

Abstract
Objective
To compare the clinical features of patients showing a classical phenotype of facioscapulohumeral muscular dystrophy (FSHD) with genetic and epigenetic characteristics of the FSHD1 and FSHD2 loci D4Z4 and SMCHD1.
Methods
This is a national multicenter cohort study. We measured motor strength, motor function, and disease severity by manual muscle testing sumscore, Brooke and Vignos scores, clinical severity score (CSS), and age-corrected CSS, respectively. We correlated these scores with genetic (D4Z4 repeat size and haplotype; SMCHD1 variant status) and epigenetic (D4Z4 methylation) parameters.
Results
We included 103 patients: 54 men and 49 women. Among them, we identified 64 patients with FSHD1 and 20 patients with FSHD2. Seven patients had genetic and epigenetic characteristics of FSHD1 and FSHD2, all carrying repeats of 9–10 D4Z4 repeat units (RU) and a pathogenic SMCHD1 variant. In the remaining patients, FSHD was genetically excluded or remained unconfirmed. All clinically affected SMCHD1 mutation carriers had a D4Z4 repeat of 9–16 RU on a disease permissive 4qA haplotype. These patients are significantly more severely affected by all clinical scales when compared to patients with FSHD1 with upper-sized FSHD1 alleles (8–10 RU).
Conclusion
The overlap between FSHD1 and FSHD2 patients in the 9–10 D4Z4 RU range suggests that FSHD1 and FSHD2 form a disease continuum. The previously established repeat size threshold for FSHD1 (1–10 RU) and FSHD2 (11–20 RU) needs to be reconsidered.
Clinicaltrials.gov identifier
Facioscapulohumeral dystrophy (FSHD) is associated with epigenetic derepression of the polymorphic D4Z4 repeat on chromosome 4q.1–3 In the common form FSHD type 1 (FSHD1; MIM158900), derepression is caused by a reduction of D4Z4 repeat units (RU) to a size of 1–10 RU.1,4–7 In the rare FSHD type 2 (FSHD2; MIM 158901), D4Z4 derepression is caused by defects in D4Z4 chromatin repressors, mostly SMCHD1.8,9 Few patients have been reported with a combination of a D4Z4 repeat size reduction and a mutation in D4Z4 chromatin modifier (FSHD1+2). FSHD1 and FSHD2 mutations can be epistatic: individuals with a FSHD1 allele and SMCHD1 mutation typically show an early disease onset and rapid progression.10,11 The DUX4 polyadenylation signal, present only on 4qA chromosomes, is mandatory to develop FSHD by facilitating stable misexpression of the D4Z4 encoded DUX4 gene toxic to skeletal muscle.8,12–16
FSHD is hallmarked by a recognizable pattern of muscle wasting and variability in disease onset and progression.17–19 Early onset and rapid progression are typically associated with the shortest D4Z4 repeats.20 In families with intermediate repeats, clinical variability is more prominent, while families with the largest FSHD alleles have a high incidence of nonpenetrant carriers.21,22 In FSHD2, the D4Z4 repeat size and the residual SMCHD1 functionality may contribute to clinical variability.23,24 Factors like age, sex,25 degree of kinship,26 hormones,27 and telomere shortening28,29 have also been associated with clinical variability in FSHD. Despite these advances, clinical variability remains partly explained. This prompted us to clinically and (epi)genetically characterize a large cohort of patients with FSHD.
Methods
This is a national multicenter study conducted in 4 neuromuscular disease specialized centers (University Hospital of Nice, Myology Institute of Paris, University Hospital of Lyon, and University Hospital of Caen). Other centers referred patients to one of these specialized centers. Patients were recruited between 2013 and 2016.
Study participants
We included patients aged 18 years and older with a typical clinical FSHD phenotype, according to the FSHD main diagnostic criteria established by Padberg et al.17,18 Patients fulfilled at least 3 of the following features: (1) facial weakness; (2) shoulder girdle muscle involvement; (3) forelimb muscle involvement; and (4) asymmetry of clinical manifestations. All patients underwent clinical examination, which included clinical history and collection of age-corrected clinical severity score (CSS), Brooke and Vignos scores, and Medical Research Council score for manual muscle testing (MMT). More specifically, motor function was evaluated by MMT sumscore of 34 individual facial, axial, and upper and lower limbs muscles (facial: orbicularis oculi and orbicularis oris; axial: neck flexor and extensor muscles; upper limbs: shoulder flexion, global abduction, abduction with scapula manually maintained, scapula retraction, elbow flexion and extension, wrist extension, fingers flexion and extension; lower limbs: hip flexion, knee flexion and extension, dorsal flexion, plantar flexion) that were scored from 0 to 5 based on the effective performance of a movement in relation to the forces of gravity and manual resistance. Patients presenting comorbidities, including obesity (body mass index ≥ 30), malignancy, joint diseases, chronic infectious diseases, other neuromuscular diseases, and any chronic or acute clinical condition that can influence the clinical evaluation, were excluded from the analysis.
