

Abstract
Tuberculosis (TB) is the deadliest infectious disease, and yet accurate diagnostics for the disease are unavailable for many subpopulations. In this study, we investigate the possibility of using human breath for the diagnosis of active TB among TB suspect patients, considering also several risk factors for TB for smokers and those with human immunodeficiency virus (HIV). The analysis of exhaled breath, as an alternative to sputum-dependent tests, has the potential to provide a simple, fast, non-invasive, and readily available diagnostic service that could positively change TB detection. A total of 50 individuals from a clinic in South Africa were included in this pilot study. Human breath has been investigated in the setting of active TB using the thermal desorption-comprehensive two-dimensional gas chromatography-time of flight mass spectrometry methodology and chemometric techniques. From the entire spectrum of volatile metabolites in breath, three machine learning algorithms (support vector machines, partial least squares discriminant analysis, and random forest) to select discriminatory volatile molecules that could potentially be useful for active TB diagnosis were employed. Random forest showed the best overall performance, with sensitivities of 0.82 and 1.00 and specificities of 0.92 and 0.60 in the training and test data respectively. Unsupervised analysis of the compounds implicated by these algorithms suggests that they provide important information to cluster active TB from other patients. These results suggest that developing a non-invasive diagnostic for active TB using patient breath is a potentially rich avenue of research, including among patients with HIV comorbidities.
Keywords: VOCs, metabolomics, pulmonary tuberculosis, comprehensive two-dimensional gas chromatography, machine learning, human exhaled breath
1. Introduction
Tuberculosis (TB) is an infectious disease which has been present in humans since ancient times [1]. The disease is caused by the bacterium Mycobacterium tuberculosis (Mtb) and primarily infects the lungs (pulmonary TB represents ~85% of TB cases). [1, 2]. The World Health Organization estimates that new infections occur in about 1% of the population each year, which in 2016 resulted in more than 10 million cases of active TB. There are several factors that increase the risk of active Mtb infection, such as malnutrition, tobacco smoking, and several copathologies, the most important being co-infection with human immunodeficiency virus (HIV). People living with HIV are anywhere from 26–31 times more likely to develop active TB than persons without HIV [3]. Symptoms of active TB disease include at least of one or a combination of the following: cough, fever, night sweats, or weight loss, which are not specifically diagnostic and may be mild for months prior to clinical evaluation.
Diagnosis of pulmonary TB, particularly at the primary care level, depends on obtaining an adequate expectorated sputum sample. The gold standard for diagnosis of active TB (bacteriological culture), as well as nucleic acid amplification (NAA) and smear microscopy, are all sputum-dependent. However, up to one-third of TB cases cannot reliably produce an adequate biological sputum sample [4]. This can lead to more invasive sampling approaches, including induced sputum or gastric aspirate or a lack of diagnosis altogether, which occurs in many low resource settings. Moreover, risk factors, particularly HIV, can decrease the accuracy of several diagnostic tests, leading to challenges in both the diagnosis and treatment. Therefore, alternative non-invasive samples, such as urine [5] and exhaled breath [6], may be useful alternatives for adjuncts in TB diagnosis.
Several research groups, using gas chromatography (GC) linked to mass spectrometry (MS), have investigated the volatile molecules present in breath during Mtb infection in active pulmonary TB, reporting different panels of marker compounds [7–12]. This lack of overlap is likely due to a multitude of considerations, including use of different sampling methods and analytical tools as well as patient population heterogeneity, patient comorbidities (or lack thereof), different control groups, and statistical approaches used. A first step to address this lack of standardization was the development of technical standards for breath collection, published recently by Horvath et al [13]. In general, classical clinical parameters, food, drug medications, and smoking habits can also influence breath content. Age and gender may affect breath profiles [14], but their effect is more subtle than smoking behaviors, that can influence the breath profile creating subpopulations [15]. In addition, the profile of volatile organic compounds possibly produced during Mtb infection maybe modified by the host at different times during infection [16] and can be variable during the progression/regression of TB disease [11].
In this study, exhaled breath was evaluated from a pilot cohort of 50 patients living in an endemic TB region who were suspected of having TB and includes smokers and subjects with HIV infection. Breath volatile molecules were collected using a multiple-bed sorbent trap and then desorbed, separated, and detected by comprehensive two-dimensional GC (GC × GC) coupled to a time of flight mass spectrometer (TOF MS). Using a variety of machine learning algorithms, we were able to determine volatile metabolic patterns that could be helpful to discriminate between Mtb-infected and TB-suspect individuals. TB status was confirmed by GeneXpert MTB/RIF® (an NAA test), in combination with bacteriological culture in the case of patients with HIV infection.
