

SUMMARY
Maize (Zea mays ssp. mays) was domesticated in southwestern Mexico ~9,000 years ago from its wild ancestor, teosinte (Zea mays ssp. parviglumis) [1]. From its centre of origin, maize experienced a rapid range expansion and spread over 90°of latitude in the Americas [2–4] which required a novel flowering time adaptation. ZEA CENTRORADIALIS 8 (ZCN8) is the maize florigen gene and has a central role in mediating flowering [5, 6]. Here, we show that ZCN8 underlies a major quantitative trait locus (qDTA8) for flowering time that was consistently detected in multiple maize-teosinte experimental populations. Through association analysis in a large diverse panel of maize inbred lines, we identified a single nucleotide polymorphism (SNP-1245) in the ZCN8 promoter that showed the strongest association with flowering time. SNP-1245 co-segregated with qDTA8 in maize-teosinte mapping populations. We demonstrate that SNP-1245 is associated with differential binding by the flowering activator ZmMADS1. SNP-1245 was a target of selection during early domestication which drove the pre-existing early-flowering allele to near fixation in maize. Interestingly, we detected an independent association block upstream of SNP-1245, wherein the early-flowering allele that most likely originated from Zea mays ssp. mexicana introgressed into the early-flowering haplotype of SNP-1245 and contributed to maize adaptation to northern high latitudes. Our study demonstrates how independent cis-regulatory variants at a gene can be selected at different evolutionary times for local adaptation, highlighting how complex cis-regulatory control mechanisms evolve. Finally, we propose a polygenic map for the pre-Columbian spread of maize throughout the Americas.
RESULTS AND DISCUSSION
ZCN8 underlies a major flowering time QTL detected in multiple maize-teosinte mapping populations
To identify genetic factors contributing to maize flowering time adaptation, we previously conducted QTL mapping for days to anthesis (DTA) in a large population of 866 maize-teosinte BC2S3 recombinant inbred lines (RILs) derived from a cross between the temperate maize inbred line W22 and CIMMYT accession 8759 (a typical accession of Zea mays ssp. parviglumis, hereafter referred to as 8759)[4, 7]. Among the mapped loci, a large-effect flowering time QTL, qDTA8, was detected at bin 8.05 on chromosome 8[4, 7] (Figure 1A). Interestingly, qDTA8 was consistently detected in five additional newly developed maize-teosinte BC1S4 RIL populations derived from crosses between five teosinte inbred lines (TIL01, TIL03, TIL11, TIL14 and TIL25) and the common maize parent W22 (Figure 1A). TIL01, TIL03, TIL11 and TIL14 belong to Zea mays ssp. parviglumis, while TIL25 belongs to Zea mays ssp. mexicana. The teosinte alleles at qDTA8 in these six maize-teosinte experimental populations consistently exhibited the effect of delaying flowering under long days relative to the maize allele (Figure 1B). It is interesting to note that Liu et al. (2016)[8] performed QTL mapping for flowering time in a large set of near-isogenic introgression lines from 10 teosinte accessions in the B73 background and also detected a large-effect QTL around qDTA8, with teosinte alleles exhibiting the effect of delaying flowering relative to the B73 allele. In addition to maize-teosinte populations, significant flowering time QTLs around qDTA8 were also detected in various maize germplasm[3, 9–12]. These consistent detections of qDTA8 in distinct maize-teosinte and maize populations indicated that qDTA8 might be an important locus regulating maize flowering time variation and adaptation.
Figure 1. ZCN8 underlies flowering time QTL qDTA8.
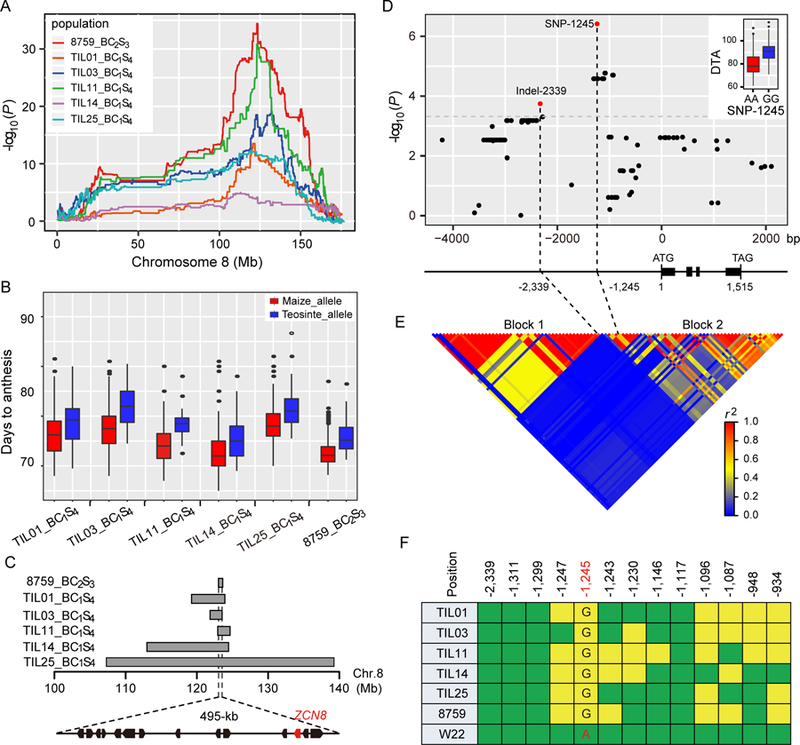
(A) qDTA8 was consistently mapped in six maize-teosinte experimental populations. The x-axis shows the physical position along chromosome 8, and the y-axis represents the logarithm of odds (LOD) score of each scanning position. Different coloured lines indicate different maize-teosinte populations. (B) Allele effects of qDTA8 in the six maize-teosinte populations. (C) ZCN8 is located in the overlapping region of qDTA8 in the six maize-teosinte populations. The arrows represent the 14 annotated genes in the reference genome in the overlapping region, and ZCN8 is indicated in red. (D) Association analysis of sequence variants in the 6.3-kb region of ZCN8 for DTA in a panel of 513 diverse maize inbred lines. SNP-1245 and Indel-2339, which are independently associated with DTA, are indicated by red dots. The gene structure of ZCN8 is shown below the association analysis plot. The positions of SNP-1245 and Indel-2339 relative to the start codon are indicated. The inset shows the difference in flowering time (days to anthesis) between the SNP-1245 alleles. (See also Table S1; Table S4 and Data S1). (E) Linkage disequilibrium (LD) analysis of the 6.3-kb region of ZCN8 in the maize association panel. The two LD blocks (Block 1 and Block 2) are indicated. (F) SNP-1245 co-segregates with qDTA8. The coloured squares indicate the allelic states of the six teosinte parents and the maize parent (W22) at the 13 significantly associated variants. The green squares indicate that the teosinte parents carry the same allele as W22, while the yellow squares indicate that the teosinte parents carry a different allele from W22. SNP-1245 is highlighted in red. (See also Table S2).
To identify possible candidate genes underlying qDTA8, we analyzed the 2-LOD support intervals of qDTA8 in the six maize-teosinte mapping populations. Interestingly, the support intervals of qDTA8 in the six populations overlapped in a 495-kb physical region that contains 14 annotated genes in the maize reference genome (AGPv3) (Figure 1C). Among these annotated genes, GRMZM2G179264 corresponds to ZCN8, which is homologous to Arabidopsis FLOWERING LOCUS T (FT) and has been shown to be the most likely maize florigen gene[5, 6]. ZCN8 is transcribed and translated in leaves and then moves via the phloem to the SAM where it interacts with the DELAYED FLOWERING1 (DLF1) protein[13] to activate downstream floral organ identity genes[5, 6, 14]. Due to its crucial role in mediating maize flowering, ZCN8 has been frequently assumed as the most likely candidate gene of flowering time QTLs mapped around the region in previous studies[3, 8–12] . Interestingly, several genome-wide association studies for flowering time also detected a few significant SNP associations near the ZCN8 region (28–30), which were considered as the causal factor of the Vgt2 QTL that is 8-Mb apart from Vgt1[15, 16], a major flowering time QTL that has been cloned[17]. However, how ZCN8 affects natural variation in flowering time and contributes to maize flowering time adaptation remains largely unknown.
SNP-1245 at the ZCN8 promoter shows the strongest association with flowering time
To test whether ZCN8 affects the natural variation in maize flowering time, we conducted an association analysis by sequencing ZCN8 and its upstream and downstream regions in a panel of 513 diverse maize inbred lines that were scored for DTA under natural long days in Beijing (39.9°N, 116.4°E), China[18] (Data S1). A total of 101 variants (74 SNPs and 27 Indels) with minor allele frequency (MAF) ≥ 0.05 were identified across the 6.3-kb sequenced region around ZCN8 (Figure 1D). Using a mixed linear model that corrects for population structure and pairwise relatedness among individuals[19], we tested the sequence variants for association with DTA in the maize association panel. A total of 13 sequence variants showed a significant association with DTA after Bonferroni multiple test correction (P< 4.95E-04) (Figure 1D; Table S1). Among these associated variants, a SNP (SNP-1245, G>A variant located 1.2-kb upstream of ZCN8) in the promoter region showed the strongest association with DTA (P =3.88E-07) (Figure 1D; Table S1). At SNP-1245, the SNP-1245G allele exhibited the effect of delaying flowering relative to the SNP-1245A allele (Figure 1D). We further examined whether SNP-1245 was associated with DTA under natural short days in Sanya, China (18.1°N, 109°E) and photoperiod response that is defined as the difference in growing degree days (GDD) to anthesis between long days and short days [4, 11]. SNP-1245 exhibited significant associations with DTA under short days (P=5.0E-03) and photoperiod response (P=1.0E-05).
