

Abstract
Diprovocim is a recently discovered exceptionally potent, synthetic small molecule agonist of TLR2/TLR1 and has shown significant adjuvant activity in anticancer vaccination against murine melanoma. Since Diprovocim bears no structural similarity to the canonical lipopeptide ligands of TLR2/TLR1, we investigated how Diprovocim interacts with TLR2/TLR1 through in vitro biophysical, structural, and computational approaches. We found that Diprovocim induced the formation of TLR2/TLR1 heterodimers as well as TLR2 homodimers in vitro. We determined the crystal structure of Diprovocim in a complex with a TLR2 ectodomain, which revealed, unexpectedly, two Diprovocim molecules bound to the ligand binding pocket formed between two TLR2 ectodomains. Extensive hydrophobic interactions and a hydrogen-bonding network between the protein and Diprovocim molecules are observed within the defined ligand binding pocket and likely underlie the high potency of Diprovocim. Our work shed first light into the activation mechanism of TLR2/TLR1 by a noncanonical agonist. The structural information obtained here may be exploited to manipulate TLR2/TLR1-dependent signaling.
Graphical Abstract

INTRODUCTION
As innate immune receptors for microbial molecules, Toll-like receptors (TLRs) are among the body’s first responders to infection, initiating signaling that promotes inflammation and contributes to activation of an adaptive immune response.1,2 Humans deficient in the function of one or more TLRs, or molecules that mediate TLR signaling, display elevated susceptibility to a variety of infections.3–8 Because of their critical role in the immune system, TLRs are recognized as important drug targets that may be specifically activated or inhibited in anti-infectious and anticancer therapies or in the treatment of allergic and autoimmune diseases, respectively.9–12 TLR agonists are particularly attractive as vaccine adjuvants that can promote cell-mediated immunity. Several small molecule agonists of TLRs have recently been described13–16 including the Neoseptins, the first structurally characterized class of noncanonical mouse TLR4 agonists that we discovered by screening a synthetic peptidomimetic compound library for innate immune activators.17,18
More recently, we reported the discovery of Diprovocim, a synthetic small molecule agonist of human and mouse TLR2/TLR1.19 Identified from a compound library designed to promote cell surface receptor dimerization,20,21 the precursor of Diprovocim was subjected to extensive structure—activity relationship characterization to improve potency, resulting in an enantiomerically unique drug with twofold symmetry. Diprovocim bears no structural similarity to bacterial triacylated lipoproteins and lipopeptides (e.g., Pam3CSK4), the natural ligands for the TLR2/TLR1 heterodimer, or to any other natural or synthetic TLR agonist. In contrast to the synthetic nonlipopeptide-based TLR2 agonists reported to date, which show weak potencies requiring micromolar concentrations for agonist activity at the cellular level,22–24 Diprovocim exhibits greater potency than Pam3CSK4 in human cells (EC50 = 110 pM). We demonstrated the use of Diprovocim as an adjuvant in successful anticancer vaccination against B16 murine melanoma.25 In conjunction with the immune checkpoint inhibitor anti-programmed death-ligand 1, immunizations adjuvanted with Diprovocim completely inhibited tumor growth, induced long-term antitumor memory, and significantly prolonged survival of tumor-bearing mice.25 Thus, Diprovocim is both structurally distinct and significantly more potent than other reported TLR2-dependent agonists/adjuvants.
To understand the basis for this potency, we set out to determine the structural mechanism by which Diprovocim binds and activates TLR2/TLR1 using in vitro biophysical and structural approaches. Comparative analysis was performed with the reported crystal structure of TLR2/TLR1 in a complex with Pam3CSK4, in which the two ester-linked palmitoyl lipid chains of the agonist are inserted into a pocket in TLR2, whereas the third amide-linked palmitoyl chain is inserted into a hydrophobic channel of TLR1, thereby facilitating formation of the m-shaped signaling-competent heterodimer.26 Unexpectedly, the remarkable efficiency with which Diprovocim induces homodimerization of TLR2 ectodomains precluded the isolation of Diprovocim-bound TLR2/TLR1 heterodimers in quantities sufficient for crystallization. We therefore determined crystal structures of human TLR2 in a complex with Diprovocim and human TLR1 in apo form. By combining these structural data with molecular dynamics (MD) simulation analysis and structure-based mutagenesis data, we showed that Diprovocim interacts with TLR2/TLR1 at the same binding pocket as Pam3CSK4 and demonstrated how these structurally distinct agonists elicit similar signaling.
RESULTS
Diprovocim Induces TLR2/TLR1 Heterodimerization as Well as TLR2 Homodimerization in Vitro.
The ectodomains of human TLR1 and TLR2 were separately overexpressed and purified from Hi5 insect cells. Each protein existed in solution as a monomer as shown by size exclusion chromatography (Figure 1). As expected, the canonical TLR2/TLR1 agonist Pam3CSK4 induced heterodimerization of TLR1 and TLR2 but not homodimerization of either TLR1 or TLR2 (Figure 1A). In contrast, Diprovocim induced formation of TLR2 (but not TLR1) homodimers (Figure 1B). In the presence of both TLR2 and TLR1 ectodomains, Diprovocim induced both TLR2/TLR2 homodimers and TLR2/TLR1 heterodimers detectable by immunoblot. Because TLR2/TLR2 homodimers and TLR2/TLR1 heterodimers have similar molecular weights, it is not possible to separate them by size exclusion chromatography. We tried to reconstitute TLR2/TLR1 heterodimers in the presence of Diprovocim using an excessive amount of TLR1 in the mixture and also tried to isolate Diprovocim-induced heterodimers by introducing a unique affinity tag on TLR1. In both cases, TLR2/TLR2 ectodomain homodimers persisted in the resulting dimeric mixtures. Over time, there were even increased amounts of TLR2/TLR2 homodimers and decreased amounts of TLR2/ TLR1 heterodimers. These observations with the purified proteins in solution indicate that Diprovocim must bind to the TLR2/TLR2 ectodomain homodimer with significantly higher affinity than to the TLR2/TLR1 heterodimer, and in a TLR2, TLR1, and Diprovocim mixture, the equilibrium would strongly shift to forming the TLR2/TLR2/Diprovocim homodimer. The strong tendency of Diprovocim to facilitate TLR2 ectodomain homodimerization hampered our efforts to isolate and purify Diprovocim-bound TLR2/TLR1 heterodimers. Therefore, we analyzed how Diprovocim interacts with and induces TLR2 homodimers, reasoning that the TLR2-Diprovocim interaction would likely be largely retained in the TLR2/TLR1/Diprovocim complex, which we could model based on the structural information from the active Pam3CSK4-bound TLR2/TLR1 heterodimer.26
Figure 1.