2. Materials and methods
2.1. Patient demographics and TB infection confirmation
A total of 50 individuals, including 32 with active pulmonary TB and 18 controls with TB symptoms, but confirmed Mtb negative (Johannesburg, South Africa; 2015–2016), were included in the present study. Sputum samples were collected following WHO guidelines for TB [17]. An Institutional Review Board at the collaborating sites (Wits Reproductive Health and HIV Institute) and Dartmouth approved the research. All subjects gave their signed informed consent to participate and were at least 18 years old. TB status was confirmed by the GeneXpert MTB/RIF assay (Cepheid, Sunnyvale, CA, USA). This NAA test is a rapid, automated, cartridge-based test that can detect Mtb along with rifampicin resistance directly from sputum [18]. In individuals with HIV infection, the accuracy of GeneXpert in classifying patients with Mtb infection maybe unreliable [19], therefore the standard Mycobacteria growth indicator tube bacteriological culture test was employed to confirm Mtb-negative status in HIV-positive subjects (n = 4). Patient demographic information is reported in table 1.
Table 1.
Study subject demographic information where n = 50.
| Mtb positive (+) |
Mtb negative (−) |
p-value |
|
|---|---|---|---|
| Number (%) | 32 (64%) | 18 (36%) | 0.001 |
| Age, mean (±SD) | 35 (±10) | 35 (±10) | 0.918 |
| Gender (M/F) | 18/14 | 11/7 | 0.95 |
| Active smoker | 7/25 | 4/14 | 0.591 |
| (Y/N) | |||
| HIV (Y/N) | 21/11 | 4/14 | 0.006 |
| HIV Treatment | 8/24 | 3/15 | 0.591 |
| (Y/N) | |||
2.2. Breath and room air sampling
Prior to breath collection, patients rinsed their mouth with water to avoid volatile molecule contamination from the oral cavity [20] and then exhaled normally for 2 s into the room [21]. One liter Tedlar bags (SKC Inc., Eighty Four, PA, US), pre-conditioned by flushing with pure nitrogen gas, were used for the collection of breath over 3–5 min of regular breathing. On the same day of collection, breath was drawn from the Tedlar bag through a 0.22 μm filter (for the removal of potential pathogens), and onto the thermal desorption (TD) tube at a rate of 150 ml min−1, for a final breath sampling volume of 1 L. The three-bed TD tube containing Carbopack Y, X, and Carboxen 1000 (Supelco, Bellefonte, PA), a sorbent combination previously optimized for the collection of a wide range of breath molecules, was used to concentrate and store volatile molecules [22]. TD tubes containing breath molecules were hermetically sealed and stored at room temperature until further analysis which occurred within a month from collection, as previously reported [12, 23, 24]. One liter of room air was directly collected into the TD tube on the day of collection.
2.3. Analytical instrumentation
The TD tubes were desorbed into a Pegasus 4D (LECO Corporation, St. Joseph, MI) GC × GC-TOF MS instrument with an Agilent 7890 GC equipped with a TD unit, cooled injection system (CIS), and a Multi-Purpose Sampler (MPS) autosampler (Gerstel, Linthicum Heights, MD). The solvent venting time was 10 min (30 °C; 60 ml min−1), cryofocusing time was 5 min (−100 °C), sample desorption time was 180 s, CIS temperature was 330 °C, and the injection mode was splitless. Chromatographic analysis was performed using an Rxi-624Sil (60 m × 250 μm × 1.4 μm) as the first dimension (1D)-GC column and a Stabilwax (1.5 m × 250 μm × 0.5 μm) as second dimension (2D)-GC column, both purchased from Restek (Bellefonte, PA, US). The modulation time was 2 s total and helium as the carrier gas (flowrate: 2 ml min−1). TOF MS was employed as a detector, with the following parameters: electron impact at 70 eV; acquisition range: 30–500 mz−1; acquisition rate: 200 spectra/s; and ion source temperature: 200 °C. Data acquisition and analysis was performed using ChromaTOF software, version 4.50 (LECO Corporation).