To determine whether the associated variants could explain the common flowering time QTL qDTA8 detected in the six maize-teosinte mapping populations, we sequenced the seven parents of the maize-teosinte mapping populations to examine their allelic states at the 13 significant variants. Interestingly, among the 13 significant variants, only SNP-1245 exhibited a completely consistent segregating pattern with qDTA8 across the six maize-teosinte mapping populations (Figure 1F; Table S2). The six teosinte parents consistently carried the late-flowering SNP-1245G allele, while the maize parent W22 carried the early-flowering SNP-1245A allele (Figure 1F; Table S2). This highly congruence with QTL segregating patterns strongly suggests that SNP-1245 might be the functional variant of ZCN8 that underlies the common flowering time QTL qDTA8 detected in the six maize-teosinte experimental populations.
SNP-1245 affects ZCN8 expression
SNP-1245 is located in the promoter region of ZCN8, therefore it is presumed to cause difference in ZCN8 expression. From a heterogeneous inbred family (HIF) that is heterozygous only at qDTA8, we developed two near-isogenic lines (NILs), one homozygous for W22 and one homozygous for 8759 across the qDTA8 region, designated NIL(W22) and NIL(8759), respectively. W22 and 8759 are the maize and teosinte parents of the maize-teosinte BC2S3 population, respectively. NIL(W22) and NIL(8759) were planted under natural long days to examine their differences in ZCN8 expression and flowering time. NIL(W22) flowered significantly earlier than NIL(8759) under long days (Figure 2A). ZCN8 is mainly expressed in mature leaves near the floral transition[5, 6]. Consistent with the role of ZCN8 as a florigen, ZCN8 showed higher expression in NIL(W22) than in NIL(8759) (Figure 2B; Figure S1). To examine the diurnal expression pattern of ZCN8 in NILs, we planted NIL(W22) and NIL(8759) under artificial long days and short days. As shown in Figure S1, ZCN8 was diurnally regulated and was expressed at higher levels in NIL(W22) than that in NIL(8759) at most time points under both long days and short days.
Figure 2. SNP-1245 affects ZCN8 expression and is associated with differential regulation by ZmMADS1.
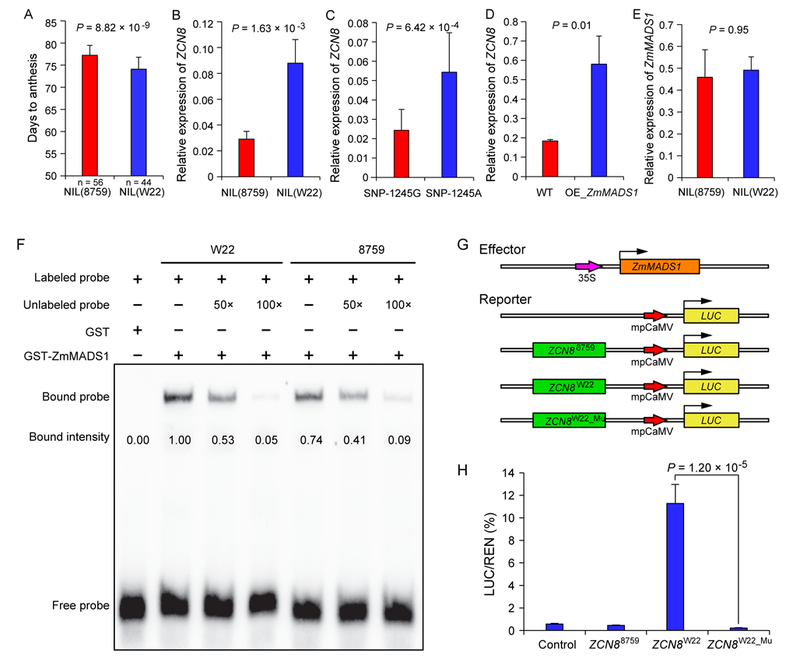
(A) The difference in days to anthesis between NIL(W22) and NIL(8759). Data represent the mean ± s.d.; P values were determined by Student’s t test. (B) Relative expression levels of ZCN8 in NIL(W22) and NIL(8759). Data represent the mean ± s.d. (n=3 biological replicates); P values were determined by Student’s t test. (See also Figure S1). (C) Relative expression levels of ZCN8 in maize inbred lines carrying different alleles of SNP-1245. For each allelic group, 10 maize inbred lines were randomly selected from the maize association panel. Data represent the mean ± s.d.; P values were determined by Student’s t test. (D) Relative expression levels of ZCN8 in ZmMADS1-overexpressing plants and wild type plants. Data represent the mean ± s.d. (n=3 biological replicates); P values were determined by Student’s t test. (E) ZmMADS1 exhibited similar expression levels in the two NILs for qDTA8. Values are presented as the means ± s.d. (n=3 biological replicates); P values were determined by Student’s t test. (F) Electrophoretic mobility shift assay (EMSA) showing that SNP-1245 is associated with differential binding by ZmMADS1. Each biotin-labelled DNA fragment was incubated with GST-ZmMADS1 or GST proteins. Competition assays for the labelled probes were performed by adding an excess of unlabelled probes. The relative bound intensity was quantified with ImageJ software (http://imagej.nih.gov/ij/). (See also Table S4). (G,H) Dual-luciferase transient expression assay in maize protoplasts. The coding sequence of ZmMADS1 driven by the 35S promoter was used as the effector. The luciferase (LUC) gene driven by a ~2-kb promoter sequence from NIL(W22) or NIL(8759) was used as the reporter. To examine the effect of SNP-1245, a mutant reporter containing the G nucleotide of SNP-1245 in the NIL(W22) promoter was constructed. Values are presented as the means ± s.d. (n=5 biological replicates); P values were determined by Student’s t test.
To further verify whether SNP-1245 is associated with differential ZCN8 expression, we randomly selected 10 maize inbred lines carrying the SNP-1245A allele and 10 maize inbred lines carrying the SNP-1245G allele from the association population to investigate ZCN8 expression. As shown in Figure 2C, the maize lines carrying the early-flowering SNP-1245A allele showed significantly higher expression of ZCN8 than the maize lines carrying the late-flowering SNP-1245G allele. Taken together, these data indicated that SNP-1245 is associated with differential expression of ZCN8, consequently resulting in a difference in flowering time.
SNP-1245 is associated with differential binding of the flowering activator ZmMADS1
Given the regulatory importance of SNP-1245 in ZCN8 expression, SNP-1245 might be associated with alteration of conserved regulatory sequences. To test this hypothesis, we analyzed the sequences surrounding SNP-1245 using PlantPAN 2.0[20]. Interestingly, SNP-1245 flanks a CArG-box-like sequence that is a putative binding site for MADS-box transcription factors[21]. We speculate that SNP-1245 might affect the binding affinity of an upstream MADS-box transcription factor.
Recently, Alter et al. (2016)[22] identified ZmMADS1, a MADS-box transcription factor that is an orthologue of the flowering regulators SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 (SOC1) in Arabidopsis[23] and OsMADS50 in rice[24]. ZmMADS1 functions as a flowering activator in maize, in which downregulation of ZmMADS1 resulted in a delay of flowering time, while overexpression caused an early-flowering phenotype[22]. We examined the expression of ZCN8 in plants overexpressing ZmMADS1 and found that the expression level of ZCN8 was significantly increased compared with that in the wild type (Figure 2D). However, in the two NILs for qDTA8, ZmMADS1 exhibited similar expression levels (Figure 2E). These results consistently suggested that ZmMADS1 functions upstream of ZCN8.
To determine whether ZmMADS1 could directly bind to the sequence surrounding SNP-1245, we performed an electrophoretic mobility shift assay (EMSA) (Figure 2F ). The results showed that the oligonucleotide probe containing the A allele (W22) robustly bound the ZmMADS1 protein, whereas the oligonucleotide probe containing the G allele (8759) exhibited decreased DNA binding affinity (Figure 2F). Increasing the amount of unlabelled probes efficiently competed with the binding of the labelled probes. These results suggested that ZmMADS1 could directly bind to the sequence surrounding SNP-1245.
To further test the effect of SNP-1245 on ZCN8 expression, we performed a dual-luciferase transient expression assay in maize protoplasts (Figure 2G). Overexpression of ZmMADS1 significantly enhanced the activity of the luciferase (LUC) gene driven by the promoter from NIL(W22) containing the A allele at SNP-1245 (Figure 2H). However, when the A nucleotide of SNP-1245 in the NIL(W22) promoter was mutated to G, luciferase activity significantly decreased to a level similar to that associated with the NIL(8759) promoter (Figure 2H). Therefore, SNP-1245 has a significant effect on the activation of ZCN8 expression by ZmMADS1.
