

Abstract
Clinical manifestations of respiratory fungal diseases in adult cystic fibrosis (CF) patients are very heterogeneous, ranging from asymptomatic colonization to chronic infections, allergic disorders, or invasive diseases in immunosuppressed CF patients after lung transplantation. In this narrative review, mainly addressed to clinicians without expertise in CF who may nonetheless encounter adult CF patients presenting with acute and chronic respiratory syndromes, we briefly summarize the most representative clinical aspects of respiratory fungal diseases in adult CF patients.
Keywords: Cystic fibrosis, ABPA, Aspergillus, Candida, Scedosporium, Exophiala, Lomentospora, CFTR, bronchitis, sensitization
Introduction
Cystic fibrosis (CF) is an autosomal recessive disease, characterized by multiorgan involvement and usually clinically dominated by chronic bacterial respiratory infection and bronchiectasis, with alternation of acute pulmonary exacerbations and interexacerbation periods, and progressive loss of lung function.1 The estimated prevalence of CF in Europe and the United States is of 20 and 50 cases per 100 000 inhabitants, respectively.2
In the last decades, a notable increase in the average life expectancy of CF patients has been registered, reaching 42 to 51 years for those born between 1985 and 1994.3 In this expanding population of adult CF patients, despite chronic and acute bacterial respiratory infections usually remain predominant, the frequency of isolation of fungi from the respiratory tract is increased compared with childhood. However, the related clinical manifestations are very heterogeneous, ranging from asymptomatic colonization to chronic infections, allergic disorders, or invasive diseases in immunosuppressed CF patients after lung transplantation.
In this narrative review, mainly addressed to clinicians without expertise in CF who may nonetheless encounter adult CF patients presenting with acute and chronic respiratory syndromes, we briefly summarize the most representative clinical aspects of respiratory fungal diseases in adult CF patients.
Methods
A MEDLINE/PubMed search was conducted in January 2019, inductively employing various combinations of the key-words “adult,” “cystic fibrosis,” “fung*,” “respiratory infection,” “aspergill*,” and “ABPA.” The full-text of retrieved papers was then reviewed and collectively discussed, with the decision about references to be included in the present narrative review being ultimately based on the subjective impression of the authors. For readers interested in an extended, in-depth description of specific clinical and pathogenetic aspects, which is beyond the scope of this article, some comprehensive reviews have been published in the past few years.4-13
Aspergillus spp
Aspergillus spp. are among the most widespread filamentous fungi in the environment, especially in areas with high humidity.14 In CF patients, the most frequently isolated species is Aspergillus fumigatus, accounting for 67%-73% of Aspergillus-positive sputum cultures.15 Isolation of other species such as A. flavus, A. niger, and A. terreus is less frequent but not rare (4%, 4%, and 2% of Aspergillus-positive sputum cultures, respectively).15 Notably, the prevalence of isolation of Aspergillus spp. from sputum cultures in CF patients increases with age, possibly reaching 46%-78% in adult CF patients, although with important interregion and intercenter variability.16-18
However, knowledge of the prevalence of Aspergillus spp. isolation from sputum does not automatically allow to infer the prevalence of the various Aspergillus-related manifestations in CF patients, which range from asymptomatic colonization to invasive diseases (especially in lung transplant recipients). In this regard, Baxter at al19 have proposed a classification based not only on sputum cultures but also on real-time polymerase chain reaction (RT-PCR) for Aspergillus, sputum level of galactomannan (a cell-wall component of Aspergillus), total IgE, specific A. fumigatus IgE, and specific A. fumigatus IgG. This classification was developed by latent class analysis in triazole-naive patients, for ultimately creating a diagnostic algorithm specific for adult CF patients.19 According to Baxter et al’s19 classification, adult CF patients may be divided into four classes with respect to Aspergillus-related diseases: (Class 1) nondiseased patients; (Class 2) allergic bronchopulmonary aspergillosis (ABPA); (Class 3) IgE-sensitized patients; (Class 4) Aspergillus airway infection or “bronchitis.” A simplified representation of Baxter’s classification is displayed in Table 1, whereas some examples of computerized tomography (CT) and bronchoscopy images of adult CF patients with Aspergillus-related diseases are shown in Figures 1 and 2.
Table 1.
Simplified classification of Aspergillus spp. diseases in adult cystic fibrosis patients, based on the categories proposed by Baxter et al.19
| Class | RT-PCR Aspergillus (sputum) | Total IgE | Specific IgE for A. fumigatus | Specific IgG for A. fumigatus | GM level (sputum) |
|---|---|---|---|---|---|
| Class 1: no disease | +/− | Normal | Normal | Normal | − |
| Class 2: ABPA | + | Increased | Increased | Increased | + |
| Class 3: Aspergillus sensitized | +/− | Increased (less than in ABPA) | Increased (less than in ABPA) | Normal | − |
| Class 4: Aspergillus bronchitis | + | Normal | Normal | Increased | + |
Abbreviations: RT-PCR, real-time polymerase chain reaction; IgE, immunoglobulin E; IgG, immunoglobulin G; GM, galactomannan; ABPA, allergic bronchopulmonary aspergillosis. Simplified from Baxter et al.19 Accurate diagnostic algorithms, together with sensitivity and specificity of the different cutoffs of the various markers, are available in the original publication.19
Figure 1.