2.4. Processing and analysis of chromatographic data
Chromatographic data were processed and aligned using ChromaTOF. For peak identification, a signal-to-noise (S/N) cutoff was set at 150:1 in at least one chromatogram and a minimum of 50:1 S/N in all others. The resulting peaks were identified by a forward search of the NIST 2011 library. For putative peak identification, a forward match score of ⩾800 (of 1000) was required. For the alignment of peaks across chromatograms, the maximum first and 2D retention time deviations were set at 6 and 0.2 s, respectively, and the inter-chromatogram spectral match threshold was stat 600. Compounds eluting prior to 300 s and artifacts (e.g., siloxane, phthalates, etc) were removed prior to statistical analysis with the support of the script tool available in ChromaTOF®, using the script reported in [25]. An additional data cleaning step was performed to remove common environmental contaminants, artifacts coming from the Tedlar® bag (e.g. phenol and N,N-dimethylacetamide), not included in the script (the complete list of compounds removed is reported in [23]). The most discriminatory features were assigned to a chemical class (Level 3) according to the criteria established by the Metabolomics Standards Initiative [26], based on mass spectral similarities to the NIST 2011 mass spectral library, with a match score ⩾750 (of 1000). Most hydrocarbons were generally assigned as ‘alkylated hydrocarbons’, as it is almost impossible to assign them a specific name based only on the mass spectra similarity, due to the intense fragmentation of this class of compounds into the MS ion source. However, the chemical class of these compounds can be assigned by considering both their location in the two-dimensional chromatogram and their mass spectral fragmentation pattern.
2.5. Statistical analysis
All statistical analyses were performed using R v3.4.3 (R Foundation for Statistical Computing, Vienna, Austria) using the ‘caret’ package [27]. Prior to statistical analyses, the relative abundance of compounds across chromatograms was normalized using probabilistic quotient normalization (PQN) [28] and peak intensities were log-transformed, mean-centered, and then unit-scaled.
Data was randomly subdivided into training (60% of samples) and validation sets (40% of samples). Three machine learning algorithms were used to identify the most discriminatory volatile metabolites and predict the class (Mtb infected versus TB suspect) to which samples in the validation set belonged: Random forest (RF) [29], support vector machines with a linear kernel (linear SVM) [30], and partial least squares discriminant analysis (PLS-DA) [31]. For each machine learning algorithm, a five-fold repeated cross validation (CV) was employed with ten repeats [32, 33]. Mean decrease in accuracy (MDA), feature specific area under the receiver operating characteristic (AUROC or AUC) curve, and the weighted sums of the absolute regression coefficients were used as the measures of variable importance for RF, linear SVM, and PLS-DA, respectively [27, 34]. Features were then selected using the ‘elbow method’ where feature importance was plotted and then a cutoff was selected in such a way that it captured the ‘elbow’ of the graph. This ensured that any large increases in feature importance were captured and eliminated features which demonstrated only incremental increases in importance [34]. Principal component analysis (PCA) [35] was used to visualize the variance between samples in the dataset given our selection of important features. Similarly, hierarchical clustering analysis (HCA) [36] was used to visualize the distance between each sample using Jaccard’s distance [37] and a heatmap is shown to visualize the relative expression of each feature.
3. Results and discussion
3.1. Breath evaluation and selected molecules
Contaminants and artifacts (e.g., siloxanes, phthalates) were removed, resulting in a reduction to 1023 features. Moreover, features present in the room air sample with a frequency of observation (FOO) ⩾50% were deleted from the matrix, reducing the number of volatile features to 251. At this point, 50% of FOO was applied within the groups to removing sparse features, leading to 128 features which were dominated by hydrocarbons (48%), followed by aromatics (11%), alcohols (8%), halogen-containing compounds (8%), esters (5%), ketones (5%), nitrogen-containing compounds (4%), sulfur-containing compounds (4%), aldehydes (3%), acids (2%), terpenes (1%), and unknowns (1%) (figure 1(b)). Prior to any further elaboration the data matrix was normalized using the PQN method, which accounts for dilution of the biological samples. This method uses median values for normalization ensuring a stability toward outliers and sampling variability, which can occur in metabolomics [28]. Then, after log-transformation and mean centering, RF, linear SVM, and PLS-DA were used to identify the most highly discriminatory volatile metabolites from the 128 features list in the discovery set and used to predict the class to which samples in the validation set belonged. A Venn diagram of the panel of 23 features obtained from each machine learning approach is reported in figure 1 (c).
Figure 1.

(a) Schematic of the feature reduction, (b) chemical class of the 128 features used for data used for statistical elaboration, and (c) Venn diagram of the panel of 23 features obtained for the three different machine learning techniques (RF, SVM, and PLS-DA).