SNP-1245 was targeted by selection and contributed to maize latitudinal adaptation
To examine the evolutionary origin of SNP-1245, we sequenced the promoter region flanking SNP-1245 in 34 diverse teosinte lines (Zea mays ssp. parviglumis, hereafter parviglumis) (Table S2). The results showed that eight parviglumis accessions (23.5%) carried the SNP-1245A allele (Figure 3A), indicating that SNP-1245 is a standing genetic variant in parviglumis. However, the frequency of the SNP-1245A allele in tropical maize and temperate maize rose to 82% and 96%, respectively (Figure 3A), indicating that the early-flowering SNP-1245A allele is nearly fixed in the maize population. This dramatic increase of the SNP-1245A allele in the maize population indicated that the early-flowering SNP-1245A allele might have been under strong selection during domestication.
Figure 3. SNP-1245 and Indel-2339 were targeted by selection and contributed to maize latitudinal adaptation.
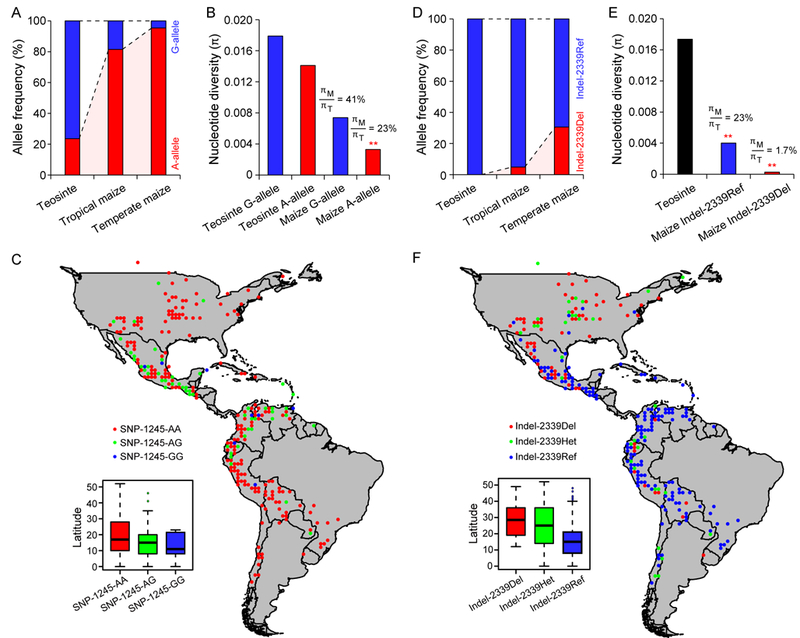
(A) The allele frequency of SNP-1245 in teosinte, tropical maize and temperate maize inbred lines (see also Table S2; Data S1). (B) Nucleotide diversity analysis of the region surrounding SNP-1245 in maize and teosinte. πM/πT indicates the amount of nucleotide diversity (π) retained in maize relative to that in teosinte.** indicates a significant departure from the neutral domestication bottleneck model, P < 0.01. (C) Geographic distribution of SNP-1245 alleles in 1,008 maize landraces native to the Americas. The inset shows the difference in latitude between the different allelic groups of SNP-1245. (See also Table S4; Data S2) (D) The allele frequency of Indel-2339 in teosinte, tropical maize and temperate maize inbred lines (see also Table S2; Data S1). (E) Nucleotide diversity analysis of the region surrounding Indel-2339 in maize and teosinte. πM/πT indicates the amount of nucleotide diversity (π) retained in maize relative to that in teosinte. ** indicates a significant departure from the neutral domestication bottleneck model, P < 0.01 (see also Figure S3). (F) Geographic distribution of Indel-2339 alleles in 1,008 maize landraces native to the Americas (see also Table S4; Data S2). The inset shows the difference in latitude between the different allelic groups of Indel-2339.
To examine whether selection has acted on SNP-1245, we analyzed the nucleotide diversity of the 2.1-kb region flanking SNP-1245 in diverse maize and parviglumis lines (Tables S2; Data S1). Maize lines carrying the late-flowering SNP-1245G allele retained 41% of the nucleotide diversity of their teosinte counterparts, whereas maize lines carrying the early flowering SNP-1245A allele retained only 23% of the nucleotide diversity of their teosinte counterparts (Figure 3B). We performed coalescent simulations that incorporated the demography of maize domestication[25] to examine whether the retained nucleotide diversity near the SNP-1245G and SNP-1245A alleles can be explained by the demography of maize domestication alone. Interestingly, we detected significant deviation from the neutral expectation for the SNP-1245A allele but not the SNP-1245G allele (Figure 3B). These results suggested that the SNP-1245A allele was targeted by selection, thereby causing its rapid accumulation in maize.
Maize originated in southwestern Mexico and was spread across over 90 degrees of latitude during pre-Columbian times[1]. To determine whether SNP-1245 contributed to maize latitudinal adaptation, we genotyped SNP-1245 in 1,008 maize landrace accessions (Data S2) representing the entire pre-Columbian range of maize landraces native to the Americas [1, 26, 27]. The results showed that only 85 landraces carried the late-flowering SNP-1245G allele, most of which are restricted to low latitudes (Figure 3C). In contrast, the early-flowering SNP-1245A allele accumulated to a predominant frequency throughout the Americas (Figure 3C). These results suggested that selection at SNP-1245 might have occurred during the early domestication of maize.
Given that flowering time is not considered a maize domestication trait, it is not clear why SNP-1245A was selected during early domestication. Several hypotheses can be considered. (1) Selection for earlier flowering could have restricted gene flow between incipient maize and teosinte (temporal genetic isolation) and thereby facilitated the domestication process. (2) Crop populations exhibit more uniform flowering time and maturity as compared to wild species population that flower over a longer time interval[28]. Bringing incipient maize closer to fixation for SNP-1245A may have promoted a more synchronous flowering time and maturity. (3) Genes that affect flowering time frequently exhibited effects on inflorescence structure[7, 29–31]. Danilevskaya et al (2011)[32]has shown that knocking down ZCN8 significantly affected inflorescence structure. Interestingly, we found that significant QTLs for ear diameter, tassel length, stem diameter and ear branch number were also detected at region around ZCN8 in the maize-teosinte BC2S3 population (Figure S2A), indicating that ZCN8 region is a QTL hotspot affecting many domestication and agronomic traits. Consistent with expectation, the maize allele exhibited effects of increasing ear size but decreasing ear branch number relative to the teosinte allele (Figure S2B). Therefore, SNP-1245A may have been selected for an effect on ear size and structure and the change in flowering time was a correlated response.
An independent association block upstream of SNP-1245
Across the 6.3-kb sequenced region around ZCN8, there exist two clear blocks of linkage disequilibrium (LD) (Block 1 and Block 2) (Figure 1E). SNP-1245 is located in Block 1, which contained the majority of the detected associations for DTA under long days. In Block 2, only one variant (Indel-2339, GAG>- variant, located 2.3-kb upstream of ZCN8) passed the Bonferroni corrected significance threshold (P=1.82E-04) (Figure 1D). Indel-2339 was not in LD with SNP-1245 (r2=0.019). Different from SNP-1245 that also exhibited significant associations with DTA under short days and photoperiod response, no significant associations were detected at Indel-2339 for the two traits. We should note that the teosinte and the maize parents of the six maize-teosinte mapping populations exhibited no allelic difference at Indel-2339 (Figure 1F). Therefore, Indel-2339 is not related to qDTA8 that was consistently detected in the six maize-teosinte populations. Indel-2339 represented another independent cis-regulatory variant of ZCN8.
Several previous genome-wide association studies for flowering time also detected a few significant SNP associations near the ZCN8 region[15, 16, 33]. For instance, Bouchet et al. (2013)[15] performed 50K SNP-based genome-wide association mapping for flowering time in an association panel including 336 tropical and temperate American and European lines. The study detected a strong association at a SNP (PZE-108070380) located 3.3-kb upstream of ZCN8. The association at PZE-108070380 was further detected in a recent study using a large European collection of 1191 maize flint lines[16]. PZE-108070380 corresponds to SNP-3366 in Block 2 in our study, which showed a marginally significant association with DTA (P= 2.9E-03) in our association panel. PZE-108070380 was in strong LD with Indel-2339 (r2=0.85). Given the stronger association of Indel-2339, Indel-2339 was used as a tag variant for Block 2 in the subsequent analysis. At Indel-2339, maize lines carrying the 3-bp deletion (Indel-2339Del) flowered significantly earlier than those carrying the B73 reference allele (Indel-2339Ref) (Figure S3A), which is consistent with the findings for PZE-108070380 from previous studies[15, 16].
To test the effect of Indel-2339 on gene expression, we cloned the promoter fragments from the W22 and Mo17 line into the reporter constructs and compared their relative activity in maize leaf protoplasts (Figure S3B). W22 carried the Indel-2339Ref allele, while Mo17 carried the Indel-2339Del allele. W22 and Mo17 carried the same SNP-1245A allele. The assay showed that the Mo17 construct exhibited higher luciferase activity than the W22 construct (Figure S3C). When nucleotides GAG at the Indel-2339 site in the W22 construct was site-directed deleted, the luciferase activity increased to a level of that of the Mo17 construct (Figure S3C). These results suggested that Indel-2339 has significant impact on gene expression.