D4Z4 allele sizing
For genetic testing, the size of the D4Z4 repeat array was determined by pulsed field gel electrophoresis followed by Southern blot analysis of EcoRI-, EcoRI/BlnI-, or HindIII-digested genomic DNA extracted from fresh blood samples using probes p13E-11, 4qA, and 4qB, as previously described.5,30
DR1 bisulfite sequencing
To establish the methylation level of the D4Z4 region proximal to DUX4, named DR1, genomic DNA was extracted with the QIAamp DNA Blood Midi Kit (Qiagen, Venlo, the Netherlands) from 4 mL of fresh blood. Bisulfite sequencing was conducted with the EZ DNA Methylation Direct kit (Zymo Research, Irvine, CA) according to manufacturer's instructions. Briefly, 200 ng of genomic DNA was diluted in CT conversion reagent buffer, denatured for 8 minutes at 98°C, and incubated for 3.5 hours at 64°C. After on-column desulfonation, DNA was eluted in 10 μL elution buffer, and subjected to 35 cycles of PCR amplification (95°C/30 seconds, 54°C/30 seconds, 72°C/20 seconds) following an initial denaturation step at 95°C for 10 minutes, targeting the DR1 region in the D4Z4 repeat with primers DR1F (5′-GAAGGTAGGGAGGAAAAG-3′) and DR1R (5′-ACTCAACCTAAAAATATACAATCT-3′), as previously described.31 Specificity for bisulfite converted DNA was verified by PCR amplification of bisulfite-treated and untreated genomic DNA. PCR products were gel-purified and cloned into pGEM-T (pGEM-T Easy Vector System, Promega, Madison, WI). Transformants containing recombinant plasmids were selected by blue/white colony screening and minimally 12 colonies were randomly picked for Sanger sequencing. Analysis was performed using BISMA online software with the default conditions (exclusion of sequences with a bisulfite conversion rate below 95% or with an identity to the reference sequence lower than 90%).32
Multiplex PCR-based next-generation sequencing (NGS)
A custom Ampliseq panel targeting the 48 exons of SMCHD1 (NM_015295) and 50 bp flanking sequences was developed using the Ampliseq Designer plugin (reference IAD52340_125, ampliseq.com/, Thermo Fisher Scientific, Waltham, MA). Targeted regions were theoretically covered by 86 amplicons of 125–175 bp average length for a total of 13.7 kb sequenced after multiplex PCR amplification. The theoretical coverage was 100% for all exons, except 48 bp missing in 5′ of exon 28.
In addition, 15 amplicons on chromosome 4q35 were dedicated to the determination of the presence of the permissive 4qA allele. These amplicons target different sequences previously described by Lemmers and collaborators5,23 that allow distinguishing between 4q and 10q. More specifically, these amplicons target the simple sequence-length polymorphism (SSLP) proximal to D4Z4 macrosatellite (1 amplicon), the P13E-11/D4F104S1 locus (5 amplicons), and the 4q subtelomeric region (AF017466 and AF085189) immediately distal to the D4Z4 repeat (2 and 7 amplicons).
Ten-nanogram genomic DNA was amplified to generate the library using the Ion AmpliSeq Library Kit 2.0 according to manufacturer's recommendations (Life Technologies, Carlsbad, CA). Amplicon synthesis by multiplex PCR, purification, amplification by emulsion PCR, and enrichment using Ion One Touch 2 System, loading on Ion 316 chips, sequencing with Ion Personal Genome Machine system (Life Technologies), and data collection were performed as described.33 A typical 24-sample run workflow was achieved in 3 days.
NGS bioinformatic analysis: Variant identification
Sequence alignment was performed with the Torrent Mapping Alignment Program (github.com/iontorrent/TMAP, Ion Torrent for Life Technologies), which was developed specifically to analyze Ion Torrent data and included in the Torrent Suite v 5.0.2. Aligned reads from BAM files were visualized using the Integrative Genomics Viewer v2.3 (broadinstitute.org/igv/) tool from the Broad Institute (Cambridge, MA). Single nucleotide variants (SNVs) and short indels detection from the BAM files was done using 2 different variant callers: we used the Torrent Suite Variant Caller version 5.0.2 (TSVC) plugin (ioncommunity.thermofisher.com/community/products/software/torrent_suite, Life Technologies), which allows a GATK-based calling for SNVs and combined it with Freebayes for complex variant or multi-nucleotide polymorphisms (MNPs) and GATK Long Indel Assembler to detect insertions or deletions from 4 to 70 bp. Candidate variants passed the filters of a minimum sequencing depth ≥d6× for single-nucleotide polymorphisms (SNPs), MNPs, or complex variants and ≥15× for short indels and 2% minimum allele frequency. They were then annotated, ranked, and interpreted using the Polydiag in-house software (Bioinformatics Paris–Descartes University Department).
In parallel, SNVs and short indels were called from BAM files using the NextGENe v2.3.4 (Softgenetics, State College, PA, softgenetics.com/NextGENe.php) and then visualized and filtered using NextGENe Viewer v2.3.4. We applied the same parameters as used with the TSVC, except for the minimum allele frequency, which we increased to 10% to facilitate discarding false-positive variants. In brief, the variants of interest were called by both variant callers, and at most in 2 samples from unrelated patients in the same sequencing run.
NGS bioinformatic analysis: Single and multiexon deletions/duplications identification
Potential complete or partial deletions of SMCHD1 were identified through examining the read coverage of the amplicons extracted from the BAM files with the plugin Coverage Analysis from the Torrent Suite. The number of reads obtained for each amplicon of candidate gene LRIF1 were used to normalize coverage data for disease gene SMCHD1 and candidate gene SUV39H1 as previously described.34 Ratios of <0.7 and >1.3 between normalized coverage data of the target gene compared to the control were indicative for deletions or duplications. Since SUV39H1 is localized on the X chromosome, its copy number variation allowed us to check the correspondence between the sex deducted from the NGS data and the one expected for each sample.