Due to the high dimensional nature of -omics data, it is essential that machine algorithms are selected which can handle cases when the number of features far outweigh the number of samples. Moreover, these algorithms need to also be able to handle highly correlated features (multicollinearity) [38, 39]. Practically, feature selection to a manageable size is necessary in order to translate biomarkers to a handheld or benchtop system in a clinic or diagnostic laboratory. [40]. RF algorithms generate many classification trees, using randomly selected subsamples of both features and data points. Features are ultimately selected based on which variables best divides the data according to class at each split [29]. RF has proven to be particularly resilient in -omics classification [38]. SVM is a non-parametric method which projects data into some highly dimensional subspace, and then identifies a hyperplane to separate the classes geometrically [41]. The objective of the PLS-DA algorithm is to maximize the covariance between samples and their dependent variable (such as case status) in high dimensional data. To achieve this, it finds a linear sub-space of explanatory variables [42, 43]. Each of these models has their own sets of parameters which require tuning. To reduce the risk of overfitting, we employed five-fold CV, where our training model was split into 5 approximately even sized pieces, and then we trained the model on 4/5 of these pieces and tested on the remaining piece. We then withhold a separate piece of the data and retrain the model on the remaining 4/5 pieces. We iterate through this process until each piece has been withheld for testing. This allows us to develop an accuracy distribution based on each model’s performance on the withheld piece of the data. We repeated our CV scheme ten times so that multiple different cuts of the training data are considered, thus reducing the variability of the results [32]. We used the entire five-fold repeated CV procedure twice—first to rank our feature importance and apply feature selection, and then again to tune our model parameters used the subset of selected features. The final models after feature selection and tuning were then used on the validation data to evaluate their performance on unseen data.
For each model, we evaluated the accuracy, sensitivity, specificity, and AUC in order to assess prediction errors [44]. Table 2 shows each statistic for each of the three final models in both the training and validation datasets. While the performance of all three models on the validation set is strong, both the SVM and PLS-DA models had slightly poorer performance in the validation data. The RF model had similar performance in both the training and validation sets. While the specificity in the validation data is low, this may be partly driven by the low number of ‘true negatives’ in our validation set (n = 5). The high level of sensitivity in the validation data may indicate that the selected volatile features maybe useful in the development of a ‘rule-out’ TB diagnostic, wherein a negative result from a diagnostic developed from these features would rule out a TB diagnosis with a high degree of certainty. This would have utility in the clinic as a tool which could be used to screen patients who have a low probability of having TB so they can avoid unnecessary invasive testing using the gold standard diagnostic [3].
Table 2.
Accuracy, sensitivity, specificity, and AUROC obtained by the machine learning techniques used.
| RF |
SVM |
PLS-DA |
||||
|---|---|---|---|---|---|---|
| Training | Validation | Training | Validation | Training | Validation | |
| Accuracy | 0.87 | 0.90 | 1.00 | 0.85 | 0.90 | 0.80 |
| Sensitivity | 0.82 | 1.00 | 1.00 | 0.87 | 0.94 | 0.87 |
| Specificity | 0.92 | 0.60 | 1.00 | 0.80 | 0.84 | 0.60 |
| AUROC | 0.93 | 0.96 | 1.00 | 0.89 | 0.99 | 0.85 |
The receiving (or relative) operator characteristic (ROC) curves for the training and validation sets in each of the three models are shown in figure 2. The final SVM model had an AUC of 1 in the training data and 0.89 in the withheld validation data. The final PLS-DA model had an AUC of 0.99 in the training data, and 0.85 in the validation data. The final RF model had an AUC of 0.93 in the training data, and 0.96 in the validation data. Given the RF models’ superior performance in the withheld validation data, we selected it as the best model for classification of active TB patients in this particular dataset.
Figure 2.

ROC curve by using SVM, PLS-DA, and RF algorithms. For each machine learning technique, the set of molecules generated in the training set (n = 30) were tested in the validation set (n = 20).
To assess whether a bias due to class imbalance was present due to the limited number of HIV−/Mtb+ samples, the accuracy of the model within this particular subgroup of data was evaluated. Overall, the RF model classified 75% of this group correctly, and hence we do not think class imbalance greatly affected our results.
There was significant overlap of selected features across our three models. In total, 23 features were selected in total from all three models, with 12 features in common to all models. The high conservation of volatile features across these three disparate models increases our confidence that these features are potentially discriminatory molecules for active TB diagnosis on this study population. In table 3, the rank of each feature for each model is given for the three machine learning techniques, the match of the feature with the NIST library, and retention time of each feature in the first and second dimensions are reported. More than 60% of volatile metabolites detected can be attributed to chemical classes related to the lipid oxidation path-ways, namely, ketones, aldehydes, alcohols, and in particular hydrocarbons (around 50%). These sorts of molecules have been reported to originate largely from free radical oxidative fragmentation of lipids due to oxidative stress [45].
Table 3.
