

Abstract
Aims:
Metformin is commonly used to treat type 2 diabetes mellitus; however, in recent years, it was found to play a potential role in the protection of myocardial injury. In this study, we intended to investigate whether metformin had protective effects on bacterial myocarditis.
Methods and Results:
We stimulated rat cardiac myoblast H9c2 cells with lipopolysaccharide (LPS) and administrated with metformin. The results showed that cell viability after LPS stimulation was greatly reduced. The expression levels of phosphorylated p38 mitogen-activated protein kinases (MAPK) and c-Jun N-terminal kinases (JNK), nuclear factor (NF)-κB (NF-κB), BAX, and cleaved Caspase3 were significantly increased, while the expression of antiapoptotic protein Bcl-2 showed a prominent decrease compared to control. Nevertheless, the cells activity increased remarkably after metformin administration, and the expression levels of intracellular related proteins showed the opposite trend to that of the LPS group.
Conclusion:
We demonstrate that LPS stimulation may activate intracellular MAPK/JNK and NF-κB signaling pathways and thus induce cell apoptosis. In contrast, metformin reduced apoptosis by inhibiting this signaling pathway and increasing the expression level of Bcl-2. Moreover, it was found that metformin could enhance the ability of cells to antagonize redox damage by regulating the activities of superoxide dismutase and lactate dehydrogenase and subsequently promote the recovery of cardiomyocyte function.
Keywords: metformin, LPS, cardiomyocyte, MAPK, NF-κB
Introduction
Bacterial myocarditis is usually caused by the direct infection of the heart muscle by bacteria or the action of toxins produced by bacteria on the myocardium or allergic reactions caused by bacterial products. With the increase in immune-compromised hosts, the number of patients with bacterial myocarditis has recently been increasing. Although the pathogenic factors are quite various, pathogens that caused bacterial myocarditis are mainly Staphylococcus aureus, Escherichia coli, Streptococcus, Pneumococcus, and Neisseria meningitides. 1 The clinical manifestations of bacterial myocarditis often depend on the extent of the lesion; while the milder can be asymptomatic, the severe one may be suddenly dead. In this study, we proposed to find a drug that can effectively treat myocardial injury caused by bacteria. During our study, we focused on metformin and attempted to find out whether metformin has a positive therapeutic effect on bacterial myocarditis.
Metformin, which was reported to have an effect on the control of blood glucose levels, is mainly used as a preferred oral antidiabetic drug in patients with type 2 diabetes mellitus.2 Metformin is a first-line drug for controlling circulating insulin levels in diabetes. Over the past decade, researchers have found that metformin has a cardiovascular protective effect, which can significantly reduce the patient’s cardiovascular events.3-6 Moreover, researches in recent years have shown that metformin has potential therapeutic effects on myocardial dysfunction, but the exact mechanism involved in this process has not been extensively elucidated. According to Sasaki et al’s study, metformin can prevent progression of heart failure in dogs.3 Subsequently, Yin et al notice that metformin improves cardiac function in a non-diabetic rat model of post-MI heart failure.4 Burn injury stimulates stress-responsive components, p38 mitogen-activated protein kinase (MAPK)/c-Jun N-terminal kinases (JNK)/nuclear factor (NF)-κB (NF-κB). p38 MAPK plays a role in postburn cardiomyocyte tumor necrosis factor-α (TNF-α) secretion and cardiac dysfunction.5 Based on their progress, in this study, we cultured rat cardiac myoblast H9c2 cells in vitro and exposed to lipopolysaccharide (LPS), in order to imitate myocarditis followed by gram-negative bacterial infection and to explore the possible response mechanisms underlying this process.
Materials and Methods
Antibodies and Regents
Lipopolysaccharide was purchased from Sigma (Sigma, Missouri), and metformin was purchased from Phoenix Pharmaceuticals Inc (California). Mouse or goat immunoglobulin G directed against p38MAPK and JNK (phosphorylated and total), NF-κB, Bcl-2, BAX, caspase 3, and p47phox were obtained from Abcam (Abcam, USA). Glyceraldehyde phosphate dehydrogenase (GAPDH) was used as loading controls. The total protein level was normalized to the GAPDH protein level.