NGS bioinformatic analysis: Detection of 4qA/4qB alleles
The sequence for 12 of the 15 informative SNPs and the SSLP fragment described by Lemmers and collaborators5 within D4F104S1 were obtained from the contiguous amplicons AMPL7153688424 (5 SNPs), AMPL7153688425 (7 SNPs), and AMPL7153742311. The corresponding primers allowed PCR amplification of these 3 amplicons from 4q and the homologous sequences from 10q. Bioinformatic analysis using an in-house algorithm (Marc Bras, PF BI-PD) based on the allele frequency of the 12 SNPs allowed discrimination of the 4q and 10q alleles. For this, alignment of the reads was first forced against the FASTA sequence of chromosome 4. The variants at the genomic position of each of the 12 SNPs were then called and their frequency determined. Our algorithm links the reads corresponding to amplicons AMPL7153688424 and AMPL7153688425 that were contiguous on the same chromosome 4 or 10 allele based on counting of the reads carrying the same contiguous SNPs. The alleles and the frequency of the SSLP determined in parallel was based on the number of CA repeats as described by Lemmers and collaborators.5 In addition, primers for AMPL7153706638 and AMPL7153742309 specifically amplify the 4qB allele (Refseq AF017466), so that a copy number analysis of the corresponding reads determined the number of 4qB alleles. This approach allowed us thus to determine the haplotypes 4qB/4qB, 4qB/4qA, or 4qA/4qA. The SNP analysis, combined with SSLP and the copy number analysis of 4qB-type sequence, allowed us to discriminate 4q and 10q alleles and in particular to identify the presence and the number of permissive 4qA alleles as described by Lemmers et al.5
Variant confirmation using Sanger sequencing
SMCHD1 variants identified by NGS were confirmed using Sanger sequencing on the corresponding exon. Sequences were aligned with Seqscape analysis software v2.5 (Applied Biosystems, Foster City, CA) to the corresponding RefSeq sequence NM_015295 (nt 18: 2655886 to 2805015) of human genome release GRCh37/hg19.
Statistical analysis
Using GraphPad Prism 7, statistical analyses were performed on data derived from 57 patients with FSHD1, 20 patients with FSHD2, and 7 patients with FSHD1+2. Four patients with FSHD1 having complex 4q rearrangements such as deletions, translocations, or mosaicism were not included in further statistical tests. Also, 3 patients with FSHD2 with undetermined number of D4Z4 RU (>11) were not included due to technical issues. Twelve patients were excluded owing to FSHD. The FSHD1 population was binned into 2 subgroups of individuals with D4Z4 repeats of 4–7 RU and 8–10 RU. After being tested for normality using a Kolmogorov-Smirnov test, the different values were analyzed for statistical significance. More precisely, if the underlying distribution was normal, an unpaired Student t test was performed. If the distribution did not pass the normality, we used a Mann-Whitney test and performed a Spearman correlation in order to analyze the relationships between the age-corrected CSS and the number of RU or the percentage of methylation of the DR1 region. The Spearman r coefficient and the p values are indicated on the corresponding figures. The clinical severity was analyzed using the age-corrected CSS, which corresponds to (CSS/age at examination) × 1,000. In each figure, when significant p values were obtained, this was indicated with an asterisk. There are no missing data. Hardy-Weinberg equilibrium was not considered.
Standard protocols approvals, registrations, and patient consent
Institutional review boards/ethics committees approved the study protocol (CPP approval number: 13.045; ANSM: 1309428-31; CNIL: 1517317V0). The study was conducted in accordance with the Declaration of Helsinki (2000) and the principles of Good Clinical Practice according to the International Conference on Harmonization. This study is registered under Clinicaltrials.gov (NCT01970735, clinicaltrials.gov/ct2/show/NCT01970735). All patients provided written informed consent before inclusion in the study.
Data availability policy
The full data are not available publicly because of participant privacy and consent. However, more information regarding the data is available from the corresponding author upon request.
Results
A total of 103 patients with FSHD were included in the study (54 male, 49 female; table 1 and figure 1). These were separated into 2 groups: those with a D4Z4 repeat of 1–10 RU (FSHD1; n = 69) and those with D4Z4 repeats of ≥11 RU (n = 34, FSHD-like) (these patients have a phenotype compatible with FSHD but without a 1–10 RU contracted D4Z4 repeat on a 4qA chromosome; they were mostly patients with FSHD2, see below). Mean age of the total population at the time of the study was of 56.2 ± 14.8 years, ranging from 19 to 77 years. The mean age at onset of symptoms was 32.2 ± 16.9. Male and female participants were equally distributed over both groups. There were no significant differences in mean age at onset of symptoms and mean disease duration between the groups. Significant differences were found in mean age at time of the study (p = 0.0028), mean age-corrected CSS (p = 0.049), and mean MMT score (p = 0.045) (table 1). All patients included in this study display facial and scapular weakness. The percentage of peroneal weakness was not significantly different among the groups.
Table 1.
Clinical and demographic data of patients included in the study

Figure 1. Breakdown of patients.

The major groups 1 (57 patients with facioscapulohumeral dystrophy [FSHD] type 1, 2 [20 patients with FSHD type 2], and 3 [7 patients with FSHD1 + FSHD2]) are indicated. RU = repeat units.
Genetic analyses
We identified 69 cases with an FSHD-sized D4Z4 repeat, of which 68 cases represented 62 families with D4Z4 repeat sizes between 4 and 10 RU on a 4qA chromosome. Of these, 4 were either mosaic for a D4Z4 repeat contraction or carried a more complex 4q rearrangement. These 4 individuals were excluded for further analyses (figure 1). One patient had a 9 RU D4Z4 repeat on a 4qB chromosome and was therefore included in the FSHD-like population. In the FSHD-like population, 34 patients representing 32 families had normal-sized D4Z4 repeats (≥11 RU) and at least one 4qA FSHD-permissive chromosome.