Machine learning model feature ranking and analytical context.
| #Feature | RF | SVM | PLS-DA | Average score | Chemical class | Forward similarity | Reverse similarity | 1tR(sec) | 2tR(sec) |
|---|---|---|---|---|---|---|---|---|---|
| 47 | 1 | 2 | 1 | 1.33 | Alkylated hydrocarbon | 823 | 847 | 1876 | 0.59 |
| 97 | 2 | 1 | 2 | 1.67 | Halogen containing | 888 | 888 | 1738 | 0.65 |
| 103 | 3 | 3 | 4 | 3.33 | Aldehyde | 903 | 903 | 1466 | 0.67 |
| 12 | 4 | 14 | 3 | 7.00 | Halogen containing | 874 | 874 | 2561 | 0.60 |
| 36 | 5 | 11 | 7 | 7.67 | Hydrocarbon | 842 | 856 | 1136 | 0.60 |
| 8 | 6 | 6 | 11 | 7.67 | Alkylated hydrocarbon | 877 | 877 | 2115 | 0.60 |
| 99 | 14 | 4 | 6 | 8.00 | Alkylated hydrocarbon | 921 | 921 | 1136 | 0.57 |
| 11 | 10 | 5 | 10 | 8.33 | Acid | 753 | 778 | 1356 | 0.64 |
| 25 | 12 | 9 | 5 | 8.67 | Alkylated aromatic | 850 | 850 | 708 | 0.67 |
| 56 | 8 | 8 | 15 | 10.33 | Alkylated hydrocarbon | 860 | 860 | 2583 | 0.61 |
| 14 | 7 | 13 | 12 | 10.67 | Alkylated hydrocarbon | 865 | 865 | 804 | 0.62 |
| 89 | 13 | 12 | 14 | 13.00 | Aldehyde | 809 | 867 | 2418 | 0.66 |
| 54 | 19 | 15 | 9 | 14.33 | Alkylated hydrocarbon | 811 | 827 | 2386 | 0.60 |
| 53 | 26 | 10 | 8 | 14.67 | Alkylated hydrocarbon | 876 | 876 | 2742 | 0.61 |
| 124 | 22 | 16 | 18 | 18.67 | Alkylated hydrocarbon | 900 | 900 | 1507 | 0.58 |
| 74 | 18 | 26 | 13 | 19.00 | Alkylated ester | 821 | 821 | 1978 | 1.80 |
| 18 | 9 | 7 | 42 | 19.33 | Alkylated alcohol | 838 | 838 | 2357 | 0.60 |
| 41 | 20 | 17 | 22 | 19.67 | Alkylated hydrocarbon | 774 | 879 | 1443 | 0.58 |
| 24 | 27 | 19 | 16 | 20.67 | Alkylated hydrocarbon | 775 | 800 | 1848 | 0.59 |
| 45 | 16 | 20 | 32 | 22.67 | Alkylated hydrocarbon | 862 | 911 | 1336 | 0.59 |
| 111 | 11 | 18 | 43 | 24.00 | Ester | 926 | 926 | 1910 | 0.69 |
| 120 | 29 | 44 | 17 | 30.00 | Alkylated alcohol | 828 | 828 | 2722 | 0.60 |
| 81 | 54 | 21 | 25 | 33.33 | Cyclo-alcohol | 881 | 886 | 1460 | 0.80 |
To visualize the ability of these features to discriminate active TB among TB suspects, we used an HCA and PCA developed using all 23 features selected by any of the three models which is shown in figure 3.
Figure3.

A heatmap showing the unsupervised clustering of all 23 features discovered across the three machine learning techniques (RF, SVM, and PLS-DA).
The HCA and subsequent heatmap shown in figure 3 shows the HCA analysis where the distance between samples was calculated using Jaccard’s index, a distance metric which has previously been shown to be resilient to noise [37]. All features selected by any of our models are shown on the vertical axis while the unique patient number and their TB and HIV status are shown on the horizontal axis. Notably, as seen by the blue and yellow annotation bar, all of the TB+ cases cluster together, with only 2 out of 14 of the TB−/HIV− cases clustering away from the TB– group. Of note, both of these cases are HIV–, which indicates that these cases are not clustering away from the other TB– cases due to confounding by HIV status. Hence, we believe that the volatile biomarkers selected by our algorithms are not sensitive to HIV status.
A PCA developed using all 23 selected features is shown in figure 4(a), where the color maps to TB/HIV case status (blue is TB–, yellow is TB+, while the darker shades are HIV– and the bright shades HIV+). While we do not observe distinct clusters by case status, a general assortment of TB– cases to TB+ cases along the PC1 axis is observed. To further examine this effect, we examine the distribution of the PC1 scores across the TB/HIV subgroups of interest using a boxplot in figure 4(b). We can clearly see differences between the TB+/TB– patients by the PC1 score. A global Kruskal–Wallis test rejected the hypothesis that these samples originated from the same distribution with a highly significant p-value of 9.1 e−7. We also conducted two-way comparisons between the various TB+/TB− subgroups using Wilcoxon’s rank-sum test. All comparisons were significant at a Benjamini–Hochberg corrected significant level of a = 0.05. With additional samples, we expect this effect to become clearer.
Figure 4.
