - | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | Pathogenic | F1, Soehn et al., 2016 |
| c.620C>T | p.T207M | 0.003 | 5.25 | 0.685 | no | 0.746 | T | T | Pathogenic | F6, Pensato et al., 2014; Kara et al., 2016 |
| c.704G>A | p.R235H | 0.001 | 5.36 | 1.0 | sd | 1.00 | D | D | Likely pathogenic | F6, F7 |
| c.785A>C | p.K262T | 0.001 | 4.83 | 1.0 | sd | 0.999 | T | D | Likely pathogenic | F13, Soehn et al., 2016 |
| c.859T>C | p.C287R | - | 0 | 0 | sd | 0.136 | T | T | Unknown significance | F7 |
| c.908G>T | p.G303V | - | 5.39 | 0.997 | sd | 1.00 | T | D | Likely pathogenic | F2 |
| c.956A>G | p.H319R | - | 4.26 | 1.0 | sd | 0.719 | D | D | Unknown significance | F9 |
| c.968C>T | p.P323L | 0.001 | 5.39 | 0.563 | sd | 1.00 | T | D | Pathogenic | F2, Rupps et al., 2013 |
cB5 = cyt. B5-like-heme-binding; CH = compound heterozygous;, fs = frameshift mutation; hom = homozygous; ms = missense mutation; n.a. = not applicable; PCH = presumably compound heterozygous; sd = sterol desaturase.
Variant coordinates refer to GRCh37 and the reference sequence ENST00000219368. Variant classification criteria are derived from the ACMG criteria (Richards et al., 2015).
Scanning electron microscopy of hair shafts
Hair samples were mounted to aluminium sample holders with Leit-Tabs G3347 (Plano). The probes were sputtered with gold (15-nm thickness) prior to imaging. Pictures were taken at a magnification of 250× by using secondary electrons with an S800 scanning electron microscope (Hitachi). In Patient F8, pictures were taken using a CS2 scanning electron microscope (CamScan).
Statistics
Descriptive statistics and survival analysis (Kaplan-Meier analysis) were performed using SPSS v25 for Windows (IBM, Armonk, NY).
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary material. Raw data regarding human subjects (e.g. genetic raw data, MRI datasets) are not shared freely to protect the privacy of the human subjects involved in this study; no consent for open sharing has been obtained.
Results
Genetic findings
Screening of a large cohort of movement disorder cases
To appraise the clinical spectrum of FA2H mutations, we screened exome data available from a large cohort of 2774 cases from 1482 families with various motor neuron and movement disorder phenotypes and family history compatible with autosomal recessive disease for mutations in FA2H [Genesis database: www.genesis-app.com (Gonzalez et al., 2015)]. Phenotypes included the following (numbers based on families): amyotrophic lateral sclerosis (n = 620), HSP (n = 139; pure/complicated 45/94), ataxia (n = 62), dystonia (n = 9), Parkinson’s syndrome (n = 67), neuromuscular diseases (n = 354), epileptic encephalopathy (n = 115), mitochondrial disease (n = 84), and others (n = 32).
This approach uncovered nine families with homozygous or compound heterozygous FA2H mutations (Families F1–F6, F11, 12 and F14; Supplementary Table 1). Although we included a wide spectrum of phenotypes in the screening, all positive cases were phenotypically classified as complicated HSP, indicating a strong enrichment of FA2H mutations in this phenotype.
Identification of additional FA2H families
We then reached out to colleagues to collect further families with FA2H mutations. Using this approach, we identified an additional seven families. In Families F7, F13, F15 and F16, whole exome sequencing was performed. Families F8 and F10 were examined using an NGS-based gene panel containing 98 spasticity associated genes at the University of Tübingen (technical details in Synofzik et al., 2014b). The index of Family F9 was screened for mutations in FA2H by Sanger sequencing. Altogether, we identified 19 FA2H cases in 16 families.
Evaluation of mutations
The 16 FA2H families we report here carry 19 distinct FA2H mutations (Table 1). As loss-of-function is the established mutation mechanism in FA2H (Edvardson et al., 2008; Dick et al., 2010) we considered all six truncating mutations (four deletions leading to a shift in the reading frame and two nonsense mutations) to be pathogenic. Among the 13 missense mutations we identified, four have been associated with an FAHN/SPG35 phenotype previously [c.460C>T, p.R154C (Kruer et al., 2010; Kara et al., 2016); c.620C>T, p.T207M (Pensato et al., 2014); c.704G>A, p.R235H (Magariello et al., 2017), c.968C>T, p.P323L (Rupps et al., 2013)]. The remaining nine were either absent in public databases (n = 5) or exceedingly rare (MAF < 0.1%, n = 4) (EVS6500, ExAC browser, accessed on 9 April 2018). All but one (c.859T>C, p.C287R) of the novel missense variants were highly conserved (GERP, PhastCons) and at least two of three prediction algorithms (PolyPhen-2, SIFT, MutationTaster) predicted them to be likely pathogenic. The only exception was the variant c.859T>C, p.C287R observed in Family F7. This variant, located in the sterol desaturase domain, is absent in public databases (EVS and ExAC) as well as in our in-house database containing ∼26 000 alleles; the affected amino acid is only weakly conserved and predicted to be tolerated in all three in silico prediction algorithms we used. In Family F7 this variant is compound heterozygous with c.704G>A, R235H, a previously-described strong missense variant (Magariello et al., 2017) also identified in Family F6 that affects an amino acid implicated in FAHN/SPG35 previously (c.703C>T, p.R235C; Dick et al., 2010).