SMCHD1 sequence analysis identified disease-causing variants in 27 individuals: 20/35 (57%) did not have a contracted D4Z4 repeat on a disease-permissive 4qA allele, while 7/64 (11%) also had a D4Z4 repeat of 9–10 RU on a 4qA allele (figure 1 and tables 2 and 3). We also identified rare SMCHD1 polymorphisms: 8/64 (12.5%) patients with FSHD1 (all with alleles of 4–8 RU) and 2/35 (0.06%) FSHD-like patients had such rare SMCHD1 polymorphism. The pathogenic status of SMCHD1 variants was based on the Polydiag and Alamut software suite prediction and D4Z4 methylation analysis (see below). We also included the candidate genes LRIF1 and SUV39H134,35 in our mutation screen but did not find evidence for mutations in this cohort of patients.
Table 2.
Level of the DR1 region methylation in the analyzed population

Table 3.
SMCHD1 variants identified in this study classified for the FSHD-like and FSHD1 population


Of the 35 patients with an FSHD-like phenotype, 12 patients in whom FSHD1 or FSHD2 could not be genetically confirmed were further investigated by standard diagnostic procedures. In 3 of them, FSHD was ruled out because of the presence of 2 4qB haplotypes. In 6, an alternative diagnosis was established: scleroderma (2 cases, one of them homozygous for 4qB), dermatomyositis (2 cases), myofibrillar myopathy, and Pompe disease (this patient was also homozygous for the 4qB haplotype). In the 3 remaining patients, all with a 4qA haplotype, no definite diagnosis could be made. In 3 patients with genetic data suggestive for FSHD2 (they carried a pathogenic SMCHD1 variant and a 4qA allele: table 3), the number of RU could not be accurately determined due to technical issues, and they were excluded from further studies.
Based on these findings, we distributed our patients with a positive molecular diagnosis over 3 groups. The first includes 57 patients with FSHD1 (those with D4Z4 repeats of 4–10 RU and without SMCHD1 disease causing variants), the second 20 patients with FSHD2 (those with ≥11 RU on a 4qA haplotype and a SMCHD1 disease-causing variant), and the third 7 individuals with FSHD1 and FSHD2 (FSHD1+2) (those carrying a D4Z4 repeat of 9–10 RU on 4qA and an SMCHD1 disease-causing variant) (figure 1). Binning patients with FSHD1 into those with repeats of 4–7 RU or 8–10 RU21 showed that 26 had a disease allele of 8–10 RU, while 31 had an allele of 4–7 RU. All 7 FSHD1+2 patients had repeats of 9–10 RU on a permissive 4qA allele.
Clinical correlations
We next studied correlations between D4Z4 repeat size, (age-corrected) CSS, MMT sumscore, and Brooke and Vignos clinical scores. We found that the smaller the size of the D4Z4 repeat, the lower the MMT sumscore (r = 0.59, p < 0.0001) and the higher the age-corrected CSS for patients with FSHD1 (Spearman r = −0.45, p = 0.0004), while no correlation between these 2 parameters was observed for FSHD1+2 or FSHD2 patients (data not shown). In support of these findings, Brooke (r = −0.39, p = 0.0029) and Vignos (r = −0.29, p = 0.03) functional scores showed the highest values in patients with FSHD1 carrying the smallest D4Z4 repeat sizes.
With the FSHD1 group further sorted into the 2 subgroups of patients with D4Z4 repeats of 4–7 and of 8–10 RU, all groups had a similar sex distribution and there were no significant differences between sexes. The age-corrected CSS slightly but significantly differed among the 4 groups, with the FSHD2 patient group showing the highest mean age-corrected CSS, which could be attributed to 2 outliers representing 2 young patients with a severe clinical phenotype (figure 2A). When we only considered the mean CSS, the FSHD1 patient group with 8–10 RU was significantly milder, with no significant differences between the other groups (figure 2B). Consistent observations were made with the MMT sumscore and Brooke and Vignos scores (figure 2, C–E).
Figure 2. Clinical assessments of the facioscapulohumeral dystrophy (FSHD) subpopulations.

(A) Significant differences in the age-corrected clinical severity score (CSS) between FSHD type 1 (FSHD1) (8–10) and FSHD1 (4–7) (p = 0.0009***), FSHD1+ FSHD type 2 (FSHD2) (p = 0.0002***), or FSHD2 (p < 0.0001****) subgroups are observed. A significant difference between FSHD1 (4–7) and FSHD2 is also present (p = 0.048*). (B) The CSS of the FSHD1 (8–10) subpopulation is significantly different from those of FSHD1+2 (p = 0.036*), FSHD2 (p = 0.002**), and FSHD1 (4–7) (p = 0.003**) subgroups. (C) Significant differences in the manual muscle testing (MMT) score between FSHD1 (8–10) and FSHD1 (4–7) (p < 0.0001****) or FSHD2 (p = 0.047*) subgroups are observed. (D) The Brooke score of the FSHD1 (8–10) subpopulation is significantly different from those of FSHD2 (p = 0.0002***) and FSHD1 (4–7) (p = 0.012*) subgroups. (E) Significant differences observed between Vignos scores of FSHD1 (8–10) and FSHD 1+2 (p = 0.034) or FSHD2 (p = 0.044*) subgroups.