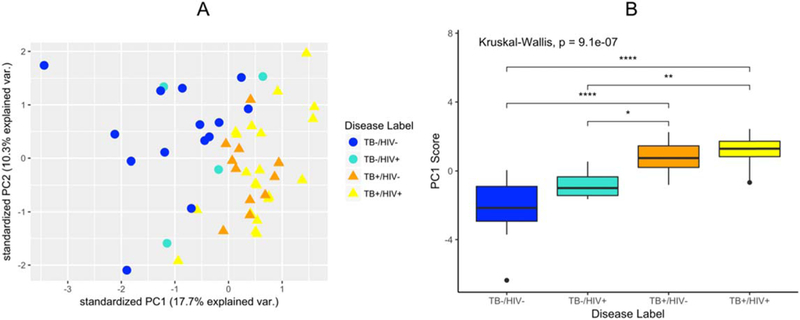
(a) PCA of the 23 discriminatory features obtained after three different machine learning techniques (RF, SVM, and PLS-DA). (b) Boxplot showing the first PC component score for each of the TB/HIV subgroups of interest, as well as a global Kruskall–Wallis p-value. Two-way comparisons between the TB+/TB−subgroups are also shown, where the number of stars indicate the significance of a Wilcoxon rank-sum test.
Similar behavior was observed using the discriminatory features obtained after CV considering the single machine learning technique applied (14 for RF, 21 for SVM, and 17 for PLS-DA), but also considering the 12 common features within each model (figure 1(c)). HCA and PCA plots for each machine learning model utilized in our analyses are available in the supplementary data (figures S1–S4 are available online at stacks.iop.org/JBR/13/016005/mmedia), while figure S4 shows HCA and PCA plots of the 12 common features for each model
3.2. Study strengths and limitations
In the present pilot study, we evaluated the potential ability of volatile molecules in the breath for discriminating between Mtb-infected and TB-suspect individuals using three different machine learning algorithms. Twenty-three discriminatory features were selected using the different algorithms (PLS-DA, SVM, and RF). Although a good match with the library was obtained (20 out of 23 features had a match >800/1000 and the other 3 > 750/1000), we preferred to not report a putative identification of these possible biomarkers, since a large cohort study is necessary to validate the biomarkers. Future studies should include a greater proportion of patients who are TB suspects that end up being negative for Mtb infection, but who are also co-infected with HIV, as well as a higher number of co-infected subjects. In addition, other comorbidities in the patient population, e.g., diabetes, would also assist in generating universal biomarkers. Despite the limitations, we plan to evaluate the panel of 23 breath molecules in future studies and hopefully confirm and validate them as biomarkers by using an external dataset. It is important to highlight that the percentage of chemical classes of the 23 breath molecules reported as discriminatory in this pilot study (table 3) is in accordance with previous GC-based techniques studies on human exhaled breath in the setting of TB disease [7–12].
4. Conclusion
This pilot study (n = 50) is part of a larger, ongoing TB breath biomarker initiative. Here, we demonstrated that volatile metabolites present in human exhaled breath can also be used to discriminate between individuals with a positive Mtb infection and people with one or more TB symptoms, but with a confirmed negative Mtb infection. In the validation set, the accuracy value was about 0.8–0.9 for all the three machine learning techniques applied, with an AUROC between 0.85 (PLS-DA), and 0.96 (RF). Although all three models showed great prediction power to discriminate those infected with Mtb and TB suspect individuals, the RF model was the most consistent, showing similar performance in both the training and validation sets. This study, along with others, reiterate that exhaled human breath in diseased individuals contains useful data which should be developed as a non-invasive clinical tool to be deployed in efforts to curb the spread of Mtb.
Supplementary Material
Acknowledgments
Dartmouth College holds an Institutional Program Unifying Population and Laboratory Based Sciences award from the Burroughs Wellcome Fund, and C. Bobak and Mavra Nasir were supported by this grant (Grant #1014106). Christiaan A. Rees was supported by a National Institutes of Health training grant (Grant #T32LM012204).