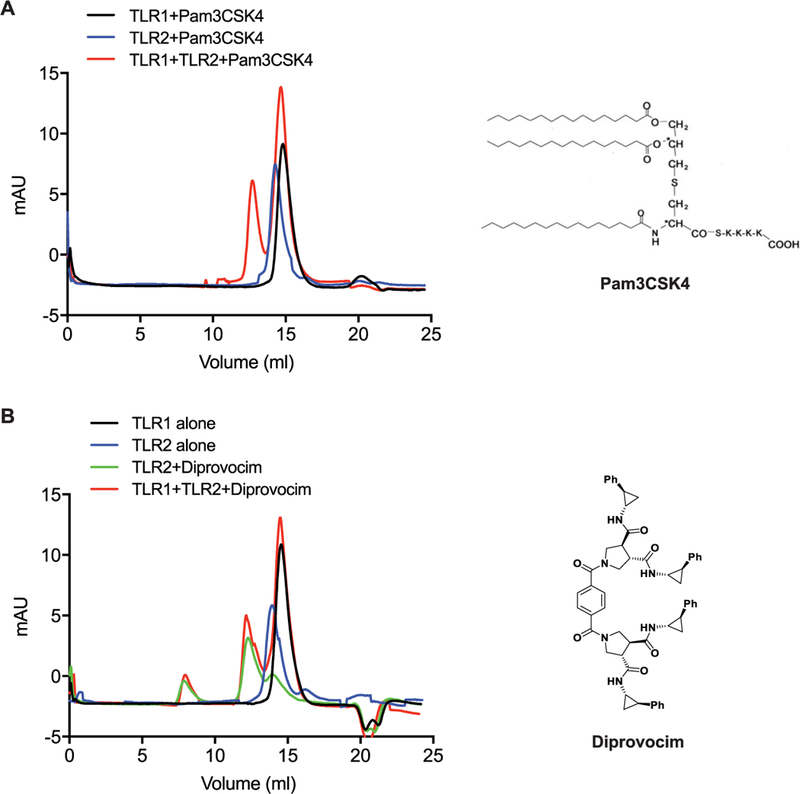
Diprovocim induces heterodimerization of TLR2/TLR1 as well as homodimerization of TLR2/TLR2. Size exclusion chromatography profiles of TLR1, TLR2, and TLR2/TLR1 in the presence of (A) Pam3CSK4 and (B) Diprovocim.
Crystal Structure of the TLR2/Diprovocim Complex.
We determined the crystal structure of Diprovocim bound to a TLR2 ectodomain homodimer at 2.35 Å resolution (Figure 2 and Table 1). Inspection of the electron density map revealed, unexpectedly but unambiguously even at early stages of the refinement, that two Diprovocim molecules were bound to the receptor at the TLR2–TLR2 homodimer interface (Figure 2A). Although the two Diprovocim molecules adopt different conformations (Figure 2B), the two halves of each molecule have nearly perfectly symmetric structure and interact with the same set of protein groups on the two TLR2 ectodomains, resulting in the homodimerization of the two TLR2 ectodomains with a twofold symmetry. The two Diprovocim molecules stack snuggly on top of each other with the two central terephthalate rings interacting in a side-to-face fashion. A number of hydrogen bonds are formed between the amide and carbonyl groups on both the protein main chains and those on the Diprovocim molecules (Figure 2C), mimicking the hydrogen-bonding patterns between protein β-strands. There are also extensive hydrophobic interactions between Diprovocim and the hydrophobic side chains of both TLR2 molecules (Figure 2D). As a result, the two Diprovocim molecules act to “staple” the two TLR2 ectodomains together in a highly specific manner (Figure 3A).
Figure 2.
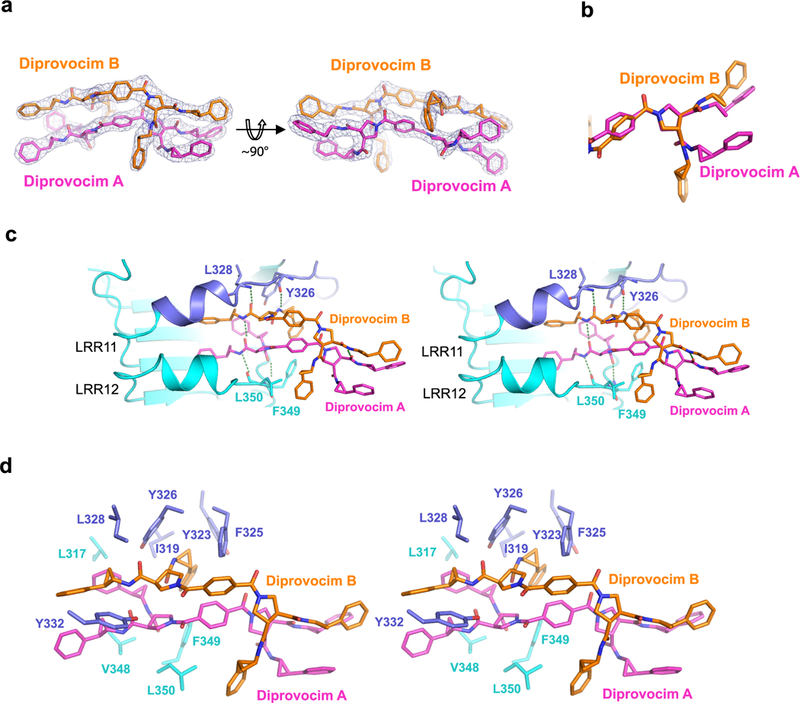
Crystal structure of Diprovocim bound to TLR2. (A) Electron density for the two Diprovocim molecules (Diprovocim A and B) bound to TLR2/TLR2. Two orthogonal views are presented. (B) Superposition of the two TLR2-bound Diprovocim molecules at the pyrrolidine ring. Only one-half of the twofold symmetrical molecule is displayed. (C) Stereo view of the hydrogen-bonding network between Diprovocim and TLR2. Only one TLR2 molecule is shown representing half of the Diprovocim binding site. (D) Stereo view of the hydrophobic interactions between TLR2 and Diprovocim. Residues from only one TLR2 molecule are shown. The same residues from the second TLR2 molecule in the homodimer make similar interactions with the other halves of the two Diprovocim molecules.
Table 1.
Data Collection and Refinement Statisticsa
| hTLR2/Diprovocim | hTLR1 | |
|---|---|---|
| data collection | ||
| space group | P21 | P21 |
| cell dimensions | ||
| a, b, c (Å) | 66.10, 201.25, 109.36 | 79.03, 74.38, 106.08 |
| α, β, γ (deg) | 90.00, 94.26, 90.00 | 90.00, 96.97, 90.00 |
| resolution (Å) | 50.00–2.34 | 50.00–2.25 |
| completeness (%) | 94.7 (79.2) | 99.1 (98.8) |
| redundancy | 4.3 (3.8) | 8.5 (7.4) |
| Rmerge (I) | 0.095 (1.447) | 0.071 (0.745) |
| I/σ(I) | 26.56 (1.09) | 20.42 (1.23) |
| CC1/2 | 0.944 (0.758) | 0.946 (0.711) |
| refinement | ||
| resolution (Å) | 37.76–2.35 | 46.40–2.30 |
| no. of reflections | 111 956 | 40 719 |
| Rwork/Rfree | 19.37/25.86 | 21.11/28.19 |
| no. of atoms | ||
| protein | 17 413 | 8387 |
| ligand | 272 | |
| water | 43 | 167 |
| B-factors | ||
| protein | 84.93 | 49.54 |
| ligand | 64.37 | |
| water | 78.34 | 33.32 |
| RMSD | ||
| bond length (Å) | 0.014 | 0.009 |
| bond angles (deg) | 1.614 | 1.092 |
Values in parentheses are for the highest resolution shells.