Escherichia coli Strain and Mice
The E coli strain JM109 was recovered from lyophilized powder by resuspending in LB medium and culturing overnight on an LB agar plate at 37°C. Single colonies of E coli on the plate were used as a group of original seed E coli.
ICR mice were 6 to 8 weeks old, weighed 20 ± 2 g, and were obtained from Jilin University Animal Center (Chang Chun, China). They were fed in the laboratory for 1 week before the experiment. Mice were kept in standard pathogen-free and temperature-controlled units. Food and water were supplied ad libitum. The animal protocol was approved by the Scientific Investigation Board of Science and Technology (Jilin).
MTT Assay
Cell viability was examined by using 3-4,5-dimethylthiazol-2-yl-2,5-diphenyl-2 H-tetrazolium bromide (MTT) assay. H9c2 cells were plated into 96-well plates and incubated with 20 mL of MTT (0.5 mg/mL) for 4 hours. After removing the media gently, 200 mL of dimethyl sulfoxide were added into each well to dissolve formazan. After shaking the samples for 10 minutes, the absorbances were measured at 570 nm by a microplate reader.
Western Blotting
Western blotting was done using standard protocol. Briefly, the samples were lysed and denatured in 5× sample buffer. Same amounts of proteins were separated on a 10% sodium dodecyl sulfate-polyacrylamide gel and transferred to a nitrocellulose membrane. The nitrocellulose membrane was then incubated with 5% nonfat milk in Tris-buffered saline (150 mM NaCl, 20 mM Tris-HCl, pH7.4) with primary antibody at 4°C overnight. After washing, the membrane was further incubated with secondary antibody for 45 minutes, and proteins were detected with an electrochemiluminescence (ECL; Thermo Pierce) detection system. Image J software was used to measure the band intensity.
Enzyme-linked Immunosorbent Assay
Lactate dehydrogenase (LDH), malondialdehyde (MDA), superoxide dismutase (SOD), enzyme-linked immunosorbent assay kits and glutathione (GSH) assay kit were purchased from Sigma (Shanghai, China). The absorbance was measured at a wavelength of 450 nm by a microplate reader, and the sample concentration was calculated.
Immunofluorescence Microscopy
The cells were fixed with 4% formaldehyde in phosphate-buffered saline (PBS) for 30 minutes, blocked, permeabilized in 5% goat serum in PBS with 0.1% Triton X-100 (15 minutes), and labeled with primary antibody for 2 hours. Cells were then washed 3 times and labeled with fluorescence-conjugated secondary antibody for 1 hour. Immunofluorescence was visualized with confocal laser scanning microscopy. All images were analyzed using a background subtraction method off-line.
Statistical Analysis
All experiments were performed 3 times, and the results reported as mean ± standard error (SE). Statistical differences among multiple groups were analyzed by analysis of variance. Comparisons between the 2 groups were performed by unpaired Student t test. A value of P < .05 was considered statistically significant.
Results
Injury Dose Detection of LPS on H9c2 Cell Activity
To construct the model of myocardial injury, we first cultured rat cardiac myoblast H9c2 cells and stimulated them with different concentrations of LPS to identify a proper administration dose. As shown in Figure 1B, we detected the viability of H9c2 cells by MTT assay at different concentrations of LPS for 24, 48, and 72 hours, respectively. With the increase in LPS stimulation concentration from 1 to 10 μg/μL, the cell viability decreased by 20% after 24 hours of treatment, 30% for 48 hours, and 40% for 72 hours. The apoptotic rate increased obviously after LPS stimulation, so we evaluated the expression levels of Caspase3 and p47phox which possibly related to apoptosis by Western blot. It can be inferred from Figure 1C that the expression levels of these 2 proteins showed a dose-dependent increase in H9c2 cells exposed to LPS compared to control. At the same time, we collected cell supernatant fraction to measure the activity of LDH, MDA, SOD, and GSH. The activity of SOD in LPS-stimulated H9c2 cells decreased by a large margin compared to untreated cells, which indicates that LPS does exert large injury on H9c2 cells. On the contrary, the activity of LDH showed the opposite change tendency to SOD, which also suggested the decline of H9c2 cell viability in LPS-treated groups (Figure. 1C). Thus, we decided to stimulate H9c2 cells with 5 μg/μL LPS for 48 hours to model cardiomycyte injury. The subsequent experiments are all based on this concentration.