In 12 of 16 families, DNA of both parents was available, allowing segregation analysis, in one family (Family F11), DNA of one parent was available and in three families, parental DNA could not be obtained (Families F2, F6 and F9). Segregation analysis revealed uniparental disomy in four families (Families F1, F8, F10 and F13; reported in Soehn et al., 2016) and identified a de novo mutation (c.460C>T, p.P154C), compound heterozygous with a paternally inherited missense mutation in Family F16. The remaining mutations were confirmed to be either homozygous or compound heterozygous, as expected (Supplementary Fig. 1). Together with this study the number of published FA2H mutations increased to a total of 43 (Fig. 1 and Supplementary Table 2). The vast majority of FA2H mutations are private and only eight mutations occur in more than one family (c.21del, p.A8Pfs*91; c.160_169del, p.A54Tfs*42; c.232G>A, p.E78K; c.460C>T, p.P154C; c.509_510delAC, p.Y170*; c.620C>T, p.T207M; c.704G>A, p.R235H; c.968C>T, p.P323L).
Figure 1.

Mutation spectrum in FA2H/SPG35. Structure of the FA2H coding sequence (NCBI Reference Sequence: NM_024306.4) is shown with numbered exons 1–7 in alternating grey shades. For each variant, cDNA change and the presumed effect on the resulting protein are given (where possible). For changes observed in this study, the family number is given in square brackets and turquoise font; the letter ‘h’ indicates (compound)-heterozygous occurrence. Missense mutations (black) and inframe deletions (brown) are listed above, truncating mutations including nonsense mutations (orange), frameshift insertions or deletions (purple) and splice changes (green) below the graph. The cDNA is overlain by the two known functional protein domains (cytochrome B5-like heme-binding domain and sterol desaturase domain), indicated as black boxes.
Description of the phenotype
Detailed clinical data on 19 cases with FAHN/SPG35 from 16 families are provided in Supplementary Table 1. Gait problems started almost invariably in early childhood at a median age of 4 years [interquartile range (IQR) 3–4.5 years]; all but two individuals (Patient F9, age at onset 20 years and Patient F14, age at onset 10 years) showed symptoms before the age of 5 years. Patients became wheelchair dependent after median disease duration of 7 years (IQR 3–12 years) (Kaplan-Meier analysis). At the time of examination (median disease duration 5.5 years), all cases had a spastic tetraparesis with lower limb predominance, and varying degrees of truncal instability (Fig. 2A). Mild cognitive deficits were noted from childhood on in the majority of cases (93%) and were considered to be progressive (information provided by parents based on loss of cognitive abilities over time) in all but two cases. Oculomotor abnormalities with cerebellar characteristics—as determined by clinical examination—were also present in all cases: the most frequent feature was saccadic pursuit (81%), accompanied by gaze-evoked nystagmus in 27%. Horizontal saccades were markedly slowed in 57%. Furthermore, 60% of the cases had exotropia in the absence of visual impairment.
Figure 2.

Phenotypic spectrum of FAHN/SPG35 and relevant differential diagnosis. (A) The phenotypic spectrum of FAHN/SPG35 with prevalence rates (in per cent) of specific features. MRI findings are coloured in yellow, clinical signs and symptoms are highlighted in blue. (B) The FAHN/SPG35 phenotype overlaps the clinical presentation of a range of movement disorders. Main differential diagnoses of FAHN/SPG35 from the spectrum of hereditary spastic paraplegias (cHSPs), NBIA and giant axonal neuropathy that shares some features of the ultrastructural hair phenotypes with FAHN/SPG35 are depicted. Discriminating features are highlighted. CoPAN = COASY protein-associated neurodegeneration; GAN = giant axonal neuropathy; MPAN = mitochondrial membrane protein-associated neurodegeneration.
Figure 3.