Footnotes
Supplementary material for this article is available online
References
- [1].Lawn SD and Zumla AI 2011. Tuberculosis Lancet 378 57–72 [DOI] [PubMed] [Google Scholar]
- [2].Southwick F 2007. Chapter 4: Pulmonary infections Infectious Diseases: A Clinical Short Course 2nd edn (New York: McGraw-Hill Medical Publishing Division; ) pp 104 313–4 [Google Scholar]
- [3].World Health Organization. Global Tuberculosis Report 2017 2017 [Google Scholar]
- [4].Parsons LM, Somoskövi Á, Gutierrez C, Lee E, Paramasivan CN, Abimiku A, Spector S, Roscigno G and Nkengasong J 2011. Laboratory diagnosis of tuberculosis in resource-poor countries: challenges and opportunities Clin. Microbiol. Rev. 24 314–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lawn SD 2012. Point-of-care detection of lipoarabinomannan (LAM) in urine for diagnosis of HIV-associated tuberculosis: a state of the art review BMC Infect. Dis. 12 103–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Boots AW, van Berkel JJBN, Dallinga JW, Smolinska A, Wouters EF and van Schooten FJ 2012. The versatile use of exhaled volatile organic compounds in human health and disease J. Breath Res. 6 027108 [DOI] [PubMed] [Google Scholar]
- [7].Phillips M, Cataneo RN, Condos R, Ring Erickson GA, Greenberg J, La Bombardi V, Munawar MI and Tietje O 2007. Volatile biomarkers of pulmonary tuberculosis in the breath Tuberculosis 87 44–52 [DOI] [PubMed] [Google Scholar]
- [8].Phillips M, Basa-Dalay V, Bothamley G, Cataneo RN, Lam PK, Natividad MPR, Schmitt P and Wai J 2010. Breath biomarkers of active pulmonary tuberculosis Tuberculosis 90 145–51 [DOI] [PubMed] [Google Scholar]
- [9].Phillips M et al. 2012. Point-of-care breath test for biomarkers of active pulmonary tuberculosis Tuberculosis 92 314–20 [DOI] [PubMed] [Google Scholar]
- [10].Kolk AH, van Berkel JJBN, Claassens MM, Walters E, Kuijper S, Dallinga JW and van Schooten F 2012. Breath analysis as a potential diagnostic tool for tuberculosis Int. J. Tuberc. Lung Dis. 16 777–82 [DOI] [PubMed] [Google Scholar]
- [11].Dang NA, Janssen HG and Kolk AH 2013. Rapid diagnosis of TB using GC–MS and chemometrics Bioanalysis 5 3079–97 [DOI] [PubMed] [Google Scholar]
- [12].Beccaria M et al. 2018. Preliminary investigation of human exhaled breath for tuberculosis diagnosis by multidimensional gas chromatography—time of flight mass spectrometry and machine learning J. Chromatogr. B 1074–75 46–50 [DOI] [PubMed] [Google Scholar]
- [13].Horváth I et al. 2017A European Respiratory Society technical standard: exhaled biomarkers in lung disease Eur. Respir. J. 49 1600965 [DOI] [PubMed] [Google Scholar]
- [14].Das MK, Bishwal SC, Das A, Dabral D, Varshney A, Badireddy VK and Nanda R 2014. Investigation of gender-specific exhaled breath volatome in humans by GCxGC-TOFMS Anal. Chem. 86 1229–37 [DOI] [PubMed] [Google Scholar]
- [15].Blanchet L, Smolinska A, Baranska A, Tigchelaar E, Swertz M, Zhernakova A, Dallinga JW, Wijmenga C and van Schooten FJ 2017. Factors that influence the volatile organic compound content in human breath J. Breath Res. 11 016013 [DOI] [PubMed] [Google Scholar]
- [16].Bean H, Jiménez-Díaz J, Zhu J and Hill JE 2015. Breathprints of model murine bacterial lung infections are linked with immune response Eur. Respir. J. 45 181–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].World Health Organization (WHO) 1999. Guidelines for the Prevention of Tuberculosis in Health Care Facilities in Resource-limited Settings (Geneva: World Health Organization; ) [Google Scholar]
- [18].World Health Organization 2015. Automated real-time nucleic acid amplification technology for rapid and simultaneous detection of tuberculosis and rifampicin resistance: Xpert MTB/RIF assay for the diagnosis of pulmonary and extrapulmonary TB in adults and children: policy update (Geneva: World Health Organization; ) http://apps.who.int/iris/handle/10665/112472 [PubMed] [Google Scholar]
- [19].Lawn SD et al. 2013. Advances in tuberculosis diagnostics: the xpert MTB/RIF assay and future prospects for a point-of-care test Lancet Infect. Dis. 13 349–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Caddy GR, Sobell MB and Sobell LC 1978. Alcohol breath tests: criterion times for avoiding contamination by ‘mouth alcohol’ Behav. Res. Methods Instrum. 10 814–8 [Google Scholar]
- [21].Mochalski P, Wzorek B, Sliwka I and Amann A 2009. Suitability of different polymer bags for storage of volatile sulphur compounds relevant to breath analysis J. Chromatogr. B 877 189–96 [DOI] [PubMed] [Google Scholar]