(A) Bilateral bronchiectasis with mucoid plugs in a 55-year-old male patient with cystic fibrosis and allergic bronchopulmonary aspergillosis (ABPA). (B) Monolateral pseudonodular infiltrates in a 41-year-old male patient with ABPA and a mild form of Aspergillus bronchitis triggering a diagnostic workflow ultimately leading to a late diagnosis of cystic fibrosis.
Figure 2.
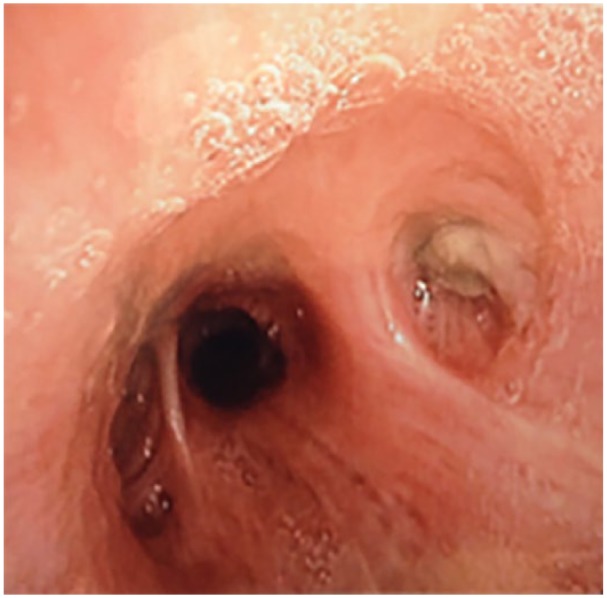
Bronchoscopy of a 41-year-old cystic fibrosis patient with allergic bronchopulmonary aspergillosis and Aspergillus bronchitis; a dense mucoid plug is responsible for obstruction of the apical segmental bronchus of the right lower lobe.
Nondiseased patients
Notably, nondiseased patients may also include those subjects in whom Aspergillus spp. is isolated from sputum cultures in the presence of negative sputum galactomannan and no measurable immunological response to Aspergillus spp.19 These patients could be defined as nondiseased patients with Aspergillus colonization, although it should be acknowledged that no standardized definition exists for Aspergillus colonization in CF patients. In any case, it seems that in these patients, the isolation of Aspergillus spp. is not associated with increased risks of pulmonary exacerbation, a more rapid decline of pulmonary function, and development of other Aspergillus-related diseases in comparison with noncolonized nondiseased patients,20 and treatment of colonization is currently not recommended.21
Allergic bronchopulmonary aspergillosis
Allergic bronchopulmonary aspergillosis refers to a complex hypersensitivity reaction which often occurs in patients affected by CF or asthma.22 Allergic bronchopulmonary aspergillosis is the most common Aspergillus-related syndrome in CF, with prevalence ranging from 3% to 25% and being more common in adult than in pediatric CF patients.20,23
The clinical presentation of ABPA is heterogeneous, ranging from acute symptoms such as dyspnea, chest pain, wheezing, and/or productive cough with blackish sputum to more subtle forms with progressive reduction of forced expiratory volume in one second (FEV1).24-26 Non-specific symptoms such as fever and myalgias may also coexist.24-26 Respiratory function tests often identify airflow obstruction, while chest radiographic imaging could show parenchymal infiltrates that disappear a few days after starting steroid therapy. According to Baxter et al’s19 criteria, the diagnosis of ABPA is serological and can be made in the presence of positive sputum galactomannan, high serum IgE levels in the absence of steroid therapy, presence of specific IgG to A. fumigatus. The classical presentation of ABPA also included eosinophilia and immediate positive skin test for A. fumigatus.27
The treatment of ABPA is aimed at the attenuation of the immune response, which is usually achieved by corticosteroid therapy.28 However, the association with azole antifungals, albeit remaining somewhat controversial, is not uncommon and aimed to contain the antigenic burden and minimize the exposure to steroid therapy.28
Aspergillus sensitization
Aspergillus sensitization is frequent in CF patients (up to 35% of cases) and is defined by the presence of elevated serum levels of specific IgE for A. fumigatus and slightly elevated total IgE independent of the presence of a positive culture or RT-PCR for Aspergillus spp.19 According to immunological studies, sputum galactomannan is usually negative, with also low levels of Aspergillus-specific IgG.19
Notably, Aspergillus sensitization is considered as a pre-ABPA condition, as it reflects a predisposition to developing an immunological reaction in the absence of the pertinent antigen/s.19 Differently from colonization in nondiseased patients, sensitization may be associated with lung function decline and increased frequency of pulmonary exacerbations.29 However, as to whether Aspergillus sensitization should be treated for ameliorating symptoms and/or reducing lung function decline in CF patients remains controversial due to the absence of convincing evidence. Against this backdrop, a prudent wait-and-see approach based on symptoms and careful evaluation for alterations of respiratory function and biochemical signs (e.g. yearly evaluation of total serum IgE levels) has been suggested by some authors to reduce unwarranted pharmacological treatment.22,30-32
Aspergillus bronchitis
In CF patients, the disease-related progressive damage of the lungs may favor the development of chronic Aspergillus infection, commonly defined as “Aspergillus bronchitis,” although aspergilloma(s) might also develop in some cases, especially in preexisting cavities or bronchiectasis.33-35 Aspergillus bronchitis has an estimated prevalence of ~2% to 8% in CF patients and may be suspected in the case of pulmonary exacerbation unresponsive to antibacterial treatment.36,37 According to Baxter et al’s19 classification, diagnosis of Aspergillus bronchitis can be made in the presence of a positive sputum galactomannan, high levels of Aspergillus-specific IgG, and negative total and Aspergillus-specific IgE.