In modern maize inbred lines, the early-flowering Indel-2339Del allele is at a low frequency (5%) in tropical maize but higher frequency (30%) in temperate maize (Figure 3D), potentially indicating that Indel-2339 might play an important role in maize adaptation to temperate regions. We analyzed the allele distribution of Indel-2339 in maize landraces. The results showed that the early-flowering Indel-2339Del allele occurs at a high frequency in northern United States (Figure 3F). In contrast, the Indel-2339Del allele showed a low frequency across South America (Figure 3F), indicating a limited contribution of this allele to the spread of maize in South America. We analyzed the nucleotide diversity surrounding Indel-2339 and found that maize lines retained only 23% of the nucleotide diversity of teosinte in this region (Figure 3E), showing a typical signature of selection. This selection signal was reflected by the high LD in Block 2 (Figure 1E). Interestingly, previous studies have also found that the ZCN8 region is located in a region of selection characteristic of flint germplasm[16, 34]. Taken together, we conclude that selection in Block 2 might have arisen more recently and contributed to maize adaptation to northern high latitudes.
Selection at SNP-1245 and Indel-2339 arose in a stepwise manner
Using the third generation Zea mays haplotype map (HapMap 3) containing ultra-high-density SNPs across maize genome, we examined the nucleotide diversity around ZCN8 in maize and teosinte. Interestingly, the promoter region of ZCN8 exhibited an obvious reduction of nucleotide diversity in maize relative to teosinte (Figure S2C), potentially indicating that this region might be targeted by selection. Our in-depth analysis of SNP-1245 and Indel-2339 alleles revealed that two closely linked selection events have occurred in the promoter region of ZCN8. To determine their relationship, we analyzed their haplotypes in the maize association panel. Based on the genotypes of SNP-1245 and Indel-2339, all maize inbred lines in the association panel could be divided into three haplotype groups, G-Ref, A-Ref and A-Del, where the first and second genotype indicate the allelic state at SNP-1245 and Indel-2339, respectively (Figure 4A). Interestingly, the Indel-2339Del allele is carried only by maize lines with the SNP-1245A allele. No maize lines carrying the G-Del haplotype were observed in the population. Therefore, the Indel-2339Del allele most likely arose from the SNP-1245A background. Haplotype A-Del flowered earliest, followed by haplotypes A-Ref and G-Ref (Figure 4A). Due to the initial selection at the SNP-1245A allele and subsequent selection at the linked Indel-2339Del allele, haplotype A-Del exhibited the lowest nucleotide diversity compared with haplotypes A-Ref and G-Ref (Figure 4A).
Figure 4. Evolutionary relationship of SNP-1245 and Indel-2339 and a polygenic map for the pre-Columbian spread of maize throughout the Americas.
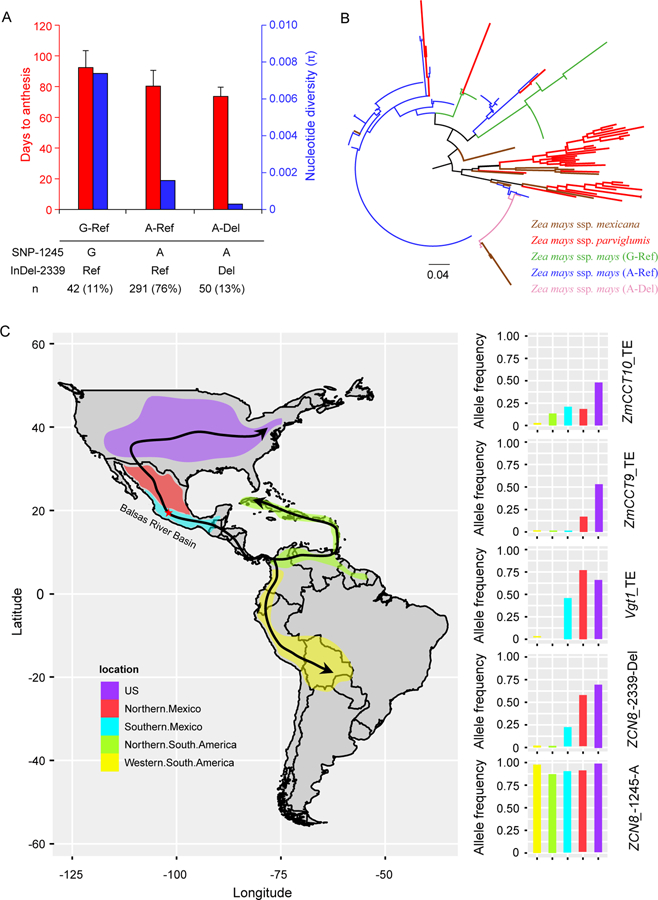
(A) Haplotype analysis of SNP-1245 and Indel-2339 in the maize association panel. According to the genotypes of SNP-1245 and Indel-2339, the maize inbred lines could be divided into three haplotype groups (x-axis) (G-Ref, A-Ref and A-Del). The left y-axis shows the phenotypic value (days to anthesis) of each haplotype (red bars); The right y-axis shows the nucleotide diversity (π) of each haplotype (blue bars). (See also Figure S2; Table S2). (B) Phylogenetic analysis of the promoter region around ZCN8 in maize and teosinte (see also Data S1). Lines belonging to G-Ref, A-Ref and A-Del are indicated in green, blue and pink, respectively. Zea mays parviglumis and Zea mays mexicana are indicated in red and saddlebrown, respectively. (See also Data S1). (C) A polygenic map for the pre-Columbian spread of maize throughout the Americas. Matsuoka et al. (2002)[1] and Vigouroux et al. (2008)[26] developed a scenario for the pre-Columbian spread of maize through the Americas. The red star indicates the central region of the Balsas River Basin, where maize domestication is thought to have occurred[1]. Arrows indicate probable routes of pre-Columbian dispersion. According to the analyses by Matsuoka et al. (2002)[1] and Vigouroux et al. (2008)[26], the 1,008 maize landraces were divided into five major geographic groups, corresponding to the US, northern Mexico, southern Mexico, northern South America and western South America which are indicated by different colours in the map. Races exhibiting post-Columbian movement[1, 26] were excluded in this analysis. The right panel shows the allele frequency of the early-flowering alleles of four genes (Vgt1, ZmCCT9, ZmCCT10 and ZCN8) in the five geographic groups across the Americas. (See also Figure S2, Table S3–S4 and Data S2).
To further reveal the evolutionary relationship of SNP-1245 and Indel-2339, we sequenced the promoter region of ZCN8 in two teosinte subspecies, including 34 accessions of Zea mays parviglumis and 11 accessions of Zea mays mexicana (hereafter mexicana) (Table S2). Zea mays parviglumis is the direct ancestor of modern maize[1], while introgression from Zea mays mexicana was important in the adaptation of maize to Mexican highlands[35, 36]. Phylogenetic analysis showed that most teosinte lines were basal to the maize lines, and maize lines separated into G-Ref, A-Ref and A-Del haplotype groups that mixed with some teosinte lines (Figure 4B). Of particularly interesting, we found that haplotype A-Del resides with mexicana lines carrying the Indel-2339Del allele, indicating possible admixture from mexicana (Figure 4B). Consistent with this observation, Indel-2339 existed as standing variation in mexicana (Table S2). However, all sequenced parviglumis lines carried the same Indel-2339Ref allele (Table S2). Different from Indel-2339, SNP-1245 existed as standing variation in both parviglumis and mexicana (Table S2). These results indicate that the early flowering allele at Indel-2339 might originate from the introgression from mexicana.
Taken together, our data showed that the selection at SNP-1245 and Indel-2339 arose in a stepwise manner. The selection at SNP-1245 most likely arose during early domestication of maize, which fixed the previously existed low-frequency SNP-1245A allele. When maize was spread into northern zones, requiring a shorter growth period, the early-flowering allele at Indel-2339 that most likely originated from mexicana introgressed into the SNP-1245A haplotype and was targeted by selection. This stepwise cis-regulatory evolution has been reported in the evolution of animal morphology[37–39]. Rebeiz et al.(2009)[38] showed that the adaptive evolution of melanism in a Ugandan population of D. melanogaster occurred through multiple, stepwise substitutions in one enhancer of the ebony locus. Recently, Glaser-Schmitt and Parsch (2018) demonstrated that three SNPs within the CG9509 enhancer were targeted by selection, and they occurred in a stepwise manner to increase CG9509 expression and, as a result, reduce wing loading as D. melanogaster expanded out of sub-Saharan Africa. Our study highlights how different cis-regulatory variants at a gene can be selected at different evolutionary times for local adaptation in plants.
Additionally, our findings further enhance the notion that FT homologs played important roles in crop domestication or local adaptation[40–42]. For example, Blackman et al. (2010)[40] demonstrated that changes affecting the expression, sequence, and gene interactions of Helianthus annuus FT paralogs have played key roles during the early domestication of sunflower.