Correlations with D4Z4 methylation
Virtually all FSHD1+2 and FSHD2 patients had hypomethylation of the DR1 region, and the level of DR1 methylation was significantly different between the FSHD1 (4–7 RU and 8–10 RU) and FSHD1+2 or FSHD2 subgroups (figure 3A), while no statistical differences were detected between FSHD1+2 and FSHD2 subgroups (figure 3A). A total of 15/57 patients with FSHD1, 19/20 patients with FSHD2, and 7/7 patients with FSHD1+2 had D4Z4 hypomethylation (table 2 and figure 3A) defined by levels of DR1 methylation ≤30% of controls.31 The one FSHD2 case with DR1 methylation >30% showed profound hypomethylation based on δ1 D4Z4 methylation score (data not shown), which corrects for the repeat size.36 DR1 hypomethylation close to the threshold of 30% was observed also in one patient with dermatomyositis and in one patient without definite diagnosis. We did not observe a correlation between the DR1 methylation and the CSS or age-corrected CSS in the FSHD1 population, even when grouped into patients with 4–7 or 8–10 RU (data not shown), but the severity of the disease measured by age-corrected CSS was increased in patients showing low levels of DR1 methylation in the total FSHD population (Spearman r = −0.37, p = 0.0005) (figure 3B).
Figure 3. Analysis of the DR1 region in the D4Z4 repeats methylation level in the facioscapulohumeral dystrophy (FSHD) subgroups.

(A) The level of methylation of the DR1 region in the D4Z4 repeats is significantly different between FSHD type 1 (FSHD1) (8–10) patients and FSHD1+ FSHD type 2 (FSHD2) (p = 0.005**) or FSHD2 subgroups (p < 0.0001****). We also observe a significant difference in methylation between FSHD1 (4–7) and FSHD1+2 (p = 0.0002***) or FSHD2 subgroups (p < 0.0001****). FSHD1+2 and FSHD2 patients show a hypomethylation status at the DR1 region (mean level < 30%). (B) A significant correlation between the methylation level of the DR1 region and the age-corrected clinical severity score (CSS) is obtained (Spearman r = −0.37, p = 0.0005***). The disease severity is increased in patients with low methylation levels (significant p values only are indicated [*]).
As expected, in all patients with FSHD2 (20/20), 18 independent variants in SMCHD1 were identified that are classified as pathogenic, consistent with the low DR1 methylation values in all but one. Conversely, in 57 individuals with FSHD1, no disease-causing variants in SMCHD1 were identified. Fifteen of them had methylation levels ≤30%, suggestive for a disease-causing variant in SMCHD1, but with the exception of 3 (methylation levels between 6% and 12%; no δ1 scores available), for the others, all of the methylation values were close to the threshold of 30%.
SMCHD1 mutations
In the FSHD1+2 group, most mutations are predicted to disrupt the open reading frame: 3 patients from 2 families are haploinsufficient for SMCHD1, and in 3 other cases the mutation is predicted to introduce a frameshift leading to a premature stop codon. Only one variant is predicted to result in the production of a mutant SMCHD1 protein. In the FSHD2 group, of the 18 independent pathogenic variants, variants that are predicted to disrupt the open reading frame can be found with approximately equal frequency to those that are predicted to maintain the open reading frame. We did not observe any correlation between DR1 methylation levels and the predicted consequence of the SMCHD1 variant at the protein level (data not shown).
FSHD1 and FSHD2 form a disease continuum
Figure 4 shows the distribution of FSHD1, FSHD1+2, and FSHD2 patients with respect to repeat size and methylation of the D4Z4 repeat. With increasing D4Z4 repeat size, the proportion of individuals having reduced DR1 methylation as a consequence of a pathogenic SMCHD1 variant increases, creating an overlap between FSHD1 and FSHD2 in the range of 9–10 RU.
Figure 4. Facioscapulohumeral dystrophy type 1 (FSHD1) and facioscapulohumeral dystrophy type 2 (FSHD2), a disease continuum.
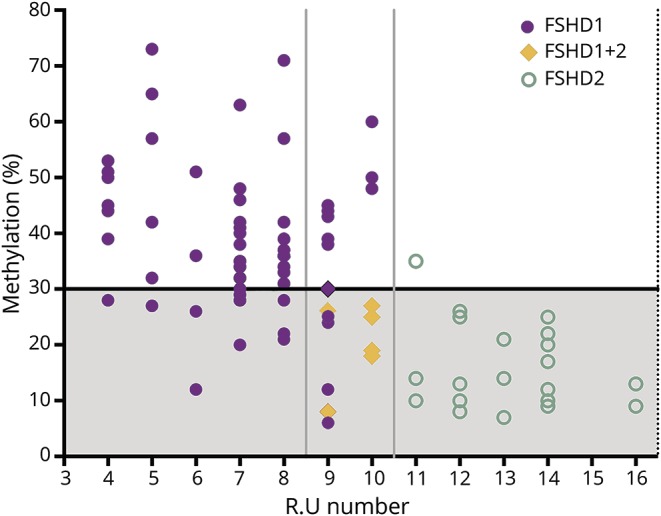
Patients with FSHD1 have a contraction of the D4Z4 repeat to a size of 1–10 repeat units (RU) (X-axis). Patients with FSHD2 show hypomethylation of the DR1 region in the presence of a normal sized D4Z4 repeat of 11–20 RU (Y axis). FSHD1+2 can be considered part of a FSHD disease continuum characterized by a contraction of the D4Z4 repeat (9–10 RU) and a hypomethylation status of the DR1 region because of mutations in D4Z4 chromatin modifiers.