Figure 1.

Effects of lipopolysaccharide (LPS) on H9c2 cells. (A) Microscopic images of H9c2 cells under different treatments. The morphology of H9c2 cells was worse after stimulation with 5 μg/μL LPS compared to control, which was improved after 10 mM metformin administration. Additionally, the cell status under the preadministration of metformin was comparable to that of the control group. (B) Cells’ viability detection of H9c2 using MTT assay after incubation with different concentrations of LPS (0, 1, 3, 5, 10 μg/μL) for 24, 48, 72 hours, respectively. There are significant differences in cells viability administrated with different concentrations of metformin for different times, (C) Western blot was used to detect the expression levels of several proteins in H9c2 cells under different concentrations of LPS stimulation (using glyceraldehyde phosphate dehydrogenase [GAPDH] as internal reference). Expression levels of cleaved Caspase3 and p47phox showed a dose-dependent increase. (D) The activities of lactate dehydrogenase (LDH), malondialdehyde (MDA), superoxide dismutase (SOD), and glutathione (GSH) in H9c2 cells under different concentrations of LPS treatment were evaluated by microplate reader to determine the appropriate dose to construct the model of cardiomyocytes injury. The activities of LDH and MDA showed an LPS dose-dependent increase, while those of SOD and GSH showed the contrary trend. *P < .05 vs control group, n = 3 for each group.
Effect of Metformin Administration on Myocardial Cells
To determine whether metformin has a protective effect on cardiomyocytes against bacterial myocardial dysfunction in vitro, we have cultured H9c2 cells and stimulated with LPS as described earlier. By setting the concentration gradient of metformin to detect the cells viability at different administrative doses, we found that when 10 mM metformin was used after LPS stimulation, H9c2 cells displayed the optimal viability. With the increasing concentration of metformin up to 30 mM, the percentage of survival in H9c2 cells remains high. However, it has a sharp descend nearly 40% when used over 50 mM metformin (Figure. 2A). We can consequently identify the ideal dose of metformin for treating the LPS-induced myocardial cell injury, that is, 10 mM. As a supplemental control, the H9c2 cells without LPS stimulation were treated with metformin (10 mM) alone to determine whether it has a cytotoxic effect on cells. The results are shown in Figure 1A; from the microscopic image we can see that cell morphology of the group treated with metformin alone showed no difference with control, in which H9c2 cells were densely distributed and grew well. However, it was displayed small, shrunken, and sparsely distributed when incubated with 5 μg/μL LPS. Surprisingly, administration with 10 mM metformin after LPS stimulation, the survival status of H9c2 cells got recovered greatly, and the cells density had increased to a large degree. These results indicate that the decreased viability of H9c2 cells stimulated by LPS could be restored by metformin, which preliminarily proved that metformin had a commendable reversal effect on LPS-induced cardiomyocyte apoptosis.
Figure 2.