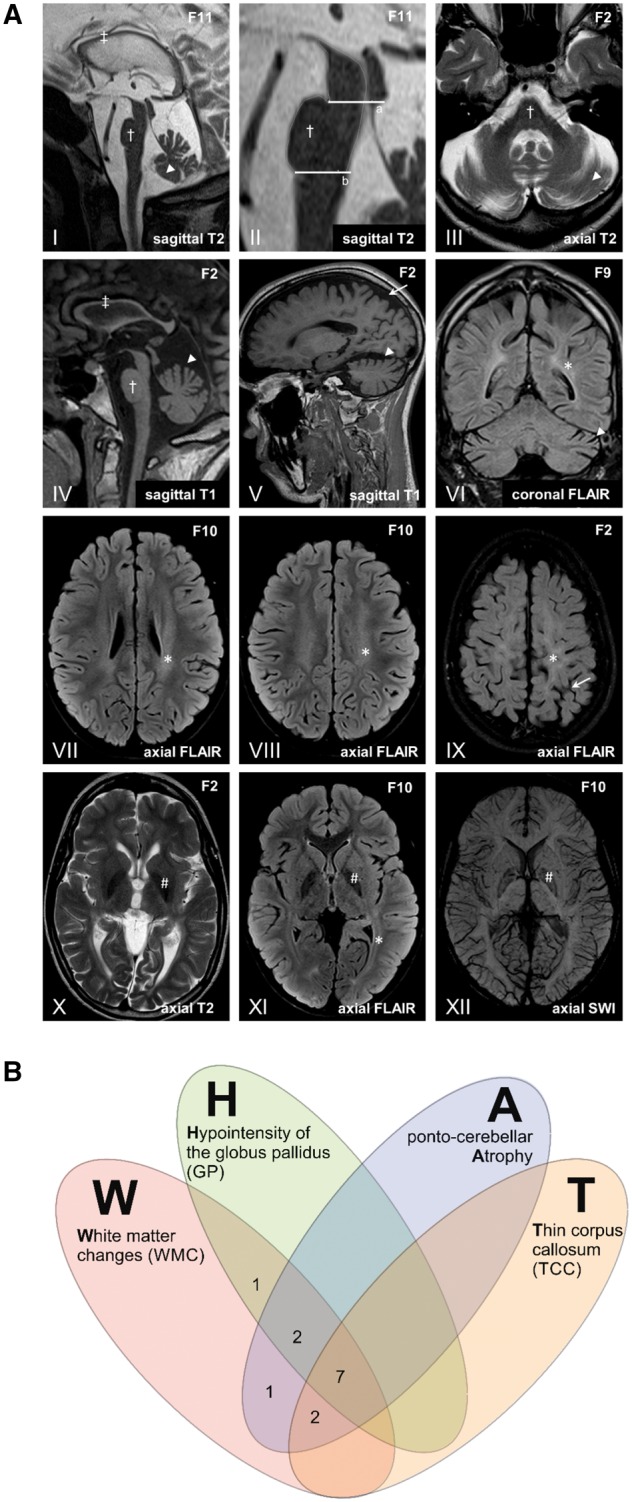
MRI findings in FAHN/SPG35. (A) The most frequent imaging findings in FAHN/SPG35 can be summarized by the acronym ‘WHAT’ (i) white matter changes (asterisk in VI–IX and XI); (ii) hypointensity of the globus pallidus in T2/T2-FLAIR/T2*/SWI (hash symbol in X–XII); (iii) ponto-cerebellar atrophy (dagger and arrowhead in I–VI); and (iv) thin corpus callosum. Sagittal images show a general corpus callosum thinning predominantly affecting the (dorsal) body (double dagger in I and IV). Pontine atrophy (dagger symbol in I–IV) was measured on midsagittal MRI (II) with the upper border a line (‘a’) through the superior pontine notch and the inferior edge of the quadrigeminal plate, and with the lower border (‘b’) parallel to the upper border and through the inferior pontine notch. Ponto (dagger)- cerebellar (arrowhead) atrophy is depicted in III with widening of the fourth ventricle, affecting the upper vermis and the hemispheres (I, III-VI). Predominantly mild supratentorial atrophy with a focus on the parietal lobe is depicted in V and IX (arrows). Periventricular white matter abnormalities are shown on T2-FLAIR images most affecting the parietal white matter (asterisk in VI–IX and XII). Hypointense signal of the globus pallidus associated with iron accumulation is shown on T2, T2-FLAIR and susceptibility weighted images (hash symbol in X–XII). (B) Venn diagram depicting the occurrence of MRI features contributing to the ‘WHAT’ imaging phenotype. In 85% of cases (11/13), at least three of four ‘WHAT’ features were found, in 54% (7/13), all four features were present.
Limb ataxia was present in 71%. Dysarthria was almost universally present (88%) and was clinically classified as pseudobulbar and/or cerebellar (Darley et al., 1969). Three patients became anarthric during the course of disease (>20 years after disease onset). Dysphagia was a frequent finding (71%). Movement disorders were present in 50% of cases with rigidity and dystonia (both present in four cases) being most common. One patient presented with resting tremor. Sensory involvement was mild if present at all. Other signs and symptoms included optic atrophy (31%) and seizures (focal semiology in two cases, one case with febrile seizures).
Neurophysiological examination of the CNS showed absent motor evoked potentials (MEP) to the lower limbs in 57% (performed in seven individuals) and normal central conduction times in the remaining cases, indicating a primarily axonal type of damage. Sensory evoked potentials were abnormal in two of nine cases (22%). Thirty-eight per cent of cases had abnormal visually evoked potentials (VEP) with prolonged P100 latency in three of eight cases. Peripheral neuropathy was present in 27% (3/11) of cases (Supplementary Table 1). This analysis was performed on retrospective data.
Since four uniparental disomy (UPD) cases are included in this series we also looked for imprinting phenotypes of chromosome 16 with intrauterine growth restriction (mUPD16) and/or alveolar capillary dysplasia with misalignment of pulmonary veins (ACDMPV) (pUPD16/mat del 16q24). However, no case was reported to be underweight at birth or had any early pulmonary problems.