Figure 3.

Overall structure of the TLR2 homodimer induced by binding to Diprovocim. (A) Diprovocim-bound TLR2 homodimer. The two TLR2 monomers are colored in two different shades of cyan. The two bound Diprovocim molecules are colored orange and magenta, respectively. (B) Stereo view of the superposition of the Diprovocim-bound TLR2 homodimer (cyan) with the Pam3CSK4-bound TLR2/TLR1 heterodimer (yellow). Only the TLR2 in both structures is superimposed.
Comparison of the active Pam3CSK4-bound TLR2/TLR1 heterodimer structure (Protein Data Bank (PDB) 2Z7X)26 and the Diprovocim-bound TLR2 homodimer revealed several differences. First, the C-termini of the TLR2 ectodomains in the homodimer are much farther apart than the C-termini in the TLR2/TLR1 heterodimer structure (Figure 3B), suggesting that such a TLR2 homodimer is likely inactive because it may not be able to bring the C-terminal intracellular Toll/ interleukin-1 receptor (TIR) domains into proximity to dimerize and initiate downstream signaling. Second, we found that Diprovocim interacts with TLR2 more extensively than Pam3CSK4. Specifically, although Diprovocim occupies only part of the ligand binding pocket on TLR2, it forms an extensive hydrogen-bonding network with TLR2 that is absent in the Pam3CSK4–receptor complex. Assuming that similar interactions occur between Diprovocim and TLR2 within the TLR2/TLR1 heterodimer, these data could explain the greater potency of Diprovocim in activating TLR2/TLR1-mediated signaling in cells.
“Open, Inactive” vs “Closed, Active” Leucine-Rich Repeat (LRR)10/11 Conformations of TLR2.
The ectodomains of TLRs are formed by 16–28 leucine-rich repeats (LRRs), each one containing a highly conserved β-strand and a more variable loop structure. The ligand binding site of TLR2 is located at a large cleft between LRR11 and LRR12, which is the boundary of the central and the C-terminal subdomains of TLR2 (Figure 4B).26,27 Superposition of the TLR2/TLR2/Diprovocim and TLR2/TLR1/Pam3CSK4 structures revealed that the two ligands bind to the same site on TLR2 (Figures 3B and 4A). The two cyclopropyl benzene arms of the first Diprovocim molecule (Diprovocim A) overlap well with the two palmitoyl chains of Pam3CSK4 that are inserted in the TLR2 lipid binding pocket (Figure 4A). The second Diprovocim molecule (Diprovocim B) also has one cyclopropyl benzene arm inside the pocket, whereas the second cyclopropyl benzene substituent of Diprovocim B points about ~100° away and binds at the edge of the pocket, in contact with the second TLR2 ectodomain.
Figure 4.
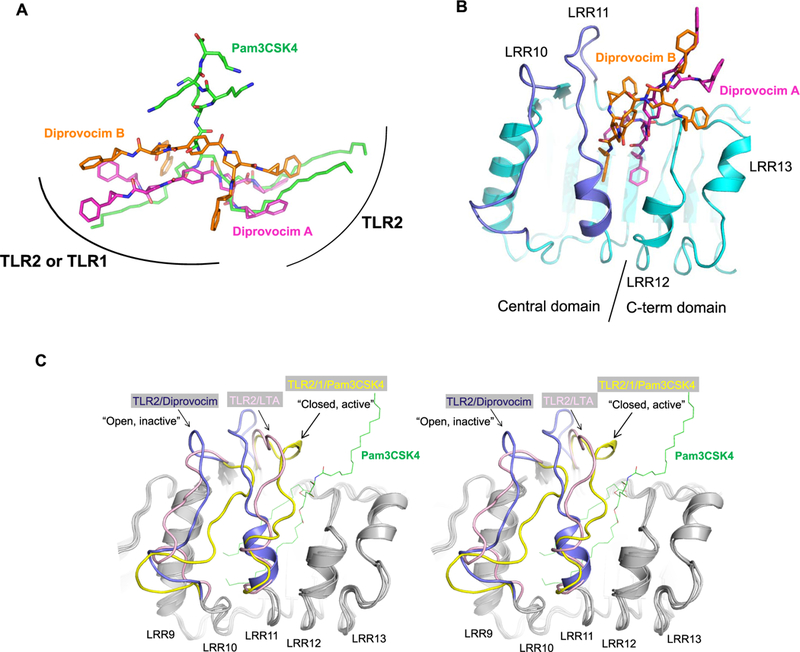
Comparison of Diprovocim and Pam3CSK4 binding modes in TLR2. (A) Positions of Diprovocim and Pam3CSK4 upon superposition of the TLR2 proteins from the TLR2/TLR2/Diprovocim complex and the TLR2/TLR1/Pam3CSK4 complex. Curved lines indicate general contact areas of TLR2 and TLR1 proteins relative to the agonists. (B) Two Diprovocim molecules bind to TLR2 (cyan) at the boundary of the central and the C-terminal subdomains between LRR11 and LRR12. (C) Stereo view of the superposition of the TLR2 structures bound to Diprovocim (dark blue), Pam3CSK4 (yellow), and lipoteichoic acid (LTA) (pink) illustrating the “open, inactive” and “closed, active” confirmations of TLR2 LRR10 and LRR11. For clarity, only Pam3CSK4 (with the peptide head groups removed) is shown in green thin lines in the ligand binding pocket of TLR2. The regions with similar conformations in all three structures are colored gray.
Although the overall conformations of individual TLR2 receptors in the Pam3CSK4- and Diprovocim-complexed states are very similar (root-mean-square deviation (RMSD) of Cα’s ~0.5A), there are large local structural rearrangements around the bound ligands. Specifically, the conformations of LRR10 and LRR11 of TLR2 are very different in the two complex structures (Figure 4B,C). In the TLR2/TLR1/Pam3CSK4 complex structure, the loops of LRR10 and LRR11 close down on the two ester-linked lipid chains inserted in the TLR2 lipid binding pocket. Residues on LRR11 also interact with the peptide head group of Pam3CSK4 and with the residues from the heterodimerizing TLR1. The structural rearrangement induced by Pam3CSK4 binding facilitates protein-protein interactions between TLR2 and TLR1, leading to the formation of the active heterodimer. This “closed, active” conformation is also observed in mouse TLR2 within the mTLR2/TLR6/Pam2CSK4 complex structure.27 In the TLR2/TLR2/Diprovocim complex structure, the loops of LRR10 and LRR11 are much more open and thereby able to accommodate two Diprovocim molecules in the cleft (Figure 4B). Inspection of the crystal packing indicates that the conformational changes of the LRR10 and LRR11 loops in the TLR2/ TLR2/Diprovocim complex structure are not caused by contact with the adjacent receptor molecules.