Effects of metformin on mitogen-activated protein kinase (MAPK) signal transduction pathway in H9c2 cells. (A) The percentage of survival in H9c2 cells was measured at different concentrations of metformin (0, 1, 5, 10, 20, 30, 50, 100, and 150 mM). When the concentration was over 50 mM, the cells viability decreased significantly, indicating that metformin excess had a toxic effect on H9c2 cells. *P < .05 vs control group, n = 3 for each group. (B) Western blot was used to detect the expression levels of several proteins in H9c2 cells under different groups of control, metformin preadministration, LPS stimulation as well as metformin administration after LPS treatment (using glyceraldehyde phosphate dehydrogenase [GAPDH] as internal reference protein). The expression level of Bcl-2 was significantly reduced after stimulation with 5 μg/μL LPS for 48 hours, while administration with 10 mM metformin reversed this. Changes in expression levels of other proteins showed the opposite trend as Bcl-2.
Based on the previous study and accessed to relevant literature, we hypothesized that metformin may function by inhibiting BAX gene expression and upregulating Bcl-2 at the same time possibly by activating MAPK/JNK signaling pathway. Thus, we aimed to evaluate the expression levels of NF and several proteins in H9c2 cells involved in this signaling pathway using Western blot and immunofluorescence; it is shown that the expression of phosphorylated p38MAPK appears to show remarkable differences among groups of model, preadministration, administration and control. In detail, the group of metformin preadministration reveals nearly the same levels of P-P38MAPK compared to control, while LPS-induced model group shows increasing trend, worthy to notice that the expression level of phosphorylated p38MAPK decreased in low amplitude after the administration of metformin. Consistently, the change in expression levels of phosphorylated JNK, NF-κB, cleaved Caspase3, p47phox among these 4 groups showed the same trend as P-P38MAPK (Figure 2B and 3).
Figure 3.

Effects of metformin on mitogen-activated protein kinase (MAPK) signal transduction pathway in H9c2 cells. Immunostaining was used to detect the expression levels of several proteins in H9c2 cells under different groups of control, metformin preadministration, lipopolysaccharide (LPS) stimulation as well as metformin administration after LPS treatment.
As the antiapoptotic protein, Bcl-2, however, shows the opposite trend as phosphorylated P38MAPK, that is, to say, in LPS-treated H9c2 cells, its expression level is much lower than the other 3 groups. Another marker of apoptosis, BAX, as is known, its expression level, is negatively correlated with cells viability. In LPS-treated H9c2 cells, the expression level of Bcl-2 reduces prominently compared to control, while BAX shows a remarkable increase. Twelve hours later after metformin administration, the expression level of Bcl-2 showed a substantial increase, while BAX showed the contrary tendency. These findings suggest that metformin encounters the cardiomyocytes and reverses LPS-induced cell apoptosis by altering the expression levels of related proteins such as Bcl-2 and BAX. The possible mechanism of this process will be discussed later.
Effects and Possible Mechanism of Metformin on Bacterial Myocarditis in Mice
In the previous step, we determined that metformin could inhibit LPS-mediated cardiomyocyte injury by activating a series of intracellular signaling pathways through in vitro experiments in H9c2 cells.
Given the results of cell experiments, we detected the expression levels of several proteins related to MAPK/JNK signaling pathway in cardiomyocytes of mice by Western blot. The results showed that, consistently with the H9c2 cells experiments in vitro, the expression of phosphorylated p38MAPK was much higher in the serum of mice with myocarditis than that of the control group, and the expression of phosphorylated JNK also showed the same trend as well as NF-κB and C-Caspase3. After injection of metformin, the expression levels of the 4 proteins above were significantly reduced, especially p-JNK, decreased to the comparable level as in the control group. It is also momentous to consider the expression levels of Bcl-2 and BAX. The content of Bcl-2 was significantly increased after the administration of metformin compared to the model group. In contrast, the expression of BAX protein decreased even more pronounced (Figure. 4A).
Figure 4.