Genotype-phenotype correlation
Genetically, missense and truncating mutations in FA2H have been reported to be pathogenic. When considering all published and the novel mutations reported here, there are 50 distinct mutations in FA2H that have been associated with a FAHN/SPG35 phenotype. These include 14 truncating (frameshift, splice, nonsense) and 36 non-truncating (missense, inframe deletion) changes. The latter, thus, are the predominant mutation type in FA2H (Fig. 1). As suggested previously, missense mutations in FA2H preferentially target one of the two known functional domains of FA2H (Edvardson et al., 2008). Among all known 33 missense changes, 10 are located in the cytochrome B5-like heme-binding domain (codons 15–85, encompassing 8.8% of the protein) and 17 in the sterol desaturase domain (codons 210–367, 19.6% of the protein), while only six mutations are located outside of these known domains (71.6% of the protein). Detailed comparative analysis of the phenotype (Supplementary Table 2) revealed no apparent genotype phenotype correlations depending on the mutation type present.
Imaging findings
We re-evaluated retrospective MRI data from 13 FA2H mutation carriers. In all cases, T2 hyperintense white matter abnormalities were found in the periventricular white matter with parietal predominance. The globus pallidus showed an overall mild T2/T2-FLAIR/SWI/T2* hypointense signal in 77%. Mild to moderate cerebellar atrophy of the vermis and the hemispheres was seen in 85% of patients. In 62% of patients, there was supratentorial atrophy, which was mild and predominantly parietal in most, but global in older cases. A substantial atrophy of the pons could be demonstrated in 85%. Another common MRI feature in FAHN/SPG35 was a thin corpus callosum (69%) ranging from focal thinning of the dorsal body to a general thinning (Supplementary Table 1 and Fig. 3A).
Hair abnormalities
Several patients presented with unusually bristly hair that differed from the hair in other family members. Often the hair was kept short by patients or caregivers due to the frizzy appearance (Fig. 4A). As FA2H has a known function in keratinocytes [responsible for epidermal permeability barrier homeostasis (Uchida et al., 2007)] and sebaceous glands [required for synthesis of 2-hydroxylated glucosylceramide and a fraction of type II wax diesters (Maier et al., 2011)], we decided to perform scanning electron microscopy of hair shafts. Between 10 and 16 hair shafts in each of four patients (Patients F1, F2, F7 and F8) were examined. Strikingly, hair shafts of all four patients demonstrated subtle to pronounced longitudinal grooves (Fig. 4C); hereby, between 20 and 100% of hair shafts in each patient were affected by this abnormality. No longitudinal grooves were observed in control hair samples.
Figure 4.

Hair analysis: macroscopy and electron microscopic finding. (A) Photograph of Patient F8 with bristle like hair structure, which is kept short by the caregivers. (B–H) Scanning electron microscopy images at 250× (B–F) and 400× (G and H) of representative hair shafts. (B) Healthy control hair. (C and D) Patient F1 exemplary hair shafts present with longitudinal grooves in C and adhesive plaques in D. (E) Hair of Patient F2 shows longitudinal grooves but no adhesive plaques. (F) Longitudinal grooves in hair of Patient F7. (G and H) Hair of Patient F8 shows longitudinal grooves and adhesive plaques with a chunky appearance.
Three of four patients also had adhesive plaques on their hair shafts (Fig. 4D). Two distinct plaque formations could be observed: small roundish shaped plaques and larger chunky plaque formations (Fig. 4G and H).
Discussion
FA2H mutations have been associated with three distinct disease categories in the past: neurodegeneration with brain iron accumulation, leukodystrophy, and hereditary spastic paraplegias. Does this indicate an (unusually) large phenotypic variability of FA2H disease or reveal the challenge to classify phenotypically complex diseases unambiguously? Our standardized clinical workup of a large cohort of 19 FAHN/SPG35 patients and the high degree of congruence with previously published studies (Edvardson et al., 2008; Dick et al., 2010; Kruer et al., 2010; Mari et al., 2018) supports the latter. First, a large phenotypic variability of FA2H mutations is unlikely given that we screened >2700 cases with a broad range of autosomal recessive movement disorder and motor neuron phenotypes, and identified biallelic mutations in FA2H exclusively in patients diagnostically labelled as HSP. Second, the clinical phenotype observed in our FAHN/SPG35 cases was rather uniform across the cohort with lower limb predominant spastic tetraparesis accompanied by truncal instability, dysarthria, dysphagia, cerebellar ataxia, and cognitive deficits present in ∼90% of cases. Other frequent symptoms, present in at least half of all cases, were exotropia and movement disorders, particularly dystonia, rigidity, or resting tremor. The FAHN/SPG35 phenotype we were able to deduce from our unbiased cohort confirms findings from previous smaller studies (Edvardson et al., 2008; Dick et al., 2010; Kruer et al., 2010; Mari et al., 2018) that describe very similar clinical characteristics of FAHN/SPG35. Pathophysiologically, neurogenic exotropia is considered to be a consequence of fastigial nucleus dysfunction (Leigh and Zee, 2015) and thus part of the cerebellar syndrome seen in FAHN/SPG35. The age of onset spectrum was narrow with an onset in early childhood (<5 years) in ∼90% of cases and thus also supports a rather conserved phenotype and disease manifestation, although later disease manifestations rarely occur (Tonelli et al., 2012). These features are compatible with and can even be considered typical for each of the three disease categories FA2H mutations have been associated with. Moreover, the categories NBIA, leukodystrophy and HSP are not divisive, as the former two are based on the non-exclusive and often overlapping presence of imaging findings while HSP is a clinical classification concept. We therefore suspect that the classification of patients with FA2H mutations may be primarily influenced by the experience and background of the diagnosing clinician, probably modified by the individual severity of specific signs or symptoms in a given patient but bears little systematic value. Similar observations have been made for other genes associated with complex phenotypes in recent years [e.g. PNPLA6 (Synofzik et al., 2014a), ATP13A2 (Estrada-Cuzcano et al., 2017), POLR3A (Minnerop et al., 2017), PLA2G6 (Synofzik and Gasser, 2017), and SPG7 (Synofzik and Schule, 2017)] and prompts currently used classification systems to be reconsidered (Marras et al., 2016; Schule, 2017).