Since Aspergillus bronchitis does reflect infection and not an immune-mediated response as in ABPA, corticosteroids are not used for treatment. Treatment with azole derivatives is the current standard of care, although the overall duration of treatment is still not univocal.28
Invasive aspergillosis
Invasive aspergillosis is observed when the infection progresses across tissues and invades the vessels, with subsequent necrosis.38 Usually, invasive aspergillosis is observed in severely-immunocompromised non-CF populations, such as hematology patients with prolonged neutropenia and patients receiving high dosages of corticosteroids or other immunosuppressant agents. Patients commonly report fever, chest pain, shortness of breath, and/or cough.38 Hemoptysis and pneumothorax might also develop in some cases.39,40 Invasive aspergillosis is rarely seen in patients affected by CF. However, it could develop in end-stage CF patient in intensive care units and in immunosuppressed CF patients after lung transplant, frequently in the form of Aspergillus tracheobronchitis, occurring mainly in the first 3 months after transplants and associated with high mortality (39%).41,42
Frequent symptoms of Aspergillus tracheobronchitis are severe dyspnea, cough, and wheezing.28 The histopathological examination may show different features: (1) obstructive bronchial aspergillosis; (2) ulcerative tracheobronchitis (characterized by the invasion of the tracheobronchial mucosa and cartilage); (3) pseudomembranous tracheobronchitis (characterized by inflammation and invasion of the tracheobronchial tree). Invasive aspergillosis is usually treated with systemic azole therapy (possibly associated with nebulized amphotericin B in some cases of tracheobronchitis).28 The U.S. guidelines recommend a minimum of 6 to 12 weeks of therapy for patients with invasive pulmonary aspergillosis, and at least 3 months in case of Aspergillus tracheobronchitis.28
Scedosporium apiospermium Complex
The Scedosporium apiospermium complex comprises three human pathogenic species: Scedosporium apiospermium sensu strictu, Scedosporium boydii, and Scedosporium aurantiacum.43 Other species have been rarely associated with human disease.44
These filamentous fungi are ubiquitous and are mainly found in soil, polluted waters, and sewage.45 They are the second most prevalent filamentous fungi isolated from the respiratory tract of CF patients, with the prevalence ranging from 0.7% to 9%.44,46,47 However, the clinical significance of isolating S. apiospermium complex from the respiratory tract of CF patients is still a matter of debate.
In most cases, the contact with the host results in asymptomatic colonization, which can be either transient or permanent.44 Colonization of the airways by S. apiospermium complex occurs more frequently in adolescents and adults than in children with CF, and often later than colonization by Aspergillus spp.47
Although colonization is the most frequently encountered situation, some cases of S. apiospermium-related disease have been described. Cimon at al48 reported on two cases of allergic bronchopulmonary mycoses (ABPM) linked to this organism, resolved after combination treatment with itraconazole and oral corticosteroids. Other similar reports have prompted the suggestion by some authors to implement a regular screening for ABPM in patients colonized by S. apiospermium complex.47
S. apiospermium complex has also been reported to possibly cause invasive, disseminated disease in immunocompromised CF patients after lung transplantation. For example, Symoens et al49 reported on a patient colonized in the pretransplant period who developed a postoperative S. apiospermium infection characterized by moderate fever, subcutaneous nodules, and bilateral chorioretinitis, with subsequent complete loss of vision, meningeal involvement and unfavorable outcome. S. apiospermium was isolated from both vitreous humor and cerebrospinal fluid.49 Considering the reported low susceptibility of S. apiospermium complex to polyenes, azoles are usually employed as first-line therapy.50
Lomentospora prolificans
Formerly known as Scedosporium prolificans, this mold has much in common with its ex-cospecies, in terms of epidemiology and pathogenicity, although it greatly differs in terms of antifungal susceptibility, being resistant to amphotericin B, flucytosine, most azoles, and echinocandins.51 This mold has a certain geographical distribution, being more frequently isolated in Australia, Northern Spain, and in the United States, although isolation from the respiratory tract of CF has also been reported in other countries such as Germany.52-54
As observed for other molds, Lomentospora prolificans usually causes transient or permanent colonization of the respiratory tract of CF patients, but an invasive disease with high mortality (>65%) may develop in a state of severe immunosuppression, with some centers considering isolation of L. prolificans as a possible contraindication to transplantation,10,55,56 due to its multidrug-resistant profile.