Effects of metformin on mitogen-activated protein kinase (MAPK) signal transduction pathway in mice. (A) The expression levels of several proteins in cardiomyocytes of mice was detected by Western blot under different groups of control, metformin preadministration, Escherichia coli injection as well as metformin administration after bacterial myocardial injury (using glyceraldehyde phosphate dehydrogenase [GAPDH] as internal reference). Changes in expression levels of these proteins in vivo showed the same trend as those of the cell experiments. (B) The activities of lactate dehydrogenase (LDH), malondialdehyde (MDA), superoxide dismutase (SOD), and glutathione (GSH) in the serum of mice under different treatment were evaluated by microplate reader. The activities of LDH and MDA showed a remarkable augment after intraperitoneal injection of E·coli, while it had a reduction after metformin administration in vivo. In contrast, changes in the activities of SOD and GSH displayed the opposite trend. # P < .05 vs model group, n = 6 for each group.
Besides, we measured the activities of LDH, SOD, MDA, and GSH in serum collected from mice tail vein using microplate reader to determine the recovery of myocardial injury before and after metformin administration. As can be seen from Figure 4B, the LDH activity in mice injected intraperitoneally with E coli was greatly increased (about 50%) compared to that of the control group. However, the activity of LDH decreased after metformin administration (more than 10%). Although slightly higher than that in the control group, it was sufficient to show that metformin reduced the degree of myocardial injury and attenuated the apoptosis of cardiomyocytes. Similarly, the activity of MDA changed in the same trend as LDH, which indirectly reflected the severity of the myocardial cells being attacked by free radicals. Nevertheless, the change in SOD activity was opposite to that of LDH. While the activity of SOD was decreased by more than 25% after injection of E·coli, it was significantly increased after metformin administration, which also indicates to some extent that the ability for cardiomyocytes scavenging oxygen free radicals increased after metformin administration. The trend of activity changes in GSH is the same as SOD. The results above suggest that the myocardial injury induced by intraperitoneal injection of E·coli in mice can be reduced by administration of metformin, and the apoptotic rate of cardiomyocytes can be decreased as well.
Discussion
In this study, we found that stimulating rat cardiac myoblast H9c2 cells with LPS at a certain dose could induce apoptosis, which may be a major cause of myocarditis caused by bacterial infection, and metformin administration could greatly alleviate this injury. In order to explore the potential mechanism of metformin in the treatment of myocardial injury, we detected the expression levels of related proteins in H9c2 cells and found that the expression of P-p38MAPK, p-JNK, and NF-κB increased significantly after LPS stimulation. It is well known that JNK induces NF-κB activation in response to a variety of stress stimuli.6-9 Therefore, we believe that LPS can induce H9c2 cells apoptosis by activating the MAPK/JNK signaling pathway and upregulating the expression of NF-κB. Interestingly, this process can be reversed by metformin, which can inhibit the activation of MAPK/JNK signaling pathway by decreasing the phosphorylation levels of p38MAPK and JNK, and inhibit the activation of downstream signal NF-κB, eventually preventing H9c2 cells from apoptosis (as shown in Figure 5).
Figure 5.
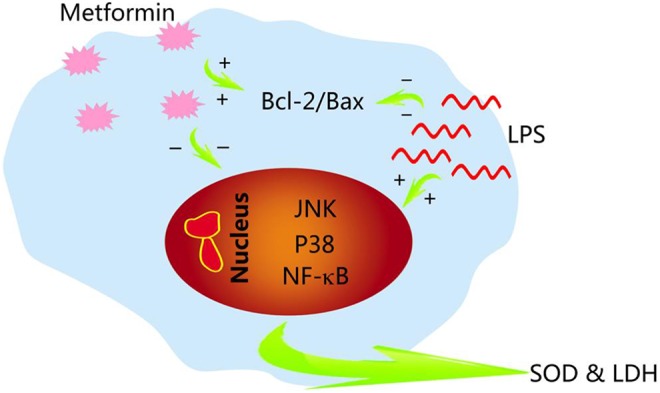
Schematic protection of metformin. Lipopolysaccharide (LPS) stimulation may activate intracellular mitogen-activated protein kinase (MAPK)/ c-Jun N-terminal kinases (JNK) and nuclear factor (NF)-κB (NF-κB) signaling pathways and thus induce cell apoptosis. In contrast, metformin reduced apoptosis by inhibiting this signaling pathways and increasing the expression level of Bcl-2. Moreover, metformin could enhance the ability of cells to antagonize redox damage by regulating the activities of superoxide dismutase (SOD) and lactate dehydrogenase (LDH).