Discussion
FSHD1 and FSHD2 are closely related disorders in terms of genetic and epigenetic underpinnings, pathophysiology, and clinical manifestations.37,38 Although initially described as separate entities based on their genetics,8 recent insight suggests that both may represent opposite ends of a molecular disease spectrum in which the impairment of genetic and epigenetic factors that govern DUX4 repression in skeletal muscle have different weights in both forms of the disease.10,24
In this study we clinically, genetically, and epigenetically characterized a cohort of patients referred to our centers because of a clinical suspicion of FSHD. Of 103 patients, 57 had classical FSHD1 defined by a D4Z4 repeat contraction to 1–10 RU on a disease permissive 4qA allele; 20 had FSHD2 defined by a clinical phenotype consistent with FSHD, the presence of at least one 4qA chromosome, and D4Z4 hypomethylation in the absence of a D4Z4 repeat contraction. Seven patients met the genetic criteria for FSHD1 and FSHD2, 4 patients with FSHD1 had a nontypical 4q rearrangement, in 6 patients an alternative diagnosis was established, and in 3 of the remaining 6 patients FSHD was ruled out because of the presence of 2 nonpermissive 4qB haplotypes. In the remaining 3 cases, the DNA quality was insufficient to reach a diagnosis. The high proportion of patients with FSHD2 in this study likely reflects a selection bias since we recruited only adult patients who met clinically defined criteria for typical FSHD. FSHD2 was originally defined by a phenotype compatible with FSHD, pan-D4Z4 hypomethylation, and the presence of a normal (albeit often smaller)–sized D4Z4 repeat on a 4qA allele.8 As not all patients who meet these criteria can be explained by mutations in SMCHD1 or DNMT3B, it is not inconceivable that additional FSHD disease genes exist. We therefore also considered SUV39H1 and LRIF1 as candidate genes for idiopathic FSHD since these protein products are known to cooperate with SMCHD1 in the repression of the inactive X chromosome.35,39 However, it seems to be an unlikely or very rare cause of FSHD.
Our diagnostic protocol could confirm the diagnosis in the majority of patients: only in 6/103 (5.8%) individuals could FSHD not be confirmed. None of them have D4Z4 DR1 hypomethylation. However, we cannot exclude that some cases, including the undiagnosed cases with a 4qA chromosome, harbor a mutation in DNMT3B9 or another yet unidentified FSHD2 gene, or have an atypical SMCHD1 disease-causing variant that can be easily missed by our sequencing strategy such as a deep intronic variant. However, D4Z4 methylation analysis does not suggest an involvement of SMCHD1 or DNMT3B since these patients generally have moderate–severe D4Z4 hypomethylation.
We confirm that in FSHD1, the size of the D4Z4 repeat inversely correlates with disease severity: patients with 8–10 RU had a milder disease than those with 4–7 RU. We did not identify patients with D4Z4 repeats <4 RU except for one mosaic patient with a repeat of 3 D4Z4 units. Patients with 1–3 RU are rare in the European population and it may reflect selection bias since we did not recruit pediatric patients. On the other hand, none of our FSHD2 patients had D4Z4 repeats >16 RU, indicating that there is probably a genetic and epigenetic threshold above which SMCHD1 disease-causing variants cannot sufficiently derepress DUX4 in skeletal muscle with relevant clinical consequences. This is consistent with the recent observation that affected SMCHD1 mutation carriers with unusually long D4Z4 repeats carry small D4Z4 duplications.40,41 It is also consistent with observations that DUX4 derepression in FSHD2 depends on the functional consequence of the SMCHD1 mutation and the size of the D4Z4 repeat on the permissive 4q allele, with patients with FSHD2 homozygous for disease-permissive alleles being more frequently symptomatic,23 and with the observation that patients carrying an 18p deletion that includes SMCHD1 can develop FSHD only when carrying a permissive 4qA allele with a D4Z4 repeat <20 RU.24,42,43 Larger patient cohorts are required to determine the upper limit of D4Z4 repeat size in which SMCHD1 disease-causing variants can still cause FSHD and to determine the penetrance.
From a clinical perspective, all groups were similar, with the exception of patients with FSHD1 with 8–10 RU, who generally presented with a milder phenotype. This is consistent with earlier reports demonstrating increased nonpenetrance, increased quadriceps strength by quantitative myometry, and reduced wheelchair dependency in this size range.21 Nevertheless, there is also large variability in clinical presentation in this group. In general, we observed that correlation studies among CSS, MMT sumscore, and the Brooke and Vignos scores showed consistent results. We did observe, however, that the age-corrected CSS has limitations as it deflates the severity of early-onset cases with long disease durations, especially after reaching wheelchair dependency. We could not establish correlations between DR1 methylation and the clinical scores in the FSHD1 population. This is different from the δ1 score, which generally correlates well with disease presentation in the 8–10 RU group.36 Indeed, some of the FSHD1 and FSHD2 patients with discordant DR1 score showed reduced δ1 scores, if available (data not shown). This difference might be explained by the correction for the D4Z4 repeat size methylation effect in the δ1 score, which is absent from the DR1 score. Nevertheless, despite these limitations, we found that lower levels of DR1 methylation was significantly associated with higher values of age-corrected CSS, i.e., a more severe and progressive disease, in the entire FSHD population (figure 3B).
It is interesting that none of the 45 patients with FSHD with 4–8 RU have a pathogenic variant in SMCHD1, while we could identify a pathogenic variant in 7 of the 19 patients with FSHD with 9–10 RU (p < 0.0001). The milder phenotype in the FSHD1 8–10 RU group, combined with the higher prevalence of disease-causing SMCHD1 variants in patients with 9–10 RU, suggests that this D4Z4 size range is a risk factor or disease modifier for FSHD, rather than a high penetrant disease-causing repeat size. This is consistent with the high prevalence of 8–10 RU 4qA alleles in the European population.5–7 Indeed, this study strongly suggests that the historically defined thresholds of 1–10 RU being FSHD1 and 11–20 units being FSHD2 need revision. Rather, it supports the notion that FSHD1 and FSHD2 form a continuum with increasing prevalence of genetic variants that affect the function of epigenetic modifiers of the D4Z4 repeat from 8 RU onwards.