Crystal Structure of Apo TLR1.
We co-crystallized human TLR1 in the presence of Diprovocim and collected X-ray diffraction data to a resolution of 2.3 Å. However, after the structure was determined by the molecular replacement method, no Diprovocim molecules were found in the electron density map. This represents the first reported structure of human TLR1 in its ligand-free apo conformation. The overall conformation of apo TLR1 is very similar to that in the TLR2/TLR1/Pam3CSK4 complex, except for the LRR11 loop (residues 311–318) (Figure 5). In apo TLR1, this loop collapses toward the interior of the protein. As a result, the narrow lipid binding channel as observed in the TLR2/TLR1/Pam3CSK4 complex is nearly completely filled. In particular, the side chain of Phe314 is now pointed inside and forms part of the hydrophobic core, overlapping with the lipid binding site in the active receptor. Any ligand binding to TLR1 must induce a near 180° rotation of the peptide backbone of the LRR11 loop to make the lipid binding channel accessible. This finding suggests that the conformational flexibility of LRR11 loop is important for ligand binding. Inspection of the sequences of LRR11 in TLR1 versus TLR6 showed that there are differences in the amino acid conservation between TLR1 and TLR6.27 TLR1 has a small residue Gly or Ser at position 313 in the motif 312F(G/S)FP, whereas TLR6 has a Leu at the corresponding position, which may render the LRR11 loop less flexible and contribute to the inability of TLR6 to accommodate a ligand at this site.
Figure 5.

Conformational flexibility of LRR11 in TLR1. Super-position of apo TLR1 (cyan with LRR11 loop colored in dark blue) and Pam3CSK4-bound TLR1 (yellow).
Modeling of Diprovocim Binding to TLR2/TLR1 Heterodimers.
The observation that two Diprovocim molecules bind to the TLR2/TLR2 homodimer in the crystal structure raised the question whether the TLR2/TLR1 heterodimer would bind to and be activated by a single Diprovocim molecule or by two Diprovocim molecules. Since we were not yet able to obtain sufficient quantities of TLR2/ TLR1/Diprovocim complexes for crystallization experiments due to the strong propensity of Diprovocim to induce TLR2 homodimers, we performed MD simulation studies to investigate the potential binding modes of Diprovocim to TLR2/TLR1, in particular whether the TLR2/TLR1 heterodimer is able to bind two Diprovocim molecules.
Structures of both TLR2/TLR2/Diprovocim and TLR2/TLR1/Pam3CSK4 (PDB 2Z7X)26 complexes were used to define the ligand binding pocket and construct the model of two Diprovocim molecules bound to TLR2/TLR1. Flexible ligand docking was performed to obtain the complex structures of one Diprovocim molecule bound to TLR2/TLR1. MD simulations were performed for both one and two Diprovocim molecules bound to the TLR2/TLR1 heterodimer. Although stable trajectories could be obtained for both complexes, the root-mean-square deviations (RMSDs) of TLR2/TLR1 bound to a single Diprovocim were smaller than those of TLR2/ TLR1 bound to two Diprovocim molecules (compared to the TLR2/TLR1/Pam3CSK4 structure; Figure S1, Supporting Information (SI)). The molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) binding free energies were calculated for both TLR2/TLR1/Diprovocim-2X and TLR2/TLR1/Diprovocim-1X complex models.28–30 The most energetically favorable MD snapshots were the ones that maintained the active heterodimer conformation with the C-termini of the two subunits close to each other (Table S1, SI). For binding one Diprovocim molecule, the overall binding free energy (ΔGMM-PBSA) was –27.6 kcal/mol. For binding two Diprovocim molecules, ΔGMM-PBSA was –52.3 kcal/mol, which is less than double the ΔGMM-PBSA for single Diprovocim binding. Therefore, binding two Diprovocim molecules was slightly less energetically efficient than binding a single Diprovocim molecule.
The TLR2/TLR1/Diprovocim-1X model predicts three stable hydrogen bonds between Diprovocim and receptor residues (Ser309 and Tyr320 of TLR1; Tyr376 of TLR2) (Table S2, SI). Consistent with this model, mutation of each of these residues greatly reduced or abolished TLR2/TLR1-dependent cellular innate immune signaling induced by Diprovocim (see mutagenesis studies below). In contrast, none of the residues predicted to form hydrogen bonds with Diprovocim in the TLR2/TLR1/Diprovocim-2X model (Table S2, SI) were confirmed to be important for activation by the mutagenesis experiments (see below). Testing of hotspot residues predicted by molecular mechanics-generalized Born surface area (MM-GBSA) free-energy decomposition analysis by mutagenesis also favored the TLR2/TLR1/Diprovocim-1X model (Tables S3 and S4, SI). These molecular modeling studies suggest that TLR2/TLR1 activation and signaling in vivo are likely initiated by the binding of a single Diprovocim molecule, although the possibility of activation by binding to two Diprovocim molecules cannot be excluded completely.
We examined the model of TLR2/TLR1 binding to a single Diprovocim molecule in more detail. In this model, the conformations around the ligand binding sites are somewhat different from that in the Pam3CSK4 complex, which is not unexpected since Diprovocim has a structure different from that of Pam3CSK4. Figure 6 shows one representative model of the TLR2/TLR1/Diprovocim complex. In the model, LRR10 and LRR11 loops of TLR2 partially close down on Diprovocim and are no longer in the “open, inactive” conformation (Figure 6A). On TLR1, the conformation of the LRR11 loop is more open than in the Pam3CSK4 complex. One of the cyclopropyl benzene arms of the bound Diprovocim would overlap with the single lipid chain of Pam3CSK4 that binds to TLR1, whereas the second arm of Diprovocim is pointing sideways toward an opening between the loops of LRR10 and LRR11 as there is not enough room to accommodate both arms in the TLR1 ligand binding pocket (Figure 6B). Nevertheless, a similar set of residues is involved in the inter-subunit interactions to form the active heterodimer.
Figure 6.

Structural model of a TLR2/TLR1/Diprovocim complex. The crystal structures of human TLR2/TLR1/Pam3CSK4 (PDB 2Z7X) and TLR2/TLR2/Diprovocim (this work) were used as the starting models.26 (A) Comparison of the TLR2 conformations in the TLR2/TLR1/ Diprovocim complex model (dark gray) and in the TLR2/TLR1/Pam3CSK4 complex (yellow). (B) Comparison of the TLR1 conformations in the TLR2/TLR1/Diprovocim complex model (dark gray) and the TLR2/TLR1/Pam3CSK4 complex (yellow). The structure of apo TLR1 is also shown. (C) Diprovocim binding site on TLR1 in the TLR2/TLR1/Diprovocim complex model.
Structure-Based Mutagenesis Analysis of the Diprovocim Binding Site in TLR2 and TLR1.