Additionally, as is universally investigated, Bcl-2 family proteins are central regulators of apoptosis in mammals which include proteins both promoting and inhibiting apoptosis.10 We also detected the expression levels of antiapoptotic protein Bcl-2 and apoptotic protein BAX, and found that the expression of Bcl-2 in H9c2 cells was significantly decreased after LPS stimulation, accompanied by an increase in the expression of BAX. It indicated that LPS could activate BAX gene and inhibit Bcl-2, thus decreasing Bcl-2–Bax ratio and eventually promoting cell apoptosis. Metformin, in contrast, could increase Bcl-2–BAX ratio and consequently inhibit LPS-induced H9c2 cell apoptosis. Moreover, the synchronous changes between BAX–Bcl-2 ratio and the phosphorylation level of p38MAPK and JNK probably means that BAX/Bcl-2 may act as the upstream regulatory effector of MAPK–JNK signaling pathway to promote or inhibit apoptosis.
In recent years, many studies have shown that metformin not only serves as a specific drug for the treatment of type 2 diabetes mellitus in clinic but also has a better therapeutic effect on diseases such as myocardial infarction and myocardial ischemia–reperfusion injury. There are also many arguments about the mechanism of metformin in the treatment of different myocardial injuries. Xu Chen et al believe that metformin can significantly inhibit the opening of mitochondrial permeability transition pore (mPTP) and the production of reactive oxygen species (ROS) by activating AMPK and regulating JNK-mediated NF-κB signal transduction pathway, thereby weakening the inflammation that occurs during ischemia–reperfusion in H9c2 cells.11 In addition, Rita Zilinyi et al found that doxorubicin treatment impairs the autophagic processes and that damaged macromolecules cannot be degraded in the cells. Coadministration of metformin with doxorubicin could normalize the autophagy activity and confer cardioprotective effects, thereby inhibiting DOX-induced myocardial cytotoxicity.12 What’s more, it was found that metformin administration decreased the level of the apoptotic effector caspase 3 and enhanced the level of anti-apoptotic Bcl-2 proteins, suggesting that metformin possesses antiapoptotic properties in doxorubicin-induced cardiotoxicity in adult male albino rats.13
In addition, we also found the obviously increasing level of cleaved caspase 3 in H9c2 cells stimulated with LPS. Studies have shown that caspase 3 is activated into cleaved caspase 3, and its expression level is positively correlated with apoptosis.14 Research by Marta Olivera Santa-Catalina et al indicated that JNK inhibition efficiently blocked sorbitol-induced caspase 3 activation, suggesting that JNK signaling pathway is an upstream regulator of both sorbitol-promoted Tau proteolysis and apoptosis by targeting caspase 3 activation in SH-SY5Y.15 As a branch of apoptosis, we believe that metformin may also reduce the expression level of C-Caspase 3 by inhibiting its activation, thereby inhibiting caspase 3-dependent apoptosis and alleviating LPS-induced myocardial injury. Meanwhile, we found that metformin significantly reversed the decrease in SOD and GSH activity and downregulated the activities of both LDH and MDA induced by LPS. As is known, SOD is the vital antioxidant enzyme found in cell fluids, which can reduce superoxide anions and protects cells from superoxide anion damage. This indicates that metformin can enhance the ability of cells to antagonize redox damage by regulating the activities of SOD, LDH, and other enzymes, and it can significantly promote the recovery of myocardial function.