Various regimens with available antifungals have been used in patients with invasive L. prolificans infection, frequently with unsatisfactory results.57 Novel antifungals with possible activity against L. prolificans are under study.58-61
Exophiala dermatitidis
Exophiala dermatitidis is a thermophilic black yeast that grows as a filamentous fungus at room temperature. It is found in plant debris and soil in open environments, as well as in moist and hot environments such as bathrooms, saunas, and dish washers in closed environments.44 Its prevalence as a colonizer of the respiratory tract in CF patients is not well defined, ranging from 1% to 15% in current literature.62,63
Its clinical significance remains uncertain, although recently Kondori et al64 demonstrated the presence of serum IgG specific for E. dermatitidis in colonized CF patients, with inflammation markers being higher than those observed in patients without specific serum IgG. In colonized CF patients with severe respiratory symptoms not responding to antibiotic therapy, some authors have suggested that treatment with antifungals and clearance of E. dermatitidis from the sputum may be associated with an improvement in clinical conditions, but this possibility has yet to be confirmed. Voriconazole and posaconazole are usually active against this fungus.65
Candida spp
The prevalence of isolation of Candida yeasts from the respiratory tract of adult CF patients may be as high as to 93%, making it the most commonly isolated fungus in this population.66 Prolonged use of antibiotic therapy seems to be one of the main factors associated with Candida spp. isolation.67 Candida albicans is the most common isolated species in CF patients, although non-negligible frequencies of C. dubliniensis, C. glabrata, C. parapsilosis, and C. tropicalis have also been reported.66,68,69
Candida spp. may cause transient and permanent colonization of the respiratory tract of CF patients, but also oral and genital candidosis.67 Furthermore, the development of candidemia during acute pulmonary exacerbation has been reported in central-venous devices carriers, including some severe cases.70,71
Although some authors have suggested that respiratory Candida colonization could be associated with a long-term decrease in FEV1 overtime, its role as a respiratory pathogen remains uncertain, and the presence of other agents/causes should always be investigated/suspected in the presence of respiratory symptoms.72
Other Fungi
The lung microbiome of CF patients comprises various permanent and transient fungi, whose growth and interactions are influenced by multiple factors, for example, antibiotic therapy.73 In this complex environment, the isolation of many other fungi (e.g. Pneumocystis jirovecii, Malassezia spp., Clavispora spp., Paecilomyces spp., Rasamsonia spp., Alternaria spp., Cladosporium spp., Penicillium spp., Trichosporon spp., and Talaromyces spp.) has been described in the literature.74 Although their clinical significance remains uncertain, noteworthy is that Rasamsonia argillacea, Trichosporon spp., and a few others have been possibly associated with long-term declines in lung function, and with invasive diseases after lung transplantation.75-77
Conclusions
The true burden of fungal disease in CF remains largely unknown. Chronic respiratory infection and acute pulmonary exacerbations caused by different bacteria, together with the possible overlap of clinical and radiological features of bacterial and fungal diseases, might interfere with both diagnosis and follow-up of fungal diseases in adult CF patients.
The classification of Aspergillus diseases proposed by Baxter is certainly useful, but it should be kept in mind that it might not always be applicable in clinical practice, owing to logistic and technical reasons, such as the the unavailability of specific A. fumigatus IgG in many centers, and the nonunivocal interpretation of nonstandardized methods such as PCR and sputum galactomannan. Of note, the diagnostic role of another fungal antigen, (1,3)-β-D-glucan (BDG), is uncertain in CF patients,78,79 although the detection of positive BDG levels in the blood of patients with a consistent clinical picture may be of help for the diagnosis of candidemia, as in other patient populations.80,81
Furthermore, different forms of fungal diseases might overlap (e.g. ABPA and Aspergillus bronchitis), interfering with a clear classification and standard treatment protocols. This is not a rare situation, since the isolation of molds in adult CF patients is more frequent than in childhood.
Following the advent of cystic fibrosis transmembrane conductance regulator (CFTR) modulators (ivacaftor, lumacaftor, and tezacaftor), drug interactions between these novel agents and azole antifungals have been reported, that may require strict therapeutic drug monitoring.66 Whether or not the reduction of airway inflammation following the use of CFTR modulators could impact the development or the progression of fungal diseases in CF patients deserves further investigation.82,83
Notably, the increase in A. fumigatus resistance to azoles84,85 might also complicate the therapeutic approach to Aspergillus diseases in CF patients, especially in adults, in whom they are expected to be more prevalent.
Finally, there is a need for standardized processing of respiratory specimens, as well as for repeated cultures over time (both because of possible suboptimal sensitivity and for differentiating between transient and chronic isolation).11,36
In conclusion, an updated knowledge of fungal respiratory diseases in adult CF patients can be of help for prompting an adequate early diagnostic and therapeutic workflow, as well as for referral to specialized centers for completion of all the necessary diagnostic procedures and the adequate management of immediate therapy and follow up.
Footnotes
Funding:The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Outside the present work, DR Giacobbe reports honoraria from Stepstone Pharma GmbH and an unconditioned grant from MSD Italia. The remaining authors have no conflicts of interest to disclose.
Author Contributions: All the authors contributed equally to the writing of the manuscript. In addition, ED and DRG provided critical revision for intellectual content, and oversight. All the authors approved the final version of the manuscript.