A polygenic map for the pre-Columbian spread of maize throughout the Americas
Through characterizing a large sample of landrace accessions covering almost the entire set of ~350 landraces native to the Americas, Matsuoka et al (2002)[1] and Vigouroux et al. (2008)[26] developed a scenario for the pre-Columbian spread of maize through the Americas. Until now, only four QTLs controlling maize flowering time, including ZmCCT9, ZmCCT10, Vgt1 and ZCN8, which we report in this study, had been well-characterized to play important roles in maize flowering time adaptation. A common feature of these cloned loci is that functional variation at all of them is due to cis-regulatory polymorphisms. Vgt1 is a major maize flowering time QTL, and a miniature transposon (MITE) located ~70-kb upstream of ZmRap2.7 was shown to be the causative variant of Vgt1 contributing to maize adaptation to temperate regions[17, 43, 44]. Yang et al (2013)[18] found that a CACTA-like transposon insertion within the ZmCCT10 promoter represses ZmCCT10 expression, rendering maize insensitive to long days. We recently reported a Harbinger-like transposon insertion located ~57-kb upstream of ZmCCT9 that functions as a cis-acting repressor of ZmCCT9 to enhance maize adaptation to higher latitudes[45]. In this study, we found that two natural cis-variants in the promoter of ZCN8 were targeted by selection in a stepwise manner, thereby contributing to maize flowering time adaptation.
To obtain an overall understanding of the relative contributions of the four genes during the pre-Columbian spread of maize throughout the Americas, we analyzed the allele distribution in the landraces used in the studies of Matsuoka et al. (2002)[1] and Vigouroux et al. (2008)[26] (Data S2). As shown in Figure 4C, among the five early-flowering alleles of the four genes, the early-flowering SNP-1245A allele of ZCN8 is at a predominant frequency throughout the Americas. Hence, the SNP-1245A allele of ZCN8 might be the first to be selected. In contrast, the other four early-flowering alleles rose to a high frequency when maize spread into northern United States, indicating that these alleles made specific contributions to northwards expansion in North America. Interestingly, all four of these early-flowering alleles are at a relatively low frequency in South America. These results suggest that the latitudinal adaptation of maize in South America might have involved a different set of alleles or genes.
Recent advances in ancient genomics allowed us to directly track genetic changes through time. Vallebueno-Estrada et al. (2016)[46] and Ramos-Madrigal et al. (2016)[47] sequenced four ~5000-year-old maize specimens excavated in the Tehuacan Valley of Mexico, while Swarts et al. (2017)[48] sequenced fifteen 1900-year-old maize cobs from Turkey Pen Shelter (TPS) in the southwestern United States. We examined the allelic states of the five flowering time loci in these archaeological maize specimens (Table S3). Due to the limited sequencing depth, only two Tehuacan maize specimens (SM10 and Tehuacan162) have read coverage in the five loci for the analysis (Table S3). SM10 and Tehuacan162 carried the later flowering alleles SNP-1245G and Indel-2339Ref at ZCN8 but the early flowering alleles of transposon insertions at ZmCCT9 and ZmCCT10. This result is somewhat contradictory to the patterns observed in the analysis of modern landraces, wherein the early flowering allele SNP-1245A of ZCN8 appeared to accumulate earlier than the transposon insertions at ZmCCT9 and ZmCCT10. However, this observation should be interpreted with caution. Because these loci segregated in maize and only two Tehuacan maize genomes had data for analysis, it is hard to determine the allele distribution of these loci in the Tehuacan Valley 5,000 years ago and thus make an accurate inference on their evolutionary states. More Tehuacan maize should to be examined in the future analysis. By contrast, the picture for Turkey Pen maize is much clearer due to the large sequenced samples. Notably, all Turkey Pen maize specimens carried the early flowering alleles of the five loci except Vgt1, indicating that 1,900 years ago, Turkey Pen maize already maintained the alleles conferring early flowering. Interestingly, none of the examined archaeological maize specimens carried the MITE insertion at Vgt1 that conferred early flowering, indicating the selection for MITE most likely occurred after 1900 years ago. Taken together, our data suggest that the pre-Columbian spread of maize throughout the Americas is a complex and gradual process, wherein numerous genetic loci involved and were selected at different evolutionary times for local adaptation.
In summary, we demonstrate that ZCN8 underlies a major flowering time QTL that was consistently detected in multiple maize-teosinte experimental populations. ZCN8 plays an important role in regulating the natural variation in flowering time in maize, which is mostly mediated by two independently associated variants in the promoter region of ZCN8. One of these two variants is a SNP that was found to be associated with differential binding by the flowering activator ZmMADS1. Both cis-regulatory variants were targeted by selection, consequently resulting in two closely linked selective sweeps in the ZCN8 promoter. These two selection events arose in a stepwise manner and played distinct roles as maize spread from its tropical origin to temperate zones. Our study highlights the importance of stepwise cis-regulatory evolution in local adaptation. By integrating current knowledge on maize flowering time adaptation, we propose a polygenic map for the pre-Columbian spread of maize throughout the Americas.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Feng Tian (ft55@cau.edu.cn).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
A large population of 866 maize-teosinte BC2S3 RILs derived from a cross between W22, a typical temperate maize (Zea mays ssp. mays) inbred line and CIMMYT accession 8759, a typical accession of teosinte (Zea mays ssp. parviglumis) was obtained from the Maize Genetics Cooperation Stock Center (Maize COOP). Detailed information on this population has been provided previously[4, 7, 45, 49–52]. From a heterogeneous inbred family (HIF) that is heterozygous only at qDTA8, a pair of NILs homozygous for the maize (W22) and the teosinte (8759) parents of the maize-teosinte BC2S3 population across the qDTA8 region was developed and planted under natural long days to examine the differences in flowering time and ZCN8 expression.
Five new maize-teosinte BC1S4 RIL populations derived from crosses between five teosinte (Zea mays ssp. parviglumis) inbred lines (TIL01, TIL03, TIL11, TIL14 and TIL25) and the temperate maize inbred W22 were recently developed. TIL01, TIL03, TIL11 and TIL14 belong to Zea mays ssp. parviglumis, whereas TIL25 belongs to Zea mays ssp. mexicana. Each maize-teosinte BC1S4 population contains ~200 RILs and has been genotyped for ~13,445 SNP markers using genotyping-by-sequencing technology. The SNP genotypes of these five maize-teosinte BC1S4 populations are available at the Cyverse Discovery Environment (de.cyverse.org/de/) under the directory: /iplant/home/shared/panzea/genotypes/GBS/TeosinteNAM/.
A maize association panel composed of 513 diverse maize inbred lines[18] (Data S1) was used in the association analysis of the 6.3-kb sequenced region around ZCN8. To analyze the nucleotide diversity at the promoter region of ZCN8, the maize association panel (Data S1) and 45 teosinte lines, including 34 accessions of Zea mays parviglumis and 11 accessions of Zea mays mexicana (Table S2), were used.
To investigate the geographic distribution of the two cis-regulatory variants in the ZCN8 promoter, 1,008 accessions of maize landraces representing the entire pre-Columbian range of maize races native to the Americas was used (Data S2). Detailed information on these maize landraces can be found in previous studies[26, 27, 45].
METHOD DETAILS
Sequencing the ZCN8 region
According to the B73 reference sequence (B73 RefGen_v3), six pairs of primers (Table S4) were used to sequence a 6.3-kb region around ZCN8, including a 1.5-kb coding sequence, a 4.2-kb upstream sequence and a 1-kb downstream sequence, in a diverse maize panel containing 513 inbred lines[18] (Data S1). The 4.2-kb upstream sequenced fragment included the whole intergenic region from the upstream neighboring gene (GRMZM2G179274) (Figure 1C). To understand the evolutionary relationship of SNP-1245 and Indel-2339, the promoter region of ZCN8 was sequenced in two teosinte subspecies, including 34 accessions of Zea mays parviglumis and 11 accessions of Zea mays mexicana (Table S2). Sequencing reactions were performed in both directions. Multiple sequence alignments were performed using BIOEDIT (v.7.0.9.0; North Carolina State University, Raleigh, NC, USA) and were manually edited if necessary. Polymorphic sites (SNPs and Indels) were extracted, and the levels of LD between sites were calculated using TASSEL 2.1.0[55].
ZCN8 expression analysis
ZCN8 is mainly expressed in mature leaves near the floral transition[5, 6]. Mature leaves of the two NILs for qDTA8 (NIL(W22) and NIL(8759)) were sampled in the morning at the V4 stage to examine ZCN8 expression. For each NIL, three biological replicates were included in the quantitative real-time PCR (qRT-PCR) analysis. To examine the diurnal expression patterns of ZCN8, NIL(W22) and NIL(8759) were planted under artificial long day (16 h light and 8 h dark) and short day (8h light and 16 h dark) conditions. The uppermost leaves of NIL(W22) and NIL(8759) were sampled every 4 h for over a 48-h period around floral transition. Three biological replicates were collected at each time point for each NIL. To examine ZCN8 expression in the maize population, 10 maize inbred lines carrying the SNP-1245A allele and 10 maize inbred lines carrying the SNP-1245G allele were randomly selected from the maize association panel and were grown in a greenhouse (18:6 h light:dark daily cycle;25–28 ℃ ). Mature leaves of the sampled inbred lines were collected around the V4 stage. Total RNA extraction, purification, and quantitation, first-strand cDNA synthesis and qRT-PCR analysis were performed as described previously[45]. ZmTubulin1 was used as an internal control.