With regard to NF-kB, a large body of evidence has demonstrated that the NF-κB pathway is associated with the release of several proinflammatory cytokines and chemokines.16 As is shown in the research by Lee et al, the TNF-α-induced secretion of inflammatory cytokines, IL-6, is mediated by NF-kB signaling, and the inhibition of NF-kB decreases the expression of IL-6.17 There is ample evidence that NF-kB and MAPK family plays a prominent role in LPS-induced transcriptional regulation of most inflammatory genes that contribute to the development of septic cardiac dysfunction.18-21 Therefore, LPS induces the upregulation of NF-κB, which may further induce the release of more proinflammatory factors and aggravate the severity of bacterial myocarditis, while metformin administration inhibits the release of related proinflammatory factors by inhibiting the expression of NF-κB, which indicates the important role of metformin in inhibiting inflammation and apoptosis of cardiomyocytes.
In conclusion, LPS stimulation activates a series of signaling pathways in H9c2 cells, mainly through activation of MAPK/JNK signaling pathway and NF-κB signaling pathway, which initiates apoptosis signaling to induce apoptosis and produce myocardial injury. Metformin administration reversed this process by inhibiting the phosphorylation level of p38MAPK and JNK, subsequently inhibiting the activation of the MAPK/JNK and its downstream NF-κB signaling pathway, and consequently inhibiting apoptosis. Nevertheless, there are still some shortcomings in this study, we did not detect changes in the expression levels of proinflammatory factors and chemokines related to NF-κB. Besides, there are also some aspects worth improving in this study; next, we intend to use inhibitors of caspase 3, MAPK, and NF-κB, respectively, or in combination to detect whether changes trend in the expression levels of various proteins is the same as that of metformin in rat cardiac myoblast H9c2 cells. Moreover, it is still unknown whether the MAPK/JNK signaling pathway is interlaced with other possible signaling pathways and remains to be investigated. Further research remains to be done so as to confirm the mechanism of metformin in the treatment of bacterial myocarditis.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported by Norman Bethune Program of Jilin University(2015226 and 2015302) & Science and Technology Department Foundation of Jilin Province (20160101221JC, 20160204026YY) & Jilin Province Development and Reform Commission Foundation (2017C046-4 and 2019C052-8).
ORCID iD: Wei Sun  https://orcid.org/0000-0003-0260-4484
https://orcid.org/0000-0003-0260-4484
References
- 1. Komuro J, Ueda K, Kaneko M, Nitta S, Kasao M, Yokoyama M. Various cardiac abnormalities caused by bacterial myocarditis. Int Heart J. 2018;59(1):229–232. [DOI] [PubMed] [Google Scholar]
- 2. Hostalek U, Gwilt M, Hildemann S. Therapeutic use of metformin in prediabetes and diabetes Prevention. Drugs. 2015;75(10):1071–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sasaki H, Asanuma H, Fujita M, et al. Metformin prevents progression of heart failure in dogs role of AMP-activated protein kinase. Circulation. 2009;119(19):2568–2577. [DOI] [PubMed] [Google Scholar]
- 4. Yin M, van der Horst IC, van Melle JP, et al. Metformin improves cardiac function in a nondiabetic rat model of post-MI heart failure. Am J Physiol Heart Circ Physiol. 2011;301(2):H459–H469. [DOI] [PubMed] [Google Scholar]