ORCID iD: Daniele Roberto Giacobbe  https://orcid.org/0000-0003-2385-1759
https://orcid.org/0000-0003-2385-1759
References
- 1. Ratjen F, Doring G. Cystic fibrosis. Lancet. 2003;361:681-689. [DOI] [PubMed] [Google Scholar]
- 2. Farrell PM. The prevalence of cystic fibrosis in the European Union. J Cyst Fibros. 2008;7:450–453. [DOI] [PubMed] [Google Scholar]
- 3. Dalcin PdT, Abreu E, Silva FA. Cystic fibrosis in adults: diagnostic and therapeutic aspects. J Bras Pneumol. 2008;34:107–117. [DOI] [PubMed] [Google Scholar]
- 4. Chotirmall SH, Martin-Gomez MT. Aspergillus species in bronchiectasis: challenges in the cystic fibrosis and non-cystic fibrosis airways. Mycopathologia. 2018;183:45–59. [DOI] [PubMed] [Google Scholar]
- 5. Dupont L. Lung transplantation in cystic fibrosis patients with difficult to treat lung infections. Curr Opin Pulm Med. 2017;23:574–579. [DOI] [PubMed] [Google Scholar]
- 6. Hamprecht A, Morio F, Bader O, Le Pape P, Steinmann J, Dannaoui E. Azole resistance in Aspergillus fumigatus in patients with cystic fibrosis: a matter of concern. Mycopathologia. 2018;183:151–160. [DOI] [PubMed] [Google Scholar]
- 7. Janahi IA, Rehman A, Al-Naimi AR. Allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. Ann Thorac Med. 2017;12:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. King J, Brunel SF, Warris A. Aspergillus infections in cystic fibrosis. J Infect. 2016;72:S50–S55. [DOI] [PubMed] [Google Scholar]
- 9. Rivosecchi RM, Samanta P, Demehin M, Nguyen MH. Pharmacokinetics of azole antifungals in cystic fibrosis. Mycopathologia. 2018;183:139–150. [DOI] [PubMed] [Google Scholar]
- 10. Schwarz C, Hartl D, Eickmeier O, et al. Progress in definition, prevention and treatment of fungal infections in cystic fibrosis. Mycopathologia. 2018;183:21–32. [DOI] [PubMed] [Google Scholar]
- 11. Schwarz C, Vandeputte P, Rougeron A, et al. Developing collaborative works for faster progress on fungal respiratory infections in cystic fibrosis. Med Mycol. 2018;56:42–59. [DOI] [PubMed] [Google Scholar]
- 12. Warris A, Bercusson A, Armstrong-James D. Aspergillus colonization and antifungal immunity in cystic fibrosis patients. Med Mycol. 2019;57:S118–S126. [DOI] [PubMed] [Google Scholar]
- 13. Williams C, Ranjendran R, Ramage G. Pathogenesis of fungal infections in cystic fibrosis. Curr Fungal Infect Rep. 2016;10:163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paulussen C, Hallsworth JE, Alvarez-Perez S, et al. Ecology of aspergillosis: insights into the pathogenic potency of Aspergillus fumigatus and some other Aspergillus species. Microb Biotechnol. 2017;10:296–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mortensen KL, Jensen RH, Johansen HK, et al. Aspergillus species and other molds in respiratory samples from patients with cystic fibrosis: a laboratory-based study with focus on Aspergillus fumigatus azole resistance. J Clin Microbiol. 2011;49:2243–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bakare N, Rickerts V, Bargon J, Just-Nubling G. Prevalence of Aspergillus fumigatus and other fungal species in the sputum of adult patients with cystic fibrosis. Mycoses. 2003;46:19–23. [DOI] [PubMed] [Google Scholar]
- 17. Lipuma JJ. The changing microbial epidemiology in cystic fibrosis. Clin Microbiol Rev. 2010;23:299–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Valenza G, Tappe D, Turnwald D, et al. Prevalence and antimicrobial susceptibility of microorganisms isolated from sputa of patients with cystic fibrosis. J Cyst Fibros. 2008;7:123–127. [DOI] [PubMed] [Google Scholar]
- 19. Baxter CG, Dunn G, Jones AM, et al. Novel immunologic classification of aspergillosis in adult cystic fibrosis. J Allergy Clin Immunol. 2013;132:560–566.e10. [DOI] [PubMed] [Google Scholar]
- 20. de Vrankrijker AM, van der Ent CK, van Berkhout FT, et al. Aspergillus fumigatus colonization in cystic fibrosis: implications for lung function. Clin Microbiol Infect. 2011;17:1381–1386. [DOI] [PubMed] [Google Scholar]
- 21. Aaron SD, Vandemheen KL, Freitag A, et al. Treatment of Aspergillus fumigatus in patients with cystic fibrosis: a randomized, placebo-controlled pilot study. PLoS ONE. 2012;7:e36077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stevens DA, Moss RB, Kurup VP, et al. Allergic bronchopulmonary aspergillosis in cystic fibrosis—state of the art: Cystic Fibrosis Foundation Consensus Conference. Clin Infect Dis. 2003;37:S225–S264. [DOI] [PubMed] [Google Scholar]