Generation of ZmMADS1 overexpressed lines
The coding sequence of ZmMADS1 was amplified from B73 cDNA and cloned into the binary vector pCUNm-eGFP under control of the Ubiquitin promoter. This construct was introduced into the maize inbred line B73 via Agrobacterium tumefaciens-mediated transformation[56]. The transgenic seeds were created by the maize functional genomics project of China Agricultural University. Mature leaves of T2 transgenic plants and wild type were collected around floral transition stage to examine the expression levels of ZmMADS1 and ZCN8 using qRT-PCR method. Three biological replicates were sampled for the T2 transgenic line and the wild type.
Electrophoretic mobility shift assay (EMSA)
The full-length CDS of ZmMADS1 was amplified and subsequently cloned into the expression vector pGEX-4T-1. The resulting plasmid was introduced into Rosetta(DE3), and GST-tagged ZmMADS1 was purified with BeaverBeads™GSH (BEAVER). Oligonucleotide probes (Table S4) were synthesized and labelled according to the standard protocol by Shanghai Invitrogen Technology. EMSA was performed using the LightShift Chemiluminescent EMSA Kit (Thermo Fisher) according to the manufacturer’s instructions. Signals were visualized by X-ray film exposure.
Protoplast transient expression assay
To examine the impact of ZmMADS1 on the expression of ZCN8, we performed a dual-luciferase transient expression assay in maize leaf protoplasts. The coding sequence of ZmMADS1 was cloned into the pGreenII 62-SK vector under the control of the 35S promoter generating the effector construct. For the reporter construct, a ~2.0-kb fragment of the ZCN8 promoter from NIL(W22) and NIL(8759) was cloned into pGreenII 0800-LUC to drive the firefly luciferase (LUC) gene. In the same construct, a Renilla luciferase (REN) reporter gene under control of the CaMV 35S promoter was used as an internal control to evaluate the protoplast transfection efficiency. To test the effect of Indel-2339 on gene expression, ~3-kb promoter fragments from the W22 and Mo17 line were amplified and cloned into the reporter constructs. The site-directed mutation was introduced with specific primers using standard oligonucleotide-directed intro mutagenesis methods. Maize leaf protoplast preparation and subsequent transfection were performed as described previously[45]. Firefly LUC and REN activities were measured using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. Relative firefly LUC activity was calculated by normalizing LUC activity to REN activity. Five biological replicates, each with two technical replicates, were assayed per construct.
Genotyping SNP-1245 and Indel-2339 in landraces
To genotype SNP-1245 in the maize landraces, a cleaved amplified polymorphic sequences (CAPS) marker was developed (Table S4). The genotypes of Indel-2339 in the maize landraces were determined electrophoretically on 8% polyacrylamide denaturing gels.
Analysis of ancient genomes
The genomic sequences of archaeological maize from Vallebueno-Estrada et al. (2016)[46], Ramos-Madrigal et al. (2016)[47] and Swarts et al. (2017)[48] were downloaded from NCBI (accession number PRJEB16754, PRJNA352392 and PRJNA386191). A total of 19 archaeological maize samples were sequenced in the three studies, including four 5000-year-old maize specimens from the Tehuacan Valley[46, 47] and fifteen 1900-year-old maize cobs from Turkey Pen Shelter in the southwestern United States[48]. The read processing and mapping followed the previous studies[46–48]. Filtered reads were mapped to the maize reference genome AGPv3.29[63] using bwa mem v0.7.10[64]. The genotypes of SNP-1245 and Indel-2339 at ZCN8, the MITE insertion at Vgt1, the CACTA-like transposon insertion at ZmCCT10 and the Harbinger-like transposon insertion at ZmCCT9 in the 19 ancient samples were determined by examining the read mapping at the corresponding regions.
QUANTIFICATION AND STATISTICAL ANALYSIS
QTL mapping
Using the 866 maize-teosinte BC2S3 RILs, we previously performed QTL mapping for DTA under long day conditions[4, 7]. The five newly constructed maize-teosinte BC1S4 RIL populations were scored for DTA in Wisconsin (43°N, 89°E), USA, for two years from 2015 to 2017. For each RIL, the average flowering time over the two years was used as the phenotype in QTL mapping. A modified version of R/QTL software[53] that considers the BC2S3 pedigree of the RILs was used for QTL mapping. The procedure employed for QTL mapping was previously described[7, 49–52, 54]. Briefly, the scanone command, which implemented Haley-Knott regression, was first used to identify an initial QTL list for multiple QTL fitting[53]. The multiple QTL model was evaluated using a drop-one ANOVA. The position of each QTL in the model was refined using the refineqtl command. The likelihood ratio test was used to measure the improvement of the model. Finally, additional QTLs was searched using the addqtl command. The ANOVA and refineqtl procedures were repeated until no other significant QTLs could be added. A total of 1000 permutation tests were performed to determine a logarithm of odds (LOD) threshold for claiming QTLs at P<0.05. A 2-LOD support interval was determined for each QTL.
Association analysis
The maize association population was scored for DTA under natural long days in Beijing (39.9°N, 116.4°E) and natural short days in Sanya, China (18.1°N, 109°E) [18]. According to the previous studies[4, 11], photoperiod response was defined as the difference in growing degree days (GDD) to anthesis between long days and short days. The associations between sequence variants with an MAF≥0.05 and flowering time phenotypes were tested using a mixed linear model that corrects for population structure and family relatedness[19]in TASSEL 2.1.0[55]. The population structure and kinship matrix used in the model was the same as that used in a previous study[18]. A Bonferroni-corrected significance threshold (P ≤4.95E-04) was used to identify significant associations.
Molecular population genetic analyses
The third generation Zea mays haplotype map (HapMap 3) data [57] was downloaded from https://www.panzea.org/ and was used to examine the SNP diversity around ZCN8 in maize and teosinte. The R package PopGenome [58] was used to calculate nucleotide diversity (π) in sliding windows (window size = 1000 bp, step size = 100 bp). The nucleotide diversity of the promoter region of ZCN8 was further analysed in a diverse panel of teosinte and maize lines using DnaSP v.5.1[59]. Insertions and deletions were not included in the analysis. The amount of nucleotide diversity retained in maize relative to that in teosinte for the sequenced regions was measured as the relative ratio of π in maize to π in teosinte. To evaluate whether the observed loss of genetic diversity in maize relative to that in teosinte could be explained by the demography of maize domestication[25, 60] alone, coalescent simulations incorporating the demography of maize domestication were performed using Hudson’s ms program[61]. The parameters used in the coalescent simulations were from the demographic model of maize domestication recently established in the study of Beissinger et al. (2016)[25]. This demographic model estimates an ancestral effective population size of Na ≈ 123,000 teosinte individuals. Maize split from teosinte ≈15,000 generations ago, with an initial size of ≈5% of the ancestral Na. After its split from teosinte, maize experienced an exponential population growth, reaching a final modern effective population size of 2.98 × Na. In particular, this model provides an estimate of gene migration between maize and teosinte. The gene flow from teosinte to maize was estimated to be Mtm = 1.1 × 10−5 × Na migrants per generation, and the gene flow from maize to teosinte was estimated to be Mmt = 1.4 × 10−5 × Na migrants per generation. The population mutation and population recombination parameters were estimated from the teosinte sequences of the sequenced region. A total of 10,000 coalescent simulations were performed. Significant deviations from expectations under the demography of maize domestication indicate selection in the examined region. Phylogenetic analysis was performed with MEGA6.0 software[62]. The phylogenetic tree was constructed using a neighbour-joining approach with bootstrapping based on 1,000 pseudoreplicates.