Acknowledgment
The authors thank the patients for participating in this study and IRCAN Genomics Core Facility for sequencing (supported by FEDER, Région Provence Alpes Côte d'Azur, Conseil Départemental 06, ITMO Cancer Aviesan [plan cancer], and INSERM).
Glossary
- CSS
clinical severity score
- FSHD
facioscapulohumeral dystrophy
- FSHD1
facioscapulohumeral dystrophy type 1
- FSHD2
facioscapulohumeral dystrophy type 2
- MMT
manual muscle testing
- MNP
multi-nucleotide polymorphism
- NGS
next-generation sequencing
- RU
repeat units
- SNP
single-nucleotide polymorphism
- SNV
single nucleotide variant
- SSLP
simple sequence-length polymorphism
- TSVC
Torrent Suite Variant Caller
Appendix. Authors

Footnotes
Editorial, page 881
Study funding
This work has been developed and supported through the University Hospital Federation OncoAge (FHU OncoAge) consortium (Nice). FHU OncoAge, a Hospital-University Federation, devotes itself to the development of care, research, innovation and teaching/training in the field of cancer and aging. This work was also supported by a grant to S.S. from the Centre Hospitalier Universitaire de Nice (13AOI-01). It was also supported by the French Government (National Research Agency, ANR, ANR-13-BSV1-0004-04) through the “Investments for the Future” (LABEX SIGNALIFE, ANR-11-LABX-0028-01) and the FHU OncoAge. S.M.v.d.M. is supported by grants from the Prinses Beatrix Spierfonds (W.OP14-01, W.OB17-01), the National Institute of Neurologic Disorders and Stroke award number P01 NS069539, the National Institute of Arthritis Musculoskeletal and Skin Disease award number R01AR066248, and the Stichting Spieren voor Spieren.
Disclosure
S. Sacconi served as a consultant for Sanofi-Genzyme and BIOGEN France. A. Briand-Suleau, M. Gros, C. Baudoin, R. Lemmers, S. Rondeau, N. Lagha, P. Nigumann, C. Cambieri, A. Puma, F. Chapon, T. Stojkovic, C. Vial, F. Bouhour, M. Cao, E. Pegoraro, P. Petiot, A. Behin, M. Bras, B. Eymard, A. Echaniz-Laguna, P. Laforet, L. Salviati, M. Jeanpierre, and G. Cristofari report no disclosures relevant to the manuscript. S. van der Maarel is consultant for Fulcrum Therapeutics and co-inventor on FSHD patent applications. Go to Neurology.org/N for full disclosures.
References
- 1.Wijmenga C, Hewitt JE, Sandkuijl LA, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet 1992;2:26–30. [DOI] [PubMed] [Google Scholar]
- 2.Wijmenga C, Frants RR, Brouwer OF, Moerer P, Weber JL, Padberg GW. Location of facioscapulohumeral muscular dystrophy gene on chromosome 4. Lancet 1990;336:651–653. [DOI] [PubMed] [Google Scholar]
- 3.van der Maarel SM, Miller DG, Tawil R, Filippova GN, Tapscott SJ. Facioscapulohumeral muscular dystrophy: consequences of chromatin relaxation. Curr Opin Neurol 2012;25:614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Deutekom JC, Wijmenga C, van Tienhoven EA, et al. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum Mol Genet 1993;2:2037–2042. [DOI] [PubMed] [Google Scholar]
- 5.Lemmers RJ, Wohlgemuth M, van der Gaag KJ, et al. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am J Hum Genet 2007;81:884–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lemmers RJ, van der Vliet PJ, van der Gaag KJ, et al. Worldwide population analysis of the 4q and 10q subtelomeres identifies only four discrete interchromosomal sequence transfers in human evolution. Am J Hum Genet 2010;86:364–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scionti I, Greco F, Ricci G, et al. Large-scale population analysis challenges the current criteria for the molecular diagnosis of fascioscapulohumeral muscular dystrophy. Am J Hum Genet 2012;90:628–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lemmers RJ, Tawil R, Petek LM, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet 2012;44:1370–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van den Boogaard ML, Lemmers RJ, Balog J, et al. Mutations in DNMT3B modify epigenetic repression of the D4Z4 repeat and the penetrance of facioscapulohumeral dystrophy. Am J Hum Genet 2016;98:1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sacconi S, Lemmers RJ, Balog J, et al. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am J Hum Genet 2013;93:744–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larsen M, Rost S, El Hajj N, et al. Diagnostic approach for FSHD revisited: SMCHD1 mutations cause FSHD2 and act as modifiers of disease severity in FSHD1. Eur J Hum Genet 2015;23:808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemmers RJ, van der Vliet PJ, Klooster R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010;329:1650–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao Z, Snider L, Balog J, et al. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum Mol Genet 2014;23:5342–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geng LN, Yao Z, Snider L, et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell 2012;22:38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kowaljow V, Marcowycz A, Ansseau E, et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul Disord 2007;17:611–623. [DOI] [PubMed] [Google Scholar]
- 16.Wallace LM, Garwick SE, Mei W, et al. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann Neurol 2011;69:540–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Padberg GW, van Engelen BG. Facioscapulohumeral muscular dystrophy: Curr Opin Neurol 2009;22:539–542. [DOI] [PubMed] [Google Scholar]
- 18.Padberg GW, Lunt PW, Koch M, Fardeau M. Diagnostic criteria for facioscapulohumeral muscular dystrophy. Neuromuscul Disord 1991;1:231–234. [DOI] [PubMed] [Google Scholar]