Detailed phenotypic analysis of published cases as well as the cases reported here revealed no association between phenotype and mutation type. Recently, it has been suggested that there may be some genotype-phenotype correlation for specific missense variants in FA2H (Mari et al., 2018); the study is, however, limited by the lack of standardized clinical assessment of the reported cases and absence of comprehensive criteria for the phenotypic classification [(i) leukoencephalopathy; (ii) spastic paraplegia; (iii) cerebellar and basal ganglia involvement] the authors propose. Further prospective studies are therefore needed to address this question.
Although rather uniform in itself, the phenotype associated with FA2H mutations overlaps the clinical presentation of a number of hereditary movement disorders. Compared to other HSPs the disease onset in FAHN/SPG35 [median 4 versus 31 years in a genetically mixed cohort of >600 HSP patients (Schule et al., 2016)] is very early and the progression decidedly faster with loss of ambulation after a median of 7 years after disease onset [compared to wheelchair dependency in one-quarter of patients with genetically mixed HSP after 37 years disease duration (Schule et al., 2016)]. Among the HSPs, FA2H mutation carriers show the largest overlap with SPG11, SPG15, SPG48 (Pensato et al., 2014) and SPG78 (Estrada-Cuzcano et al., 2017). These HSPs share frequent cognitive deficits, cerebellar ataxia, and movement disorders. However, frequency and severity of dysphagia in FAHN/SPG35, which has prompted Rupps et al. (2013) to coin the term ‘complete loss of oromotor control’ in these patients, as well as the frequent occurrence of truncal hypotonia and exotropia set FAHN/SPG35 apart from SPG11, SPG15, and SPG48 (Kruer et al., 2012; Pensato et al., 2014). Compared to SPG78, characterized by an adult onset pyramidal-cerebellar syndrome with frequent cognitive deficits, parkinsonism, and axonal neuropathy due to mutations in the ATP13A2 gene, the onset of FAHN/SPG35 is considerably earlier and the typical FA2H-associated imaging findings of a thin corpus callosum and white matter changes are rarely seen in SPG78 (Estrada-Cuzcano et al., 2017).
In comparison to other subtypes of the NBIA spectrum, extrapyramidal involvement, most commonly rigidity (four patients) or dystonia (four patients), seems to be less frequent and less pronounced at least in our cohort of FAHN/SPG35 patients. Among the NBIAs, spasticity can also be a prominent feature especially in mitochondrial membrane protein-associated neurodegeneration (MPAN) and COASY protein-associated neurodegeneration (CoPAN) (Hogarth, 2015). Prominent parkinsonism and oromandibular dystonia in CoPAN may help to differentiate it from FAHN/SPG35 whereas MPAN frequently presents with optic atrophy and sometimes psychiatric features (Hogarth, 2015). Figure 2B offers a guideline for clinical and imaging differential diagnosis among these overlapping disease entities.
A unique clinical feature of FA2H disease appears to be the wiry hair that elicits the association of bristles when being touched. Interestingly, a fur phenotype was also described for Fa2h knock-out mice (Maier et al., 2011). Here, emergence of hair from sebaceous glands is believed to be blocked by sebum plugs that are formed by highly viscous sebum trapped in the dilated hair canals and released en bloc from the sebaceous gland. These changes result in delayed fur development with a less soft and glossy appearance than in wild-type littermates (Maier et al., 2011). Similar sebum plugs, although not shown in our patients directly, might explain the longitudinal grooves and adhesive plaques that were visible ultrastructurally. Sebum trapped in the hair canal might lead to deformation of emerging hair shafts and the adhesive plaques might correspond to additional sebum material sticking to the hair shafts. Longitudinal grooves have also been described in the curly hair of patients with giant axonal neuropathy (Treiber-Held et al., 1994). In FAHN/SPG35, however, the hair does not seem to have the same propensity to form curls as in giant axonal neuropathy. Since scanning electron microscopy is widely available, hair may be used as an easily accessible biomarker that can guide towards a diagnosis of FAHN/SPG35.