- 23. Maturu VN, Agarwal R. Prevalence of Aspergillus sensitization and allergic bronchopulmonary aspergillosis in cystic fibrosis: systematic review and meta-analysis. Clin Exp Allergy. 2015;45:1765–1778. [DOI] [PubMed] [Google Scholar]
- 24. Agarwal R. Allergic bronchopulmonary aspergillosis. Chest. 2009;135:805–826. [DOI] [PubMed] [Google Scholar]
- 25. Krasnick J, Patterson R, Roberts M. Allergic bronchopulmonary aspergillosis presenting with cough variant asthma and identifiable source of Aspergillus fumigatus. Ann Allergy Asthma Immunol. 1995;75:344–346. [PubMed] [Google Scholar]
- 26. Roth R, Schatz M. Allergic bronchopulmonary aspergillosis presenting as chronic cough in an elderly woman without previously documented asthma. Perm J. 2013;17:e103–e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Agarwal R, Chakrabarti A, Shah A, et al. Allergic bronchopulmonary aspergillosis: review of literature and proposal of new diagnostic and classification criteria. Clin Exp Allergy. 2013;43:850–873. [DOI] [PubMed] [Google Scholar]
- 28. Patterson TF, Thompson GR, III, Denning DW, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the infectious diseases society of America. Clin Infect Dis. 2016;63:e1–e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kraemer R, Delosea N, Ballinari P, Gallati S, Crameri R. Effect of allergic bronchopulmonary aspergillosis on lung function in children with cystic fibrosis. Am J Respir Crit Care Med. 2006;174:1211–1220. [DOI] [PubMed] [Google Scholar]
- 30. Castellani C, Duff AJA, Bell SC, et al. ECFS best practice guidelines: the 2018 revision. J Cyst Fibros. 2018;17:153–178. [DOI] [PubMed] [Google Scholar]
- 31. Fukutomi Y, Taniguchi M. Sensitization to fungal allergens: resolved and unresolved issues. Allergol Int. 2015;64:321–331. [DOI] [PubMed] [Google Scholar]
- 32. Wojnarowski C, Eichler I, Gartner C, et al. Sensitization to Aspergillus fumigatus and lung function in children with cystic fibrosis. Am J Respir Crit Care Med. 1997;155:1902–1907. [DOI] [PubMed] [Google Scholar]
- 33. Amin R, Dupuis A, Aaron SD, Ratjen F. The effect of chronic infection with Aspergillus fumigatus on lung function and hospitalization in patients with cystic fibrosis. Chest. 2010;137:171–176. [DOI] [PubMed] [Google Scholar]
- 34. Maguire CP, Hayes JP, Hayes M, Masterson J, FitzGerald MX. Three cases of pulmonary aspergilloma in adult patients with cystic fibrosis. Thorax. 1995;50:805–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pendleton M, Denning DW. Multifocal pulmonary aspergillomas: case series and review. Ann N Y Acad Sci. 2012;1272:58–67. [DOI] [PubMed] [Google Scholar]
- 36. Brandt C, Roehmel J, Rickerts V, Melichar V, Niemann N, Schwarz C. Aspergillus bronchitis in patients with cystic fibrosis. Mycopathologia. 2018;183:61–69. [DOI] [PubMed] [Google Scholar]
- 37. Shoseyov D, Brownlee KG, Conway SP, Kerem E. Aspergillus bronchitis in cystic fibrosis. Chest. 2006;130:222–226. [DOI] [PubMed] [Google Scholar]
- 38. Kousha M, Tadi R, Soubani AO. Pulmonary aspergillosis: a clinical review. Eur Respir Rev. 2011;20:156–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Betancourt BY, Garofoli AC, Sandhu JS, Boma N, Sy AM. Pulmonary aspergillosis presenting with recurrent haemoptysis [published online ahead of print July 7, 2015]. BMJ Case Rep. doi: 10.1136/bcr-2015-211249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang W, Hu Y, Chen L, Gao J, Xie L. Pleural aspergillosis complicated by recurrent pneumothorax: a case report. J Med Case Rep. 2010;4:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fernandez-Ruiz M, Silva JT, San-Juan R, et al. Aspergillus tracheobronchitis: report of 8 cases and review of the literature. Medicine (Baltimore). 2012;91:261–273. [DOI] [PubMed] [Google Scholar]
- 42. Singh N, Husain S. Aspergillus infections after lung transplantation: clinical differences in type of transplant and implications for management. J Heart Lung Transplant. 2003;22:258–266. [DOI] [PubMed] [Google Scholar]
- 43. Lackner M, de Hoog GS, Yang L, et al. Proposed nomenclature for Pseudallescheria, Scedosporium and related genera. Fungal Divers. 2014;67:1–10. [Google Scholar]
- 44. Pihet M, Carrere J, Cimon B, et al. Occurrence and relevance of filamentous fungi in respiratory secretions of patients with cystic fibrosis: a review. Med Mycol. 2009;47:387–397. [DOI] [PubMed] [Google Scholar]
- 45. Bernhardt A, Sedlacek L, Wagner S, Schwarz C, Wurstl B, Tintelnot K. Multilocus sequence typing of Scedosporium apiospermum and Pseudallescheria boydii isolates from cystic fibrosis patients. J Cyst Fibros. 2013;12:592–598. [DOI] [PubMed] [Google Scholar]