- 19.Sacconi S, Salviati L, Desnuelle C. Facioscapulohumeral muscular dystrophy. Biochim Biophys Acta 2015;1852:607–614. [DOI] [PubMed] [Google Scholar]
- 20.Goselink RJM, Voermans NC, Okkersen K, et al. Early onset facioscapulohumeral dystrophy: a systematic review using individual patient data. Neuromuscul Disord 2017;27:1077–1083. [DOI] [PubMed] [Google Scholar]
- 21.Statland JM, Donlin-Smith CM, Tapscott SJ, Lemmers RJ, van der Maarel SM, Tawil R. Milder phenotype in facioscapulohumeral dystrophy with 7–10 residual D4Z4 repeats. Neurology 2015;85:2147–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wohlgemuth M, Lemmers RJ, Jonker M, et al. A family-based study into penetrance in facioscapulohumeral muscular dystrophy type 1. Neurology 2018;91:e444–e454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lemmers RJ, van der Vliet PJ, Balog J, et al. Deep characterization of a common D4Z4 variant identifies biallelic DUX4 expression as a modifier for disease penetrance in FSHD2. Eur J Hum Genet 2018;26:94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemmers RJ, van den Boogaard ML, van der Vliet PJ, et al. Hemizygosity for SMCHD1 in facioscapulohumeral muscular dystrophy type 2: consequences for 18p deletion syndrome. Hum Mutat 2015;36:679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zatz M, Marie SK, Cerqueira A, Vainzof M, Pavanello RCM, Passos-Bueno MR. The facioscapulohumeral muscular dystrophy (FSHD1) gene affects males more severely and more frequently than females. Am J Med Genet 1998;77:155–161. [PubMed] [Google Scholar]
- 26.Ricci G, Scionti I, Sera F, et al. Large scale genotype–phenotype analyses indicate that novel prognostic tools are required for families with facioscapulohumeral muscular dystrophy. Brain 2013;136:3408–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teveroni E, Pellegrino M, Sacconi S, et al. Estrogens enhance myoblast differentiation in facioscapulohumeral muscular dystrophy by antagonizing DUX4 activity. J Clin Invest 2017;127:1531–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stadler G, Rahimov F, King OD, et al. Telomere position effect regulates DUX4 in human facioscapulohumeral muscular dystrophy. Nat Struct Mol Biol 2013;20:671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robin JD, Ludlow AT, Batten K, et al. SORBS2 transcription is activated by telomere position effect–over long distance upon telomere shortening in muscle cells from patients with facioscapulohumeral dystrophy. Genome Res 2015;25:1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lemmers RJ, de Kievit P, Sandkuijl L, et al. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat Genet 2002;32:235–236. [DOI] [PubMed] [Google Scholar]
- 31.Hartweck LM, Anderson LJ, Lemmers RJ, et al. A focal domain of extreme demethylation within D4Z4 in FSHD2. Neurology 2013;80:392–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rohde C, Zhang Y, Reinhardt R, Jeltsch A. BISMA: fast and accurate bisulfite sequencing data analysis of individual clones from unique and repetitive sequences. BMC Bioinformatics 2010;11:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pasmant E, Parfait B, Luscan A, et al. Neurofibromatosis type 1 molecular diagnosis: what can NGS do for you when you have a large gene with loss of function mutations? Eur J Hum Genet 2015;23:596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brideau NJ, Coker H, Gendrel AV, et al. Independent mechanisms target SMCHD1 to trimethylated histone H3 lysine 9-modified chromatin and the inactive X chromosome. Mol Cell Biol 2015;35:4053–4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeng W, de Greef JC, Chen YY, et al. Specific loss of histone H3 lysine 9 trimethylation and HP1γ/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet 2009;5:e1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lemmers RJ, Goeman JJ, van der Vliet PJ, et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum Mol Genet 2015;24:659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Greef JC, Lemmers RJ, Camano P, et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology 2010;75:1548–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sacconi S, Camaño P, de Greef JC, et al. Patients with a phenotype consistent with facioscapulohumeral muscular dystrophy display genetic and epigenetic heterogeneity. J Med Genet 2012;49:41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nozawa R-S, Nagao K, Igami KT, et al. Human inactive X chromosome is compacted through a PRC2-independent SMCHD1-HBiX1 pathway. Nat Struct Mol Biol 2013;20:566–573. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen K, Puppo F, Roche S, et al. Molecular combing reveals complex 4q35 rearrangements in Facioscapulohumeral dystrophy. Hum Mutat 2017;38:1432–1441. [DOI] [PubMed] [Google Scholar]
- 41.Lemmers RJ, van der Vliet PJ, Vreijling JP, et al. Cis D4Z4 repeat duplications associated with facioscapulohumeral muscular dystrophy type 2. Hum Mol Genet 2018;27:3488–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Balog J, Goossens R, Lemmers RJ, et al. Monosomy 18p is a risk factor for facioscapulohumeral dystrophy. J Med Genet 2018;55:469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Renard D, Taieb G, Garibaldi M, et al. Inflammatory facioscapulohumeral muscular dystrophy type 2 in 18p deletion syndrome. Am J Med Genet A 2018;176:1760–1763. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The full data are not available publicly because of participant privacy and consent. However, more information regarding the data is available from the corresponding author upon request.