- [22].Libardoni M, Stevens PT, Waite JH and Sacks R 2006. Analysis of human breath samples with a multi-bed sorption trap and comprehensive two-dimensional gas chromatography (GCxGC) J. Chromatogr. B 842 13–21 [DOI] [PubMed] [Google Scholar]
- [23].Mellors TR, Blanchet L, Flynn JL, Tomko J, O’Malley M, Scanga CA, Lin PL and Hill JE 2017A new method to evaluate macaque health using exhaled breath: a case study of M. tuberculosis in a BSL-3 setting J. Appl Physiol. 122 695–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mellors TR et al. 2018. Identification of Mycobacterium tuberculosis using volatile biomarkers in culture and exhaled breath J. Breath Res. 13 016004 [DOI] [PubMed] [Google Scholar]
- [25].Purcaro G, Stefanuto P, Franchina FA, Beccaria M, Wieland-Alter WF, Wright PFand Hill JE 2018. SPME-GC × GC-TOF MS fingerprint of virally-infected cell culture: sample preparation optimization and data processing evaluation Anal. Chim. Acta. 1027 158–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sumner LW, Samuel T, Noble R, Gmbh SD, Barrett D, Beale MH and Hardy N 2007. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI) Metabolomics 3 211–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kuhn M 2018. Caret: Classification and Regression Training. R package version 6.0–79 (https://CRAN.R-project.org/package=caret)
- [28].Dieterle F, Ross A, Schlotterbeck G and Senn H 2006. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures: application in 1H NMR metabolomics Anal. Chem. 78 4281–90 [DOI] [PubMed] [Google Scholar]
- [29].Breiman L 2001. Random forests Mach. Learn. 45 5–32 [Google Scholar]
- [30].Cortes C and Vapnik V 1995. Support-vector networks Mach. Learn. 20 273–97 [Google Scholar]
- [31].Barker M and Rayens W 2003. Partial least squares for discrimination J. Chemom. 17 166–73 [Google Scholar]
- [32].Mosteller F and Tukey JW 1968. Data analysis, including statistics Handbook of Social Psychology vol 1968 (Reading, MA: Addison-Wesley; ) [Google Scholar]
- [33].Kohavi R 1995. A study of cross-validation and bootstrap for accuracy estimation and model selection IJCAI 14 1137–45 [Google Scholar]
- [34].Krooshof PWT, Ustun B, Postma GJand Buydens LMC 2010. Visualization and recovery of the (bio)chemical interesting variables in data analysis with support vector machine classification Anal. Chem. 82 7000–7 [DOI] [PubMed] [Google Scholar]
- [35].Hotelling H 1933. Analysis of a complex of statistical variables into principal components J. Educ. Psychol. 24 417 [Google Scholar]
- [36].Tibshirani R and Friedman J 2001. The Elements of Statistical Learning: Data Mining, Inference, and Prediction (Berlin, Heidelberg: Springer; ) [Google Scholar]
- [37].Toldo R and Fusiello A 2008. Robust multiple structures estimation with j-linkage Lecture Notes in Computer Science (Berlin, Heidelberg: Springer; ) pp 537–47 [Google Scholar]
- [38].Lebedev A et al. 2014. Random forest ensembles for detection and prediction of Alzheimer’s disease with a good between-cohort robustness NeuroImage: Clinical 6 115–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Statnikov A, Wang L and Aliferis CF 2008. A comprehensive comparison of random forests and support vector machines for microarray-based cancer classification BMC Bioinform. 9 319–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pal NR 2007A fuzzy rule based approach to identify biomarkers for diagnostic classification of cancers Fuzzy Systems Conf.. FUZZ-IEEE 2007 IEEE Int; pp 1–6 [Google Scholar]
- [41].Suykens JA and Vandewalle J 1999. Least squares support vector machine classifiers Neural Process. Lett. 9 293–300 [Google Scholar]
- [42].Gromski PS, Muhamadali H, Ellis DI, Xu Y, Correa E, Turner ML and Goodacre R 2015A tutorial review: metabolomics and partial least squares-discriminant analysis —a marriage of convenience or a shotgun wedding Anal. Chim. Acta. 879 10–23 [DOI] [PubMed] [Google Scholar]
- [43].Pérez-Enciso M and Tenenhaus M 2003. Prediction of clinical outcome with microarray data: a partial least squares discriminant analysis (PLS-DA) approach Hum. Genet. 112 581–92 [DOI] [PubMed] [Google Scholar]
- [44].Fielding A and Bell J 1997. A review of methods for the assessment of prediction errors in conservation presence/absence models Environ. Conserv. 24 38–49 [Google Scholar]
- [45].Schulz S and Dickschat JS 2007. Bacterial volatiles: the smell of small organisms Nat. Prod. Rep 24 814–42 Haick H, Broza YY, Mochalski P, Ruzsanyi V and Amann A 2014. Assessment, origin, and implementation of breath volatile cancer markers Chem. Soc. Rev. 43 1423–49 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.