- 46. Horre R, Marklein G. Isolation and clinical significance of Pseudallescheria and Scedosporium species. Med Mycol. 2009;47:415–421. [DOI] [PubMed] [Google Scholar]
- 47. Schwarz C, Brandt C, Antweiler E, et al. Prospective multicenter German study on pulmonary colonization with Scedosporium/Lomentospora species in cystic fibrosis: epidemiology and new association factors. PLoS ONE. 2017;12:e0171485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cimon B, Carrere J, Vinatier JF, Chazalette JP, Chabasse D, Bouchara JP. Clinical significance of Scedosporium apiospermum in patients with cystic fibrosis. Eur J Clin Microbiol Infect Dis. 2000;19:53–56. [DOI] [PubMed] [Google Scholar]
- 49. Symoens F, Knoop C, Schrooyen M, et al. Disseminated Scedosporium apiospermum infection in a cystic fibrosis patient after double-lung transplantation. J Heart Lung Transplant. 2006;25:603–607. [DOI] [PubMed] [Google Scholar]
- 50. Boyle M, Moore JE, Whitehouse JL, Bilton D, Downey DG. The diagnosis and management of respiratory tract fungal infection in cystic fibrosis: a UK survey of current practice. Med Mycol. 2019;57:155–160. [DOI] [PubMed] [Google Scholar]
- 51. Lackner M, de Hoog GS, Verweij PE, et al. Species-specific antifungal susceptibility patterns of Scedosporium and Pseudallescheria species. Antimicrob Agents Chemother. 2012;56:2635–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tintelnot K, Just-Nubling G, Horre R, et al. A review of German Scedosporium prolificans cases from 1993 to 2007. Med Mycol. 2009;47:351–358. [DOI] [PubMed] [Google Scholar]
- 53. Delhaes L, Harun A, Chen SC, et al. Molecular typing of Australian Scedosporium isolates showing genetic variability and numerous S. aurantiacum. Emerg Infect Dis. 2008;14:282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lopez L, Gaztelurrutia L, Cuenca-Estrella M, et al. [Infection and colonization by Scedosporium prolificans]. Enferm Infecc Microbiol Clin. 2001;19:308–313. [DOI] [PubMed] [Google Scholar]
- 55. Troke P, Aguirrebengoa K, Arteaga C, et al. Treatment of scedosporiosis with voriconazole: clinical experience with 107 patients. Antimicrob Agents Chemother. 2008;52:1743–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rammaert B, Puyade M, Cornely OA, et al. Scedosporium complex and Lomentospora prolificans in lung transplantation: an international practice survey. Paper presented at: European Society of Clinical Microbiology and Infectious Diseases; April 21–24, 2018; Madrid, Spain. [Google Scholar]
- 57. Rodriguez-Tudela JL, Berenguer J, Guarro J, et al. Epidemiology and outcome of Scedosporium prolificans infection, a review of 162 cases. Med Mycol. 2009;47:359–370. [DOI] [PubMed] [Google Scholar]
- 58. Biswas C, Law D, Birch M, et al. In vitro activity of the novel antifungal compound F901318 against Australian Scedosporium and Lomentospora fungi. Med Mycol. 2018;56:1050–1054. [DOI] [PubMed] [Google Scholar]
- 59. Jung EH, Meyers DJ, Bosch J, Casadevall A. Novel antifungal compounds discovered in medicines for malaria venture’s malaria box. mSphere. 2018;3:e00537–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rivero-Menendez O, Cuenca-Estrella M, Alastruey-Izquierdo A. In vitro activity of APX001A against rare moulds using EUCAST and CLSI methodologies. J Antimicrob Chemother. 2019;74:1295–1299. [DOI] [PubMed] [Google Scholar]
- 61. Seyedmousavi S, Rafati H, Ilkit M, Tolooe A, Hedayati MT, Verweij P. Systemic antifungal agents: current status and projected future developments. Methods Mol Biol. 2017;1508:107–139. [DOI] [PubMed] [Google Scholar]
- 62. Lebecque P, Leonard A, Huang D, et al. Exophiala (Wangiella) dermatitidis and cystic fibrosis—prevalence and risk factors. Med Mycol. 2010;48:S4–S9. [DOI] [PubMed] [Google Scholar]
- 63. Masoud-Landgraf L, Badura A, Eber E, Feierl G, Marth E, Buzina W. Modified culture method detects a high diversity of fungal species in cystic fibrosis patients. Med Mycol. 2014;52:179–186. [DOI] [PubMed] [Google Scholar]
- 64. Kondori N, Lindblad A, Welinder-Olsson C, Wenneras C, Gilljam M. Development of IgG antibodies to Exophiala dermatitidis is associated with inflammatory responses in patients with cystic fibrosis. J Cyst Fibros. 2014;13:391–399. [DOI] [PubMed] [Google Scholar]
- 65. Kondori N, Gilljam M, Lindblad A, Jonsson B, Moore ER, Wenneras C. High rate of Exophiala dermatitidis recovery in the airways of patients with cystic fibrosis is associated with pancreatic insufficiency. J Clin Microbiol. 2011;49:1004–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Muthig M, Hebestreit A, Ziegler U, Seidler M, Müller FM. Persistence of Candida species in the respiratory tract of cystic fibrosis patients. Med Mycol. 2010;48:56–63. [DOI] [PubMed] [Google Scholar]