Based on the crystal structures of TLR2/Diprovocim (this work), TLR2/TLR1/Pam3CSK4 (PDB 2Z7X),26 and our computational modeling studies, we systematically mutated a series of residues at the ligand binding sites on both TLR1 and TLR2 and analyzed the effects of the mutations on TLR2/TLR1-mediated innate immune signaling in response to Diprovocim and Pam3CSK4 (Figure 7). On TLR1, several residues, such as I319, Y320, and W258, are centrally located in the ligand binding pocket and directly interact with the bound Pam3CSK4 or with the Diprovocim molecule in our model. Mutations of these residues to Ala abolished the responses to both Pam3CSK4 and Diprovocim (Figure 7A). More interestingly, a number of TLR1 mutants respond differently to Pam3CSK4 and Diprovocim. In particular, when the two Phe residues on LRR11, F312 and F314, are mutated to alanine, the response of the mutant TLR2/TLR1 to Pam3CSK4 is largely abolished, whereas the activation by Diprovocim is unchanged. This result is consistent with the observation that in the Pam3CSK4-complexed TLR1 structure, the two Phe side chains point toward the inside of the ligand binding pocket and interact with the inserted lipid chain of Pam3CSK4. In contrast, our modeling suggests that these two Phe residues orient away from Diprovocim and do not directly interact with the ligand (Figure 6C). A mutation of another residue on LRR11 of TLR1, Ser309, has the opposite effect: it decreased the receptor’s response to Diprovocim significantly but did not affect Pam3CSK4 activation (Figure 7A). Ser309 is not in contact with the bound Pam3CSK4 (closest distance >4.6 Å) in the crystal structure of the TLR2/TLR1/Pam3CSK4 complex but potentially forms a hydrogen bond with a carbonyl and/or amide group on Diprovocim in our model (Figure 6C and Table S2). In addition to these residues that are in contact with the agonists, mutations of V339A and H340A of TLR1 also abolished the activity completely for both Pam3CSK4 and Diprovocim. These two residues do not interact directly with either ligand; instead, they are in contact with TLR2 in the heterodimer and contribute to the agonist-induced active heterodimer formation.
Figure 7.

Structure-based mutagenesis analysis of Diprovocim binding sites on TLR1 and TLR2. NF-κB-dependent luciferase reporter activity in HEK293T cells expressing (A) wild-type (WT) TLR2 and the indicated mutant TLR1 proteins or (B) wild-type TLR1 and the indicated mutant TLR2 proteins. Results are representative of at least three independent experiments.
On TLR2, a number of residues lining the ligand binding pocket are important for both Pam3CSK4 and Diprovocim binding because mutating these residues (Y323, F325, V348, and F349) to Ala abolished responses of the receptor to both agonists (Figure 7B). Residues D327, L328, Y332, and L350 are critical for Pam3CSK4 activity but not for Diprovocim activity. A mutation of these residues to Ala largely abolished Pam3CSK4 responses but somewhat enhanced Diprovocim responses. It is possible that because Diprovocim interacts with TLR2 more extensively than Pam3CSK4, a single mutation of any one of these residues does not affect the overall binding energy for Diprovocim as drastically as for Pam3CSK4. Residues important for TLR2 to heterodimerize with TLR1, such as Y323 and Y376, are sensitive to mutations for both agonists, suggesting that Diprovocim would induce the formation of a similarly active heterodimer of TLR2/TLR1 as that induced by Pam3CSK4.
DISCUSSION AND CONCLUSIONS
TLR2 monomers have previously been crystallized in complexes with various ligands,26,27 including Pam3CSK4, Pam2CSK4, lipoteichoic acid (LTA), and a synthetic phosphatidylethanolamine derivative, PE-DTPA. In these monomeric TLR2/ligand complex structures, LRR10 and LRR11 loops also adopt an open conformation more similar to that in the TLR2/TLR2/Diprovocim structure than to the “closed, active” confirmation observed in the TLR2/TLR1/Pam3CSK4 heterodimeric structure (Figure 4C). In such an open conformation, the two lipid chains of these ligands adopt different orientations from that of Pam3CSK4 in the active complex.27 Since these ligands are not bulkier than Diprovocim, it would suggest that the “open, inactive” conformation of LRR10 and LRR11 is a more relaxed and energetically more favorable conformation of TLR2 in the absence of an agonist and heterodimerization partner. Therefore, TLR2 must be able to accommodate ligand molecules of diverse structures, such as Diprovocim, given its very flexible binding site. However, ligands that bind only to TLR2 but fail to induce proper conformational changes of LRR10 and LRR11 will not be able to recruit TLR1 or TLR6 and activate TLR2-mediated signaling. It is reasonable to envision that in the presence of TLR1 or TLR6, binding of an agonist would induce the loops on LRR10 and LRR11 of TLR2 to adopt an active conformation, facilitate interactions with the heterodimerizing partner, and form the active receptor.
Because of the stronger affinity of Diprovocim for TLR2 than for TLR1 in solution, TLR2 ectodomains predominantly form homodimers in the presence of Diprovocim to the nearly complete exclusion of TLR2/TLR1 heterodimers. However, the situation in vivo is quite different. Mice with targeted null alleles of either Tlr2 or Tlr1 failed to respond to Diprovocim,25 indicating that signaling induced by Diprovocim requires the presence of both TLR2 and TLR1. Additionally, if Diprovocim induces a full-length TLR2 homodimer in cells, it would be expected that Diprovocim would function as an antagonist at higher concentrations by sequestrating TLR2 and preventing it from dimerizing with TLR1 or TLR6. However, this was not observed in our cell-based functional assay.25 The most likely explanation for this is that there must be some feature of the full-length receptors or how they are oriented in the cell membrane that precludes TLR2 homodimerization. We speculate that within the confines of the cell membrane, full-length TLR1 and TLR2 can adopt a limited number of orientations, one of which is the active heterodimer captured by binding of agonists, such as Pam3CSK4 and Diprovocim. On the other hand, full-length TLR2 might be anchored in the membrane in such a way that the two TLR2 ectodomains are not able to dimerize when each is bound to Diprovocim. In our crystal structure of the TLR2/TLR2/Diprovocim complex, the C-termini of the TLR2 ectodomains (where the trans-membrane helices are located) in the homodimer are much farther apart than in the active TLR2/TLR1 heterodimer. We hypothesize that the TLR2/TLR2 homodimer as observed in the TLR2/TLR2/Diprovocim crystal structure may not be achievable in cells where the two receptors must be anchored in the membrane at their C-termini. Overall, the structural, computational, and mutagenesis studies presented here support that a single Diprovocim molecule binds to the TLR2/TLR1 heterodimer in the same region bound by Pam3CSK4 to induce the formation of a signaling-competent receptor. We calculated the volumes of the ligand binding pockets in the crystal structures of both TLR2/TLR2/Diprovocim and TLR2/TLR1/Pam3CSK4 and found that the TLR2 binding pocket in TLR2/TLR2/Diprovocim has the same volume as the TLR2 pocket in TLR2/TLR1/Pam3CSK4. However, the volume of the TLR1 pocket is less than 30% of that of the TLR2 pocket and unlikely to be able to accommodate two Diprovocim molecules. The extremely low EC50 (110 pM) for activation in cells also supports the notion that TLR2/TLR1 is likely activated by one Diprovocim molecule, though it cannot be completely ruled out that the TLR2/TLR1 heterodimer might be able to accommodate two Diprovocim molecules.