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Rosetta(DE3) | Shanghai Weidi biotechnology | CAT#: EC1010 |
| Biological Samples | ||
| W22 X CIMMYT accession 8759 BC2S3 RILs | John Doebley Lab | N/A |
| W22 X TIL01 BC1S4 RILs | John Doebley Lab | N/A |
| W22 X TIL03 BC1S4 RILs | John Doebley Lab | N/A |
| W22 X TIL11 BC1S4 RILs | John Doebley Lab | N/A |
| W22 X TIL14 BC1S4 RILs | John Doebley Lab | N/A |
| W22 X TIL25 BC1S4 RILs | John Doebley Lab | N/A |
| Maize association panel | Xiaohong Yang Lab | http://www.pnas.org/content/110/42/16969.long |
| Maize landraces | John Doebley Lab | https://onlinelibrary.wiley.com/doi/epdf/10.3732/ajb.0800097 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| GST-tagged ZmMADS1 | This paper | N/A |
| Critical Commercial Assays | ||
| LightShift Chemiluminescent EMSA Kit | Thermo Fisher | Prod #20148X |
| Dual-Luciferase Reporter Assay System | Promega | E1910 |
| BeaverBeadsGSH | BEAVER | 70601-5 |
| Deposited Data | ||
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Sequences reported in this paper | This paper | NCBI: MH735187–MH735686, MH735687–MH737678 |
| B73 reference genome sequence version AGPv3.29 | Ensembl Genomes | ftp://ftp.ensemblgenomes.org/pub/plants/release-29/fasta/zea_mays/dna/ |
| The third generation Zea mays haplotype map (HapMap 3) data | [49] | https://doi.org/10.1093/gigascience/gix134 |
| Ancient Genomes (SM10) | [46] | NCBI: PRJEB16754 |
| Ancient Genomes (Tehuacan162) | [47] | NCBI: PRJNA352392 |
| Ancient Genomes (Turkey Pen maize) | [48] | NCBI: PRJNA386191 |
| Oligonucleotides | ||
| Primers for sequencing, genotyping, qRT-PCR see Table S4 | This paper | N/A |
| EMSA oligonucleotide probes, see Table S4 | Shanghai Invitrogen Technology | N/A |
| Recombinant DNA | ||
| pCUNm-eGFP | Maize functional genomics project of CAU | N/A |
| pGEX-4T-1 | GE Healthcare | 27-4580-01 |
| pGreenII 62-SK | Susheng Song Lab | N/A |
| pGreenII 0800-LUC | Biovector | Biovector0800LUC |
| Software and Algorithms | ||
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| PlantPAN 2.0 | [20] | http://plantpan2.itps.ncku.edu.tw/ |
| Modified R/QTL software | [50] | http://www.biostat.jhsph.edu/∼kbroman/qtl |
| BIOEDIT v.7.0.9.0 | North Carolina State University | N/A |
| TASSEL 2.1.0 | [51] | http://www.maizegenetics.net/tassel |
| ImageJ | N/A | http://imagej.nih.gov/ij/ |
| R package PopGenome | [52] | https://cran.r-project.org/ |
| DnaSP v.5.1 | [53] | http://www.ub.edu/dnasp/ |
| ms program | [54] | http://home.uchicago.edu/∼rhudson1/source/mksamples.html |
| MEGA6.0 | [55] | http://www.megasoftware.net/ |
ACKNOWLEDGEMENTS
We thank Xiaohong Yang (National Maize Improvement Center, China Agricultural University) for providing the maize association panel. We thank Lora L. Daskalska and Aria C. Peterson (Department of Genetics, University of Wisconsin) for helping collect the flowering time data of the maize-teosinte BC1S4 populations. This research was supported by the National Key Research and Development Program of China (2016YFD0100303 and 2016YFD0100404), the National Natural Science Foundation of China (31771806 and 31322042), the Recruitment Program of Global Experts, the Fundamental Research Funds for the Central Universities, and the US National Science Foundation (grants IOS1025869 and IOS1238014).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes three figures, four tables, and two data files and can be found with this article online at https://doi.org/10.1016/j.cub.2018.07.029..
Data S1.Information on the maize association panel used in this study. Related to Figure 1, Figure 3 and Figure 4.
Data S2.Information on the maize landraces used in this study. Related to Figure 3 and Figure 4.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Matsuoka Y, Vigouroux Y, Goodman MM, Sanchez GJ, Buckler E, and Doebley J (2002). A single domestication for maize shown by multilocus microsatellite genotyping. Proc. Natl. Acad. Sci. USA 99, 6080–6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuleshov NN (1933). World’s diversity of phenotypes of maize, Jour. Amer. Soc. Agron, 25: 688–699., 1933. Agron. J 25, 688–700. [Google Scholar]
- 3.Buckler ES, Holland JB, Bradbury PJ, Acharya CB, Brown PJ, Browne C, Ersoz E, Flint-Garcia S, Garcia A, and Glaubitz JC (2009). The genetic architecture of maize flowering time. Science 325, 714–718. [DOI] [PubMed] [Google Scholar]
- 4.Hung HY, Shannon LM, Tian F, Bradbury PJ, Chen C, Flint-Garcia SA, McMullen MD, Ware D, Buckler ES, Doebley JF, et al. (2012). ZmCCT and the genetic basis of day-length adaptation underlying the postdomestication spread of maize. Proc. Natl. Acad. Sci. USA 109, E1913–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lazakis CM, Coneva V, and Colasanti J (2011). ZCN8 encodes a potential orthologue of Arabidopsis FT florigen that integrates both endogenous and photoperiod flowering signals in maize. Journal of experimental botany 62, 4833–4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meng X, Muszynski MG, and Danilevskaya ON (2011). The FT-like ZCN8 Gene Functions as a Floral Activator and Is Involved in Photoperiod Sensitivity in Maize. Plant Cell 23, 942–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li D, Wang X, Zhang X, Chen Q, Xu G, Xu D, Wang C, Liang Y, Wu L, Huang C, et al. (2016). The genetic architecture of leaf number and its genetic relationship to flowering time in maize. The New phytologist 210, 256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Z, Cook J, Meliahancock S, Guill K, Bottoms C, Garcia A, Ott O, Nelson R, Recker J, and Balintkurti P (2016). Expanding Maize Genetic Resources with Predomestication Alleles: Maize-Teosinte Introgression Populations. Plant Genome [DOI] [PubMed] [Google Scholar]
- 9.Vlăduţu C, Mclaughlin J, and Phillips RL (1999). Fine mapping and characterization of linked quantitative trait loci involved in the transition of the maize apical meristem from vegetative to generative structures. Genetics 153, 993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chardon F, Virlon B, Moreau L, Falque M, Joets J, Decousset L, Murigneux A, and Charcosset A (2004). Genetic architecture of flowering time in maize as inferred from quantitative trait loci meta-analysis and synteny conservation with the rice genome. Genetics 168, 2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coles ND, McMullen MD, Balint-Kurti PJ, Pratt RC, and Holland JB (2010). Genetic control of photoperiod sensitivity in maize revealed by joint multiple population analysis. Genetics 184, 799–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hung HY, Browne C, Guill K, Coles N, Eller M, Garcia A, Lepak N, Meliahancock S, Oropezarosas M, and Salvo S (2012). The relationship between parental genetic or phenotypic divergence and progeny variation in the maize nested association mapping population. Heredity 108, 490–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muszynski MG, Dam T, Li B, Shirbroun DM, Hou Z, Bruggemann E, Archibald R, Ananiev EV, and Danilevskaya ON (2006). delayed flowering1 Encodes a basic leucine zipper protein that mediates floral inductive signals at the shoot apex in maize. Plant Physiol. 142, 1523–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Danilevskaya ON, Meng X, Selinger DA, Deschamps S, Hermon P, Vansant G, Gupta R, Ananiev EV, and Muszynski MG (2008). Involvement of the MADS-box gene ZMM4 in floral induction and inflorescence development in maize. Plant Physiol 147, 2054–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bouchet S, Servin B, Bertin P, Madur D, Combes V, Dumas F, Brunel D, Laborde J, Charcosset A, and Nicolas S (2013). Adaptation of maize to temperate climates: mid-density genome-wide association genetics and diversity patterns reveal key genomic regions, with a major contribution of the Vgt2 (ZCN8) locus. PLoS One 8, e71377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gouesnard B, Negro S, Laffray A, Glaubitz J, Melchinger A, Revilla P, Moreno-Gonzalez J, Madur D, Combes V, Tollon-Cordet C, et al. (2017). Genotyping-by-sequencing highlights original diversity patterns within a European collection of 1191 maize flint lines, as compared to the maize USDA genebank. TAG. Theoretical and applied genetics. Theoretische und angewandte Genetik 130, 2165–2189. [DOI] [PubMed] [Google Scholar]
- 17.Salvi S, Sponza G, Morgante M, Tomes D, Niu X, Fengler KA, Meeley R, Ananiev EV, Svitashev S, Bruggemann E, et al. (2007). Conserved noncoding genomic sequences associated with a flowering-time quantitative trait locus in maize. Proc. Natl. Acad. Sci. USA 104, 11376–11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Q, Li Z, Li W, Ku L, Wang C, Ye J, Li K, Yang N, Li Y, Zhong T, et al. (2013). CACTA-like transposable element in ZmCCT attenuated photoperiod sensitivity and accelerated the postdomestication spread of maize. Proc. Natl. Acad. Sci. USA 110, 16969–16974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, et al. (2006). A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet 38, 203–208. [DOI] [PubMed] [Google Scholar]
- 20.Chow CN, Zheng HQ, Wu NY, Chien CH, Huang HD, Lee TY, Chiang-Hsieh YF, Hou PF, Yang TY, and Chang WC (2016). PlantPAN 2.0: an update of plant promoter analysis navigator for reconstructing transcriptional regulatory networks in plants. Nucleic acids research 44, D1154–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smaczniak C, Immink RG, Angenent GC, and Kaufmann K (2012). Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development 139, 3081–3098. [DOI] [PubMed] [Google Scholar]