Considering the extent and frequency of white matter changes present we were surprised to find abnormal evoked potentials (motor: 57% abnormal; sensory: 22% abnormal; visual: 38% abnormal) in only a subset of cases. The validity of our findings, however, is limited because of the retrospective nature of the available neurophysiological data, and the lack of standardization between the diagnostic laboratories.
The MRI phenotype in our cohort of FAHN/SPG35 patients was strikingly uniform. The most common findings were white matter changes (100%), followed by cerebellar atrophy (85%), pons atrophy (85%), overall mild T2/T2-FLAIR/T2*/SWI hypointense signal of the pallidum (77%), a thin corpus callosum (69%), and supratentorial atrophy (62%). Interestingly, the very same MRI features have already been identified by Kruer et al. (2010) based on MRIs obtained in two cases and review of the literature. In our larger cohort we were now able to confirm that these features are indeed typical hallmarks of FAHN/SPG35. We therefore suggest the term ‘WHAT’ to summarize the imaging findings in FA2H disease, whereby each letter represents a common MRI feature: white matter changes, hypointensity of the globus pallidus, ponto-cerebellar atrophy, and thin corpus callosum. Eighty-five per cent of patients had at least three of the WHAT features, in more than half of the patients (54%), all four features were present (Fig. 3B). The WHAT imaging phenotype therefore might assist the clinical diagnosis of FAHN/SPG35 although its sensitivity and specificity need to be determined in larger prospective cohorts.
We were surprised to find a high rate of patients with T2/T2-FLAIR/T2*/SWI hypointense signal changes indicating iron deposition in the globus pallidus (77%), as this contrasts with reports from Marelli et al. (2015). In a review of several small case studies (33 FAHN/SPG35 patients in 11 studies) they report MRI indication of iron deposits in no more than a third of cases. Several factors might contribute to this discrepancy: First, the lack of consensus on the extent of hypointense T2/T2* signals that are considered physiological or outside physiological ranges, especially when considering MRIs obtained using heterogeneous protocols at different field strengths may introduce bias in MRI interpretation; this is of particular relevance for the literature report by Marelli et al. (2015). The overall small number of patients in whom T2* or susceptibility-weighted imaging (SWI) sequences were performed and the lack of a systematic and uniform MRI analysis across the heterogeneous studies reported by Marelli et al., may have led to an underestimation of iron deposition in FAHN/SPG35. Prospective studies using standardized MRI protocols and scanners and preferably automated image analysis are necessary to overcome this bias. Second, lack of CT scans or quantitative susceptibility mapping (QSM) did not allow us to exclude basal ganglia calcifications that thus might have contributed to hypointense signals within the globus pallidus and thus may have led to an overestimation of iron deposition in our retrospective analysis.
Brainstem atrophy has been described previously in about two-thirds of FAHN/SPG35 patients (Marelli et al., 2015). here, we used cross-sectional area quantification to characterize this feature further, which we observed at even higher frequency (85%) in our cohort. Characteristically, the volume of the pons is diminished in FAHN/SPG35 while the mesencephalon is comparably preserved. Advanced MRI techniques such as diffusion tensor imaging and voxel-based morphometry may help to identify fibre tracts associated with specific clinical signs such as exotropia [e.g. dorsal visual pathway (Yan et al., 2010)] or identify additionally involved brain regions (in analogy to Lindig et al., 2015).
Conclusions
Biallelic FA2H mutations cause a narrow phenotypic spectrum characterized by early-onset spastic tetraparesis, cerebellar ataxia, dysarthria, dysphagia and cognitive deficits, frequently accompanied by exotropia, and movement disorders (dystonia, rigidity). Bristle-like hair, a clinical correlate of ultrastructural changes of the hair shafts due to altered sebum composition, are a tell-tale clinical sign. Characteristic imaging features can be summarized by the ‘WHAT’ acronym: white matter changes, hypointensity of the globus pallidus, ponto-cerebellar atrophy, and thin corpus callosum.
Supplementary Material
Acknowledgements
Scanning electron microscopy work was performed at the Department of Cellular Biophysics, Institute of Medical Physics and Biophysics, University of Münster, Münster, Germany by Prof. Jürgen Klingauf and his staff. We thank Montse Ruiz (PhD) and Agatha Schlüter (PhD) from the Pujol lab for genetic work with whole exome sequencing on Patient F13.