The crystal structure of apo TLR1 revealed the presence of a collapsed LRR11 loop that would prevent any ligand binding. Therefore, the flexibility of this LRR loop must be important for ligand binding and it would also enable TLR1 to accommodate ligands of different structures, including Pam3CSK4 and possibly Diprovocim. In fact, our modeling and mutagenesis studies suggest that LRR11 adopts different conformations when binding to Diprovocim versus to Pam3CSK4. Like Pam3CSK4, Diprovocim cannot activate TLR2/TLR6, probably because the lipid binding channel on TLR6 is truncated by several bulkier amino acid side chains at the interior, in particular Phe343 and Phe365 of TLR6; the corresponding residues in TLR1 are Met and Leu. Additionally, as revealed by this work, the LRR11 loop of TLR6 may not be flexible enough to open up and allow ligand binding.
Diprovocim has shown significant potential as an immune adjuvant, having been effectively applied in both prophylactic and therapeutic anticancer vaccination in mice.25 Further studies will examine Diprovocim as an adjuvant in infectious disease vaccination. The high-affinity binding of Diprovocim to the TLR2 ectodomain suggests that a TLR2 antagonist might also be created by generating a half Diprovocim molecule, which would bind to a TLR2 monomer. Indeed, one such compound, YM1–128-1, containing only half of the Diprovocim molecule, has been shown to antagonize both Diprovocim and Pam3CSK4 activation of TLR2/TLR1-induced signaling (Figure S2, SI). These studies will be facilitated by the ease of synthesizing and modifying Diprovocim.19
EXPERIMENTAL SECTION
Expression and Purification of Human TLR1 and TLR2.
The hybrid construct of human TLR1 (residues 1–475) fused with hagfish variable lymphocyte receptor (VLR) (residues 133–200) was cloned into plasmid pVL1393 with a protein A tag at the C-terminus. The hybrid construct of human TLR2 (27–506) fused with hagfish VLR (residues 133–200) was cloned into plasmid pAcGP67a with a protein A tag at the C-terminus.26 Both hybrid constructs of human TLR1 and TLR2 were expressed separately in Hi5 insect cells (Invitrogen) and purified by IgG Sepharose (GE Healthcare) affinity chromatography. The eluted hTLR1-protein A and hTLR2-protein A were buffer-exchanged into a buffer containing 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, pH 7.5) and 100 mM NaCl and mixed with peptide/N-glycosidase F (New England Biolabs) to partially deglycosylate the proteins at room temperature for 3 h. Thrombin was added and the reaction mixture was incubated at 4 °C overnight to allow protein A cleavage. The tag-free hTLR1 and hTLR2 were further purified by ion exchange (HiTrap Q) and gel filtration (Superdex 200 16/60) chromatography. The final protein buffer contained 25 mM HEPES (pH 8.0) and 150 mM NaCl (buffer A).
For analytical gel filtration studies, 1 μM purified hTLR1, hTLR2, or hTLR1 and hTLR2 were incubated with 30 μM Diprovocim or 30 μM Pam3CSK4 in a buffer containing 25 mM HEPES (pH 8.0), 150 mM NaCl, and 10% dimethyl sulfoxide (DMSO) at room temperature for 4 h. The mixtures were then subjected to gel filtration chromatography (Superdex 200 10/300) to analyze dimer formation induced by Diprovocim or Pam3CSK4.
Crystal Data Collection and Structure Determination.
The hTLR2/Diprovocim crystals were obtained in a hanging drop vapor diffusion setting. A reconstituted hTLR2/Diprovocim complex (13.4 mg/mL in buffer A) was mixed in equal volume with the reservoir solution containing 0.2 M ammonium citrate tribasic (pH 7.0), 0.1 M imidazole (pH 7.5), and 18% poly(ethylene glycol) (PEG)1900 monomethyl ether (MME). The crystals were cryoprotected in 0.2 M ammonium citrate tribasic (pH 7.0), 0.1 M imidazole (pH 7.5), 0.1 M NaCl, and 38% PEG1900 MME before flash-freezing in liquid nitrogen.
The apo hTLR1 crystals were obtained from one of the conditions in the JCSG+ screening kit containing 0.1 M Bis–Tris (pH 5.5), 0.2 M magnesium chloride, and 25% w/v PEG3350. The concentration of hTLR1 was 20 mg/mL in buffer A. Crystals were cryoprotected in 0.1 M Bis–Tris (pH 5.5), 0.2 M magnesium chloride, 0.1 M NaCl, 2% DMSO, and 40% w/v PEG 3350 before flash-freezing in liquid nitrogen.
X-ray diffraction data were collected at Beamlines BM19 and ID19 of Advance Photon Source, Argonne National Laboratory. The data were indexed, integrated, and scaled using the HKL3000 package.31 The initial phases for the hTLR2/Diprovocim complex and hTLR1 were determined by the molecular replacement method using the program PHASER.32 The published human TLR2/TLR1/Pam3CSK4 structure (PDB 2Z7X)26 was used as a search model for structure determinations. The electron densities for Diprovocim were evident from the early stages of refinement, and the atomic model of Diprovocim was built into the electron density map unambiguously. The manual model building was performed with COOT,33 and the crystallographic refinement was performed with Refmac534 and Phenix.Refine.35 The data collection and refinement statistics for the two structures are summarized in Table 1. Notably, the B-factors for TLR2 in the TLR2/Diprovocim complex structure are quite high, especially at the N- and C- terminal regions. High B-factors are characteristic for some TLR structures because the conformations of the horseshoe-shaped TLRs are intrinsically flexible. In some cases, the conformations of the receptors in the crystal are stabilized by crystal packing contacts and thus have relatively lower B-factors.
Modeling of Diprovocim Binding to TLR2/TLR1 and Molecular Dynamics Simulation.
Both the structures of TLR2/Diprovocim (this work) and TLR2/TLR1/Pam3CSK4 (PDB 2Z7X)26 were used to construct the complex structure of two Diprovocim molecules bound to the active TLR2/TLR1 heterodimer. Ligand flexible docking was performed for binding of one Diprovocim molecule to the active TLR2/TLR1 heterodimer. Three structure models of the TLR2/TLR1/Diprovocim-1X complex were constructed using the top docking poses that have distinct binding modes. MD simulations were performed for TLR2/TLR1 with either one or two bound Diprovocim molecules. The stability of the MD trajectories was studied first. The receptor/Diprovocim binding was further characterized by calculating the MM-PBSA binding free energies.36–38 Additionally, MM-GBSA free-energy decomposition analyses were performed to identify hotspot residues.29,30 Detailed methods are described in SI.
Mutagenesis.