- 22.Alter P, Bircheneder S, and Zhou LZ (2016). Flowering Time-Regulated Genes in Maize Include the Transcription Factor ZmMADS1. Plant Physiology 172, 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borner R, Kampmann G, Chandler J, Gleissner R, Wisman E, Apel K, and Melzer S (2000). A MADS domain gene involved in the transition to flowering in Arabidopsis. The Plant journal : for cell and molecular biology 24, 591–599. [DOI] [PubMed] [Google Scholar]
- 24.Lee S, Kim J, Han JJ, Han MJ, and An G (2004). Functional analyses of the flowering time gene OsMADS50, the putative SUPPRESSOR OF OVEREXPRESSION OF CO 1/AGAMOUS-LIKE 20 (SOC1/AGL20) ortholog in rice. The Plant journal : for cell and molecular biology 38, 754–764. [DOI] [PubMed] [Google Scholar]
- 25.Beissinger TM, Wang L, Crosby K, Durvasula A, Hufford MB, and Rossibarra J (2016). Recent demography drives changes in linked selection across the maize genome. Nat. Plants 2, 16084. [DOI] [PubMed] [Google Scholar]
- 26.Vigouroux Y, Glaubitz JC, Matsuoka Y, Goodman MM, Sanchez GJ, and Doebley J (2008). Population structure and genetic diversity of New World maize races assessed by DNA microsatellites. American journal of botany 95, 1240–1253. [DOI] [PubMed] [Google Scholar]
- 27.van Heerwaarden J, Doebley J, Briggs WH, Glaubitz JC, Goodman MM, de Jesus Sanchez Gonzalez J, and Ross-Ibarra J (2011). Genetic signals of origin, spread, and introgression in a large sample of maize landraces. Proc. Natl. Acad. Sci. USA 108, 1088–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harlan JR, de Wet JMJ, and Price EG (1973). COMPARATIVE EVOLUTION OF CEREALS. Evolution 27, 311–325. [DOI] [PubMed] [Google Scholar]
- 29.Bomblies K, and Doebley JF (2006). Pleiotropic Effects of the Duplicate Maize FLORICAULA/LEAFY Genes zfl1 and zfl2 on Traits Under Selection During Maize Domestication. Genetics 172, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wills DM, Fang Z, York AM, Holland JB, and Doebley JF (2017). Defining the role of the MADS-box gene, Zea agamous like1, a target of selection during maize domestication. Journal of Heredity [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xue W, Xing Y, Weng X, Zhao Y, Tang W, Wang L, Zhou H, Yu S, Xu C, Li X, et al. (2008). Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat. Genet 40, 761–767. [DOI] [PubMed] [Google Scholar]
- 32.Danilevskaya ON, Meng X, Mcgonigle B, and Muszynski MG (2011). Beyond flowering time: Pleiotropic function of the maize flowering hormone florigen. Plant Signaling & Behavior 6, 1267–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Romay MC, Millard MJ, Glaubitz JC, Peiffer JA, Swarts KL, Casstevens TM, Elshire RJ, Acharya CB, Mitchell SE, Flint-Garcia SA, et al. (2013). Comprehensive genotyping of the USA national maize inbred seed bank. Genome Biol 14, R55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brandenburg JT, Mary-Huard T, Rigaill G, and Hearne SJ (2017). Independent introductions and admixtures have contributed to adaptation of European maize and its American counterparts. Plos Genetics 13, e1006666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heerwaarden JV, Doebley J, Briggs WH, Glaubitz JC, Goodman MM, Gonzalez JDJS, and Ross-Ibarra J (2011). Genetic signals of origin, spread, and introgression in a large sample of maize landraces. Proc. Natl. Acad. Sci. USA 108, 1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Navarro JAR, Willcox M, Burgueño J, Romay C, Swarts K, Trachsel S, Preciado E, Terron A, Delgado HV, and Vidal V (2017). A study of allelic diversity underlying flowering-time adaptation in maize landraces. Nature Genetics 49, 476. [DOI] [PubMed] [Google Scholar]
- 37.Williams TM, Selegue JE, Werner T, Gompel N, Kopp A, and Carroll SB (2008). The regulation and evolution of a genetic switch controlling sexually dimorphic traits in Drosophila. Cell 134, 610–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rebeiz M, and Carroll SB (2009). Stepwise modification of a modular enhancer underlies adaptation in a Drosophila population. Science 326, 1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glaserschmitt A, and Parsch J (2018). Functional characterization of adaptive variation within a cis-regulatory element influencing Drosophila melanogaster growth. Plos Biology 16, e2004538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blackman BK, Strasburg JL, Raduski AR, Michaels SD, and Rieseberg LH (2010). The role of recently derived FT paralogs in sunflower domestication. Current Biology 20, 629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kong F, Liu B, Xia Z, Sato S, Kim BM, Watanabe S, Yamada T, Tabata S, Kanazawa A, and Harada K (2010). Two coordinately regulated homologs of FLOWERING LOCUS T are involved in the control of photoperiodic flowering in soybean. Plant Physiology 154, 1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao C, Takeshima R, Zhu J, Xu M, Sato M, Watanabe S, Kanazawa A, Liu B, Kong F, and Yamada T (2016). A recessive allele for delayed flowering at the soybean maturity locusE9is a leaky allele ofFT2a, a FLOWERING LOCUS T ortholog. Bmc Plant Biology 16, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ducrocq S, Madur D, Veyrieras JB, Camus-Kulandaivelu L, Kloiber-Maitz M, Presterl T, Ouzunova M, Manicacci D, and Charcosset A (2008). Key impact of Vgt1 on flowering time adaptation in maize: evidence from association mapping and ecogeographical information. Genetics 178, 2433–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castelletti S, Tuberosa R, Pindo M, and Salvi S (2014). A MITE transposon insertion is associated with differential methylation at the maize flowering time QTL Vgt1. G3 (Bethesda) 4, 805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang C, Sun H, Xu D, Chen Q, Liang Y, Wang X, Xu G, Tian J, Wang C, Li D, et al. (2018). ZmCCT9 enhances maize adaptation to higher latitudes. Proc. Natl. Acad. Sci. USA 115, E334–e341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vallebuenoestrada M, Rodríguezarévalo I, Rougoncardoso A, Martínez GJ, García CA, Montiel R, and Viellecalzada JP (2016). The earliest maize from San Marcos Tehuacán is a partial domesticate with genomic evidence of inbreeding. Proc. Natl. Acad. Sci. USA 113, 14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramosmadrigal J, Smith BD, Morenomayar JV, Gopalakrishnan S, Rossibarra J, Gilbert MT, and Wales N (2016). Genome Sequence of a 5,310-Year-Old Maize Cob Provides Insights into the Early Stages of Maize Domestication. Current Biology 26, 3195–3201. [DOI] [PubMed] [Google Scholar]
- 48.Swarts K, Gutaker RM, Benz B, Blake M, Bukowski R, Holland J, Krusepeeples M, Lepak N, Prim L, and Romay MC (2017). Genomic estimation of complex traits reveals ancient maize adaptation to temperate North America. Science 357, 512. [DOI] [PubMed] [Google Scholar]
- 49.Huang C, Chen Q, Xu G, Xu D, Tian J, and Tian F (2016). Identification and fine mapping of quantitative trait loci for the number of vascular bundle in maize stem. Journal of integrative plant biology 58, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu G, Wang X, Huang C, Xu D, Li D, Tian J, Chen Q, Wang C, Liang Y, Wu Y, et al. (2017). Complex genetic architecture underlies maize tassel domestication. The New phytologist 214, 852–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, Chen Q, Wu Y, Lemmon ZH, Xu G, Huang C, Liang Y, Xu D, Li D, and Doebley JF (2018). Genome-wide analysis of transcriptional variability in a large maize-teosinte population. Molecular Plant 11, 443–459. [DOI] [PubMed] [Google Scholar]
- 52.Xu D, Wang X, Huang C, Xu G, Liang Y, Chen Q, Wang C, Li D, Tian J, and Wu L (2017). Glossy15 plays an important role in the divergence of the vegetative transition between maize and its progenitor, teosinte. Molecular Plant 10,1579–1583. [DOI] [PubMed] [Google Scholar]
- 53.Broman KW, Wu H, Sen S, and Churchill GA (2003). R/qtl: QTL mapping in experimental crosses. Bioinformatics 19, 889–890. [DOI] [PubMed] [Google Scholar]
- 54.L, S. (2012). The genetic architecture of maize domestication and range expansion PhD thesis, The University of Wisconsin-Madison, Madison, WI, USA. [Google Scholar]
- 55.Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, and Buckler ES (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. [DOI] [PubMed] [Google Scholar]
- 56.Ishida Y, Hiei Y, and Komari T (2007). Agrobacterium-mediated transformation of maize. Nature Protocols 2, 1614–1621. [DOI] [PubMed] [Google Scholar]
- 57.Bukowski R, Guo X, Lu Y, Zou C, He B, Rong Z, Wang B, Xu D, Yang B, Xie C, et al. (2018). Construction of the third-generation Zea mays haplotype map. GigaScience 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pfeifer B, Wittelsburger U, Ramos-Onsins SE, and Lercher MJ (2014). PopGenome: an efficient swiss army knife for population genomic analyses in R. Molecular Biology and Evolution 31, 1929–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Librado P, and Rozas J (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. [DOI] [PubMed] [Google Scholar]
- 60.Wang L, Beissinger TM, Lorant A, Ross-Ibarra C, Ross-Ibarra J, and Hufford MB (2017). The interplay of demography and selection during maize domestication and expansion. Genome Biology 18, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hudson RR (2002). Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18, 337–338. [DOI] [PubMed] [Google Scholar]
- 62.Tamura K, Stecher G, Peterson D, Filipski A, and Kumar S (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Molecular biology and evolution 30, 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, and Graves TA (2009). The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115. [DOI] [PubMed] [Google Scholar]
- 64.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows–Wheeler transform, (Oxford University Press; ). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.