Glossary
Abbreviations
- HSP
hereditary spastic paraplegia
- NBIA
neurodegeneration with brain iron accumulation
- SPG
spastic paraplegia gene
Funding
This study was supported by the E-RARE JTC grant ‘NEUROLIPID’ (BMBF, 01GM1408B to R.S. and grant by the Italian ministry of Health to M.T.B.), the 7th European Community Framework Programme through funding for the NEUROMICS network (F5–2012–305121 to L.S.) and a Marie Curie International Outgoing Fellowship (grant PIOF-GA-2012–326681 to R.S.), grant 779257 ‘Solve-RD’ from the Horizon 2020 research and innovation programme to R.S.), the National Institute of Health (NIH) (grant 5R01NS072248 to R.S. and S.Z., grants 1R01NS075764, 5R01NS054132, 2U54NS065712 to S.Z.), the 5XMille Funds and grant No. RC 2014–2018 (to M.T.B.). The work was further supported by the German HSP-Selbsthilfegruppe e.V. (grant to R.S. and L.S.), the Spastic Paraplegia Foundation through funding for the Alliance for Treatment in HSP and PLS (grant to R.S.), the ISCIII and ‘Fondo Europeo de Desarrollo Regional (FEDER), Unión Europea, una manera de hacer Europa’ (FIS PI14/00581), ‘La Marató de TV3’ Foundation 345/C/2014, the Hesperia Foundation and CIBER on Rare Diseases (CIBERER) to A.P. T.W.R. receives funding from the University of Tübingen, medical faculty, for the Clinician Scientist Program Grant: #386–0–0. S.W. is supported by the Ministry of Science, Research and the Arts of Baden-Württemberg and the European Social Fund (ESF) of Baden-Württemberg (31–7635 41/67/1). A.M. was supported by the Possehl-Stiftung (Lübeck, Germany). This work was supported by the Austrian Science Fund (FWF, P27634FW to M.A-G.), the Jubiläumsfonds der Oesterreichischen Nationalbank (OeNB), Nr.16880 to M.A-G.). J.B. is supported by a Senior Clinical Researcher mandate of the Research Fund - Flanders (FWO) under grant agreement number 1805016N.
Competing interests
The authors declare no competing interests.
References
- Astudillo L, Sabourdy F, Therville N, Bode H, Segui B, Andrieu-Abadie N et al. . Human genetic disorders of sphingolipid biosynthesis. J Inherit Metab Dis 2015; 38: 65–76. [DOI] [PubMed] [Google Scholar]
- Darley FL, Aronson AE, Brown JR. Differential diagnostic patterns of dysarthria. J Speech Hear Res 1969; 12: 246–69. [DOI] [PubMed] [Google Scholar]
- Dick KJ, Eckhardt M, Paisan-Ruiz C, Alshehhi AA, Proukakis C, Sibtain NA et al. . Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35). Hum Mutat 2010; 31: E1251–60. [DOI] [PubMed] [Google Scholar]
- Donkervoort S, Dastgir J, Hu Y, Zein WM, Marks H, Blackstone C et al. . Phenotypic variability of a likely FA2H founder mutation in a family with complicated hereditary spastic paraplegia. Clin Genet 2014; 85: 393–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S, Hama H, Shaag A, Gomori JM, Berger I, Soffer D et al. . Mutations in the fatty acid 2-hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia. Am J Hum Genet 2008; 83: 643–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada-Cuzcano A, Martin S, Chamova T, Synofzik M, Timmann D, Holemans T et al. . Loss-of-function mutations in the ATP13A2/PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain 2017; 140: 287–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M, Falk MJ, Gai X, Postrel R, Schüle R, Zuchner S. Innovative genomic collaboration using the GENESIS (GEM.app) platform. Hum Mutat 2015; 36: 950–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogarth P. Neurodegeneration with brain iron accumulation: diagnosis and management. J Mov Disord 2015; 8: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kara E, Tucci A, Manzoni C, Lynch DS, Elpidorou M, Bettencourt C et al. . Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain 2016; 139: 1904–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruer M, Boddaert N, Schneider S, Houlden H, Bhatia K, Gregory A et al. . Neuroimaging features of neurodegeneration with brain iron accumulation. Am J Neuroradiol 2012; 33: 407–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruer MC, Paisan-Ruiz C, Boddaert N, Yoon MY, Hama H, Gregory A et al. . Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Ann Neurol 2010; 68: 611–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh RJ, Zee DS. The neurology of eye movements. Oxford, UK: Oxford University Press; 2015. [Google Scholar]
- Lindig T, Bender B, Hauser T-K, Mang S, Schweikardt D, Klose U et al. . Gray and white matter alterations in hereditary spastic paraplegia type SPG4 and clinical correlations. J Neurol 2015; 261: 1961–71. [DOI] [PubMed] [Google Scholar]
- Magariello A, Russo C, Citrigno L, Züchner S, Patitucci A, Mazzei R et al. . Exome sequencing reveals two FA2H mutations in a family with a complicated form of Hereditary Spastic Paraplegia and psychiatric impairments. J Neurol Sci 2017; 372: 347–9. [DOI] [PubMed] [Google Scholar]