For reasons currently unknown, overexpression of human TLR2 in cultured cells (293T and SW620) resulted in constitutive activation of innate immune responses in the absence of an added agonist. To overcome this, we generated human TLR(1–2) and human TLR(2–1) chimeras in which the hTLR1 ectodomain (residues 1–579 of hTLR1) was fused with the hTLR2 trans-membrane and intracellular TIR domains (residues 588–784 of hTLR2) to make TLR(1–2), whereas TLR(2–1) consisted of the hTLR2 ectodomain (residues 1–587 of hTLR2) fused with hTLR1 transmembrane and intracellular TIR domains (residues 580–786 of hTLR1). Overexpression of these chimeras in HEK293T cells in the absence of agonists does not activate innate immune responses. Only when both TLR(2–1) and TLR(1–2) are expressed, the cells respond to Pam3CSK4 and Diprovocim in a dose-dependent manner (Figure S3, SI). Therefore, these constructs were used to test the effects of TLR2 and TLR1 ectodomain mutations for their ability to respond to Diprovocim. For simplicity, these chimeric constructs were labeled TLR1 and TLR2 for their ectodomain identities.
Point mutations of human TLR1 and human TLR2 chimeras were introduced by standard site-directed mutagenesis following the QuickChange II site-directed Mutagenesis (Agilent Technologies) protocol. Briefly, the mutagenic primers, the corresponding DNAs encoding human TLR1 and TLR2 chimeras, 10× reaction buffer, dNTP mix, and Pfu Ultra HF DNA polymerase were mixed together and subjected to thermal cycling for 18 cycles. After the PCR amplification, 1 μL of the DpnI restriction enzyme (New England Biolabs) was added to each reaction mixture and incubated at 37 °C for 1 h. The mutated DNAs were then individually transformed into stellar competent cells (Clontech) for nick repair. DNA sequencing was performed to select the correct point mutations of human TLR1 and TLR2 for NF-κB luciferase activity assays.
Luciferase Assays for Innate Immune Activation.
HEK293T cells stably expressing an NF-κKB-driven luciferase reporter were cultured in Dulbecco’s modified Eagle’s medium. Cells were plated in 96-well plates 18 h before transfection at a density of 0.9 × 106/well. Each well of cells was co-transfected with plasmids encoding wild-type (WT) or mutant human TLR1 and TLR2, using Lipofectamine 2000 (Life Technologies). Protein expression was verified by immunoblotting (Figure S4, SI). After 24 h of transfection, cells were stimulated with 100 nM Diprovocim or either 20 or 50 ng/mL Pam3CSK4 for 6 h. Cells were lysed, and luciferase activity was measured using the Steady-Glo Luciferase Assay System (Promega).
Measurement of Cytokine Production.
Cells were seeded onto 96-well plates at 1 × 105 cells per well and stimulated with Diprovocim (dissolved in DMSO, and final DMSO concentrations (≤0.2%) were kept constant in all experiments) for 4 h. Human tumor necrosis factor in the supernatants was measured using an enzyme-linked immunosorbent assay kit according to the manufacturer’s instructions (eBioscience). Pretreatment with YM1–128-1 was for 1 h.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. James Chen and Diana Tomchick for assistance with the X-ray diffraction data collection. This work was supported by NIH grants R01GM104496 (H.Z.), AI125581 (B.B.), CA042056 (D.L.B.), AI082657 (D.L.B., B.B.), and R01 GM079383 (J.W.). This work was also supported by the Lyda Hill foundation (B.B.). The results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02–06CH11357.
ABBREVIATIONS
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- hagfish VLR
hagfish variable lymphocyte receptor
- LRR
leucine-rich repeat
- LTA
lipoteichoic acid
- MM-GBSA
molecular mechanics generalized Born surface area
- MM-PBSA
molecular mechanics Poisson–Boltzmann surface area
- pam2CSK4
dipalmitoylated lipopeptide pam2CysSerLys4
- Pam3CSIK4
tripalmitoylated lipopeptide Pam3CysSerLys4
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.8b01583.
Molecular dynamic simulation and modeling of Diprovocim binding to TLR2/TLR1; RMSDs vs simulation time curves (Figure S1); antagonistic activity of YM1–128-1 (Figure S2); dose dependent response of NF-kB reporter (Figure S3); expression of wild type and mutant TLR1 and TLR2 proteins (Figure S4); initial model of two Diprovocim molecules bound to TLR2/TLR1 (Figure S5); energy components of MM-PBSA binding free energies for different TLR2/TLR1/ligand complex models (Table S1); predicted hydrogen-bond occupancies for different TLR2/1/ligand complex models (Table S2); TLR2 hotspot residues identified by MM-GBSA binding free energy decomposition (Table S3); TLR1 hotspot residues identified by MM-GBSA binding free energy decomposition (Table S4); and references (PDF)
Coordinates of the model of TLR2/TLR1/Diprovocim (PDB)
Accession Codes
The coordinates and structure factors of human TLR2/Diprovocim and apo human TLR1 have been deposited with the Protein Data Bank (PDB) under accession codes 6NIG and 6NIH, respectively. Authors will release the atomic coordinates and experimental data upon article publication.
The authors declare the following competing financial interest(s): B.B., D.L.B., and H.Z. have financial interests in Tollbridge Therapeutics, LLC., which has licensed the patent for Diprovocim.
REFERENCES
- (1).Kang JY; Lee JO Structural biology of the Toll-like receptor family. Annu. Rev. Biochem. 2011, 80, 917–941. [DOI] [PubMed] [Google Scholar]
- (2).Moresco EM; LaVine D; Beutler B Toll-like receptors. Curr. Biol. 2011, 21, R488–R493. [DOI] [PubMed] [Google Scholar]
- (3).Casrouge A; Zhang SY; Eidenschenk C; Jouanguy E; Puel A; Yang K; Alcais A; Picard C; Mahfoufi N; Nicolas N; Lorenzo L; Plancoulaine S; Senechal B; Geissmann F; Tabeta K; Hoebe K; Du X; Miller RL; Heron B; Mignot C; de Villemeur TB; Lebon P; Dulac O; Rozenberg F; Beutler B; Tardieu M; Abel L; Casanova JL Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 2006, 314, 308–312. [DOI] [PubMed] [Google Scholar]
- (4).Zhang SY; Jouanguy E; Ugolini S; Smahi A; Elain G; Romero P; Segal D; Sancho-Shimizu V; Lorenzo L; Puel A; Picard C; Chapgier A; Plancoulaine S; Titeux M; Cognet C; von Bernuth H; Ku CL; Casrouge A; Zhang XX; Barreiro L; Leonard J; Hamilton C; Lebon P; Heron B; Vallee L; Quintana-Murci L; Hovnanian A; Rozenberg F; Vivier E; Geissmann F; Tardieu M; Abel L; Casanova JL TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007, 317, 1522–1527. [DOI] [PubMed] [Google Scholar]