- 67. Chotirmall SH, Greene CM, McElvaney NG. Candida species in cystic fibrosis: a road less travelled. Med Mycol. 2010;48:S114–S124. [DOI] [PubMed] [Google Scholar]
- 68. Muller FM, Seidler M. Characteristics of pathogenic fungi and antifungal therapy in cystic fibrosis. Expert Rev Anti Infect Ther. 2010;8:957–964. [DOI] [PubMed] [Google Scholar]
- 69. Wahab AA, Taj-Aldeen SJ, Kolecka A, ElGindi M, Finkel JS, Boekhout T. High prevalence of Candida dubliniensis in lower respiratory tract secretions from cystic fibrosis patients may be related to increased adherence properties. Int J Infect Dis. 2014;24:14–19. [DOI] [PubMed] [Google Scholar]
- 70. Grosse-Onnebrink J, Stehling F, Tschiedel E, et al. Bacteraemia and fungaemia in cystic fibrosis patients with febrile pulmonary exacerbation: a prospective observational study. BMC Pulm Med. 2017;17:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kuhl T, Langebartels G, Madershahian N, Wahlers T. [Extracorporeal life support given to a 16-year-old girl with cystic fibrosis, candida pneumonia and acute respiratory distress syndrome]. Dtsch Med Wochenschr. 2010;135:2071–2075. [DOI] [PubMed] [Google Scholar]
- 72. AbdulWahab A, Salah H, Chandra P, Taj-Aldeen SJ. Persistence of Candida dubliniensis and lung function in patients with cystic fibrosis. BMC Res Notes. 2017;10:326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Delhaes L, Monchy S, Frealle E, et al. The airway microbiota in cystic fibrosis: a complex fungal and bacterial community—implications for therapeutic management. PLoS ONE. 2012;7:e36313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hong G, Miller HB, Allgood S, Lee R, Lechtzin N, Zhang SX. Use of selective fungal culture media increases rates of detection of fungi in the respiratory tract of cystic fibrosis patients. J Clin Microbiol. 2017;55:1122–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Abdolrasouli A, Bercusson AC, Rhodes JL, et al. Airway persistence by the emerging multi-azole-resistant Rasamsonia argillacea complex in cystic fibrosis. Mycoses. 2018;61:665–673. [DOI] [PubMed] [Google Scholar]
- 76. Esther CR, Jr, Plongla R, Kerr A, Lin FC, Gilligan P. Clinical outcomes in cystic fibrosis patients with Trichosporon respiratory infection. J Cyst Fibros. 2016;15:e45–e49. [DOI] [PubMed] [Google Scholar]
- 77. Hong G, White M, Lechtzin N, et al. Fatal disseminated Rasamsonia infection in cystic fibrosis post-lung transplantation. J Cyst Fibros. 2017;16:e3–e7. [DOI] [PubMed] [Google Scholar]
- 78. Rautemaa V, Green HD, Jones AM, Rautemaa-Richardson R. High level of beta-(1,3)-d-glucan antigenaemia in cystic fibrosis in the absence of invasive fungal disease. Diagn Microbiol Infect Dis. 2017;88:316–321. [DOI] [PubMed] [Google Scholar]
- 79. Trager J, Melichar VO, Meyer R, Rauh M, Bogdan C, Held J. Serum (1–>3)-beta-D-glucan and galactomannan levels in patients with cystic fibrosis: a retrospective cohort study. BMC Pulm Med. 2018;18:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Giacobbe DR, Del Bono V, Viscoli C, Mikulska M. Use of 1,3-beta-D-glucan in invasive fungal diseases in hematology patients. Expert Rev Anti Infect Ther. 2017;15:1101–1112. [DOI] [PubMed] [Google Scholar]
- 81. Giacobbe DR, Mikulska M, Tumbarello M, et al. Combined use of serum (1,3)-beta-D-glucan and procalcitonin for the early differential diagnosis between candidaemia and bacteraemia in intensive care units. Crit Care. 2017;21:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Heltshe SL, Mayer-Hamblett N, Burns JL, et al. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin Infect Dis. 2015;60:703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hisert KB, Heltshe SL, Pope C, et al. Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am J Respir Crit Care Med. 2017;195:1617–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Prigitano A, Esposto MC, Biffi A, et al. Triazole resistance in Aspergillus fumigatus isolates from patients with cystic fibrosis in Italy. J Cyst Fibros. 2017;16:64–69. [DOI] [PubMed] [Google Scholar]
- 85. Seufert R, Sedlacek L, Kahl B, et al. Prevalence and characterization of azole-resistant Aspergillus fumigatus in patients with cystic fibrosis: a prospective multicentre study in Germany. J Antimicrob Chemother. 2018;73:2047-2053. [DOI] [PubMed] [Google Scholar]