- 5. Zhang Y, Liu X, Zhang L, et al. Metformin protects against H2O2-induced cardiomyocyte injury by inhibiting the miR-1a-3p/GRP94 pathway. Molecular Therapy: Nucleic Acids. 2018;13:189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang J, Wang Y, Wang J, et al. Antithrombin is protective against myocardial ischemia and reperfusion injury. J Thromb Haemost. 2013;11(6):1020–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pan Y, Zhang X, Wang Y, et al. Targeting JNK by a new curcumin analog to inhibit NF-kB-mediated expression of cell adhesion molecules attenuates renal macrophage infiltration and injury in diabetic mice. Plos One. 2013;8(11):e79084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stokes SE, Winn LM. NF-kappaB signaling is increased in HD3 cells following exposure to 1,4-benzoquinone: role of reactive oxygen species and p38-MAPKa. Toxicol Sci. 2014;137(2):303–310. [DOI] [PubMed] [Google Scholar]
- 9. Sheta A, Elsakkar M, Hamza M, Solaiman A. Effect of metformin and sitagliptin on doxorubicin-induced cardiotoxicity in adult male albino rats. Hum Exp Toxicol. 2016;35(11):1227–1239. [DOI] [PubMed] [Google Scholar]
- 10. Aupanun S, Phuektes P, Poapolathep S, Sutjarit S, Giorgi M, Poapolathep A. Apoptosis and gene expression in Jurkat human T cells and lymphoid tissues of fusarenon-X-treated mice. Toxicon. 2016;123:15–24. [DOI] [PubMed] [Google Scholar]
- 11. Chen X, Li X, Zhang W, et al. Activation of AMPK inhibits inflammatory response during hypoxia and reoxygenation through modulating JNK-mediated NF-κB pathway. Metabolism. 2018;83:256–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zilinyi R, Czompa A, Czegledi A, et al. The cardioprotective effect of metformin in doxorubicin-induced cardiotoxicity: the role of autophagy. Molecules 2018;23(5):1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. An D, Kewalramani G, Chan JK, et al. Metformin influences cardiomyocyte cell death by pathways that are dependent and independent of caspase3. Diabetologia. 2006;49(9):2174–2184. [DOI] [PubMed] [Google Scholar]
- 14. Cheng Q, Cao X, Yuan F, Li G, Tong T. Knockdown of WWP1 inhibits growth and induces apoptosis in hepatoma carcinoma cells through the activation of Caspase3 and p53. Biochem Biophys Res Commun. 2014;448(3):248–254. [DOI] [PubMed] [Google Scholar]
- 15. Olivera Santa-Catalina M, Caballero Bermejo M, Argent R, Alonso JC, Centeno F, Lorenzo MJ. JNK signaling pathway regulates sorbitol-induced Tau proteolysis and apoptosis in SH-SY5Y cells by targeting Caspase3. Arch Biochem Biophys. 2017;636:42–49. [DOI] [PubMed] [Google Scholar]
- 16. Ma L, Liu H, Xie Z, et al. Ginsenoside Rb3 protects cardiomyocytes against ischemia-reperfusion injury via the inhibition of JNK mediated NF-kB pathway: a mouse cardiomyocyte model. Plos One. 2014;9(8):e103628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee C, Oh JI, Park J, et al. TNF alpha mediated IL-6 secretion is regulated by JAK/STAT pathway but not by MEK phosphorylation and AKT phosphorylation in U266 multiple myeloma cells. Biomed Res Int. 2013;2013:580135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang P, Han Y, Gui L, et al. Gastrodin attenuation of the inflammatory response in H9c2 cardiomyocytes involves inhibition of NF-kB and MAPKs activation via the phosphatidylinositol 3-kinase signaling. Biochem Pharmacol. 2013;85(8):1124–1133. [DOI] [PubMed] [Google Scholar]
- 19. Niu J, Wang K, Graham S, Azfer A, Kolattukudy PE. MCP-1-induced protein attenuates endotoxin-induced myocardial dysfunction by suppressing cardiac NF-kB activation via inhibition of IkB kinase activation. J Mol Cell Cardiol. 2011;51:177–186. [DOI] [PubMed] [Google Scholar]
- 20. Peng T, Zhang T, Lu X, Feng Q. JNK1/c-fos inhibits cardiomyocyte TNF-alpha expression via a negative crosstalk with ERK and p38 MAPK in endotoxaemia. Cardiovasc Res. 2009;81(4):733–741. [DOI] [PubMed] [Google Scholar]
- 21. An J, Du J, Wei N, Guan T, Camara AK, Shi Y. Differential sensitivity to LPS induced myocardial dysfunction in the isolated brown Norway and Dahl S rat hearts: roles of mitochondrial function, NF-kB activation, and TNF-a production. Shock. 2012;37(3):325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]