- Maier H, Meixner M, Hartmann D, Sandhoff R, Wang-Eckhardt L, Zoeller I et al. . Normal fur development and sebum production depends on fatty acid 2-hydroxylase expression in sebaceous glands. J Biol Chem 2011; 286: 25922–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado EN, Alderson NL, Monje PV, Wood PM, Hama H. FA2H is responsible for the formation of 2-hydroxy galactolipids in peripheral nervous system myelin. J Lipid Res 2008; 49: 153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marelli C, Salih MA, Nguyen K, Mallaret M, Leboucq N, Hassan HH et al. . Cerebral iron accumulation is not a major feature of FA2H/SPG35. Mov Disord Clin Pract 2015; 2: 56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari F, Berti B, Romano A, Baldacci J, Rizzi R, Alessandrì MG et al. . Clinical and neuroimaging features of autosomal recessive spastic paraplegia 35 (SPG35): case reports, new mutations, and brief literature review. Neurogenetics 2018; 19: 123–30. [DOI] [PubMed] [Google Scholar]
- Marras C, Lang A, van de Warrenburg BP, Sue CM, Tabrizi SJ, Bertram L et al. . Nomenclature of genetic movement disorders: recommendations of the international Parkinson and movement disorder society task force. Mov Disord 2016; 31: 436–57. [DOI] [PubMed] [Google Scholar]
- Minnerop M, Kurzwelly D, Wagner H, Soehn AS, Reichbauer J, Tao F et al. . Hypomorphic mutations in POLR3A are a frequent cause of sporadic and recessive spastic ataxia. Brain 2017; 140: 1561–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oba H, Yagishita A, Terada H, Barkovich A, Kutomi K, Yamauchi T et al. . New and reliable MRI diagnosis for progressive supranuclear palsy. Neurology 2005; 64: 2050–5. [DOI] [PubMed] [Google Scholar]
- Pensato V, Castellotti B, Gellera C, Pareyson D, Ciano C, Nanetti L et al. . Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. Brain 2014; 137 (Pt 7): 1907–20. [DOI] [PubMed] [Google Scholar]
- Potter KA, Kern MJ, Fullbright G, Bielawski J, Scherer SS, Yum SW et al. . Central nervous system dysfunction in a mouse model of Fa2h deficiency. Glia 2011; 59: 1009–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan S, Kanfer JN. Ceramide galactoside of enriched neuronal and glial fractions from rat brain. J Biol Chem 1972; 247: 1055–6. [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al. . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupps R, Hukin J, Balicki M, Mercimek-Mahmutoglu S, Rolfs A, Dias C. Novel mutations in FA2H-associated neurodegeneration: an underrecognized condition? J Child Neurol 2013; 28: 1500–4. [DOI] [PubMed] [Google Scholar]
- Schule R. Reply: complicated hereditary spastic paraplegia due to ATP13A2 mutations: what’s in a name? Brain 2017; 140: e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schule R, Holland-Letz T, Klimpe S, Kassubek J, Klopstock T, Mall V et al. . The Spastic Paraplegia Rating Scale (SPRS): a reliable and valid measure of disease severity. Neurology 2006; 67: 430–4. [DOI] [PubMed] [Google Scholar]
- Schule R, Wiethoff S, Martus P, Karle KN, Otto S, Klebe S et al. . Hereditary spastic paraplegia: clinicogenetic lessons from 608 patients. Ann Neurol 2016; 79: 646–58. [DOI] [PubMed] [Google Scholar]
- Soehn AS, Rattay TW, Beck-Wodl S, Schaferhoff K, Monk D, Dobler-Neumann M et al. . Uniparental disomy of chromosome 16 unmasks recessive mutations of FA2H/SPG35 in 4 families. Neurology 2016; 87: 186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synofzik M, Gasser T. Moving beyond syndromic classifications in neurodegenerative disease: the example of PLA2G6. Mov Disord Clin Pract 2017; 4: 8–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synofzik M, Gonzalez MA, Lourenco CM, Coutelier M, Haack TB, Rebelo A et al. . PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain 2014a; 137 (Pt 1): 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synofzik M, Schule R. Overcoming the divide between ataxias and spastic paraplegias: shared phenotypes, genes, and pathways. Mov Disord 2017; 32: 332–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synofzik M, Schulze M, Gburek-Augustat J, Schweizer R, Schirmacher A, Gonzalez M et al. . Phenotype and frequency of STUB1 mutations: next-generation screenings in Caucasian ataxia and spastic paraplegia cohorts. Orphanet J Rare Dis 2014b; 9: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonelli A, D’Angelo MG, Arrigoni F, Brighina E, Arnoldi A, Citterio A et al. . Atypical adult onset complicated spastic paraparesis with thin corpus callosum in two patients carrying a novel FA2H mutation. Eur J Neurol 2012; 19: e127–9. [DOI] [PubMed] [Google Scholar]
- Treiber-Held S, Budjarjo-Welim H, Reimann D, Richter J, Kretzschmar H, Hanefeld F. Giant axonal neuropathy: a generalized disorder of intermediate filaments with longitudinal grooves in the hair. Neuropediatrics 1994; 25: 89–93. [DOI] [PubMed] [Google Scholar]
- Uchida Y, Hama H, Alderson NL, Douangpanya S, Wang Y, Crumrine DA et al. . Fatty acid 2-hydroxylase, encoded by FA2H, accounts for differentiation-associated increase in 2-OH ceramides during keratinocyte differentiation. J Biol Chem 2007; 282: 13211–9. [DOI] [PubMed] [Google Scholar]
- Yan X, Lin X, Wang Q, Zhang Y, Chen Y, Song S et al. . Dorsal visual pathway changes in patients with comitant extropia. PLoS One 2010; 5: e10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary material. Raw data regarding human subjects (e.g. genetic raw data, MRI datasets) are not shared freely to protect the privacy of the human subjects involved in this study; no consent for open sharing has been obtained.