- (5).Picard C; Puel A; Bonnet M; Ku CL; Bustamante J; Yang K; Soudais C; Dupuis S; Feinberg J; Fieschi C; Elbim C; Hitchcock R; Lammas D; Davies G; Al Ghonaium A; Al Rayes H; Al Jumaah S; Al Hajjar S; Al Mohsen IZ; Frayha HH; Rucker R; Hawn TR; Aderem A; Tufenkeji H; Haraguchi S; Day NK; Good RA; Gougerot-Pocidalo MA; Ozinsky A; Casanova JL Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science 2003, 299, 2076–2079. [DOI] [PubMed] [Google Scholar]
- (6).von Bernuth H; Picard C; Jin Z; Pankla R; Xiao H; Ku CL; Chrabieh M; Mustapha IB; Ghandil P; Camcioglu Y; Vasconcelos J; Sirvent N; Guedes M; Vitor AB; Herrero-Mata MJ; Arostegui JI; Rodrigo C; Alsina L; Ruiz-Ortiz E; Juan M; Fortuny C; Yague J; Anton J; Pascal M; Chang HH; Janniere L; Rose Y; Garty BZ; Chapel H; Issekutz A; Marodi L; Rodriguez-Gallego C; Banchereau J; Abel L; Li X; Chaussabel D; Puel A; Casanova JL Pyogenic bacterial infections in humans with MyD88 deficiency. Science 2008, 321, 691–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Döffinger R; Smahi A; Bessia C; Geissmann F; Feinberg J; Durandy A; Bodemer C; Kenwrick S; Dupuis-Girod S; Blanche S; Wood P; Rabia SH; Headon DJ; Overbeek PA; Le DF; Holland SM; Belani K; Kumararatne DS; Fischer A; Shapiro R; Conley ME; Reimund E; Kalhoff H; Abinun M; Munnich A; Israel A; Courtois G; Casanova JL X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat. Genet. 2001, 27, 277–285. [DOI] [PubMed] [Google Scholar]
- (8).Courtois G; Smahi A; Reichenbach J; Doffinger R; Cancrini C; Bonnet M; Puel A; Chable-Bessia C; Yamaoka S; Feinberg J; Dupuis-Girod S; Bodemer C; Livadiotti S; Novelli F; Rossi P; Fischer A; Israel A; Munnich A; Le Deist F; Casanova JL A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J. Clin. Invest. 2003, 112, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Beutler B Neo-ligands for innate immune receptors and the etiology of sterile inflammatory disease. Immunol. Rev. 2007, 220, 113–128. [DOI] [PubMed] [Google Scholar]
- (10).Kanzler H; Barrat FJ; Hessel EM; Coffman RL Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat. Med. 2007, 13, 552–559. [DOI] [PubMed] [Google Scholar]
- (11).Hennessy EJ; Parker AE; O’Neill LA Targeting Toll-like receptors: emerging therapeutics? Nat. Rev. Drug Discovery 2010, 9, 293–307. [DOI] [PubMed] [Google Scholar]
- (12).Connolly DJ; O’Neill LA New developments in Toll-like receptor targeted therapeutics. Curr. Opin. Pharmacol. 2012, 12, 510–518. [DOI] [PubMed] [Google Scholar]
- (13).Chan M; Hayashi T; Mathewson RD; Nour A; Hayashi Y; Yao S; Tawatao RI; Crain B; Tsigelny IF; Kouznetsova VL; Messer K; Pu M; Corr M; Carson DA; Cottam HB Identification of substituted pyrimido[5,4-b]indoles as selective Toll-like receptor 4 ligands. J. Med. Chem. 2013, 56, 4206–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Neve JE; Wijesekera HP; Duffy S; Jenkins ID; Ripper JA; Teague SJ; Campitelli M; Garavelas A; Nikolakopoulos G; Leone P. d. A.; Pham P; Shelton NB; Fraser P; Carroll N; Avery AR; McCrae VM; Williams C; Quinn N; Euodenine RJ A: a small-molecule agonist of human TLR4. J. Med. Chem. 2014, 57, 1252–1275. [DOI] [PubMed] [Google Scholar]
- (15).Fu J; Kanne DB; Leong M; Glickman LH; McWhirter SM; Lemmens E; Mechette K; Leong JJ; Lauer P; Liu W; Sivick KE; Zeng Q; Soares KC; Zheng L; Portnoy DA; Woodward JJ; Pardoll DM; Dubensky TW Jr.; Kim Y STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015, 7, No. 283ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhang L; Dewan V; Yin H Discovery of small molecules as multi-Toll-like receptor agonists with proinflammatory and anticancer Activities. J. Med. Chem. 2017, 60, 5029–5044. [DOI] [PubMed] [Google Scholar]
- (17).Wang Y; Su L; Morin MD; Jones BT; Whitby LR; Surakattula MM; Huang H; Shi H; Choi JH; Wang KW; Moresco EM; Berger M; Zhan X; Zhang H; Boger DL; Beutler B TLR4/MD-2 activation by a synthetic agonist with no similarity to LPS. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, E884–E893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Morin MD; Wang Y; Jones BT; Su L; Surakattula MM; Berger M; Huang H; Beutler EK; Zhang H; Beutler B; Boger DL Discovery and structure-activity relationships of the neoseptins: a new class of Toll-like receptor-4 (TLR4) agonists. J. Med. Chem. 2016, 59, 4812–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Morin MD; Wang Y; Jones BT; Mifune Y; Su L; Shi H; Moresco EMY; Zhang H; Beutler B; Boger DL Diprovocims: a new and exceptionally potent class of toll-like receptor agonists. J. Am. Chem. Soc. 2018, 140, 14440–14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Goldberg J; Jin Q; Ambroise Y; Satoh S; Desharnais J; Capps K; Boger DL Erythropoietin mimetics derived from solution phase combinatorial libraries. J. Am. Chem. Soc. 2002, 124, 544–555. [DOI] [PubMed] [Google Scholar]
- (21).Whitby LR; Boger DL Comprehensive peptidomimetic libraries targeting protein-protein interactions. Acc. Chem. Res. 2012, 45, 1698–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Guan Y; Omueti-Ayoade K; Mutha SK; Hergenrother PJ; Tapping RI Identification of novel synthetic toll-like receptor 2 agonists by high throughput screening. J. Biol. Chem. 2010, 285, 23755–23762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Murgueitio MS; Ebner S; Hortnagl P; Rakers C; Bruckner R; Henneke P; Wolber G; Santos-Sierra S Enhanced immunostimulatory activity of in silico discovered agonists of Toll-like receptor 2 (TLR2). Biochim. Biophys. Acta, Gen. Subj. 2017, 1861, 2680–2689. [DOI] [PubMed] [Google Scholar]
- (24).Cheng K; Gao M; Godfroy JI; Brown PN; Kastelowitz N; Yin H Specific activation of the TLR1-TLR2 heterodimer by small-molecule agonists. Sci. Adv. 2015, 1, No. e1400139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wang Y; Su L; Morin MD; Jones BT; Mifune Y; Shi H; Wang K; Zhan X; Liu A; Wang J; Li X; Tang M; Ludwig S; Hildebrand S; Zhou K; Siegwart D; Moresco EMY; Zhang H; Boger DL; Beutler B Adjuvant effect of the novel TLR1/2 agonist Diprovocim synergizes with anti-PD-L1 to eliminate melanoma in mice. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E8698–E8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jin MS; Kim SE; Heo JY; Lee ME; Kim HM; Paik SG; Lee H; Lee JO Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007, 130, 1071–1082. [DOI] [PubMed] [Google Scholar]
- (27).Kang JY; Nan X; Jin MS; Youn SJ; Ryu YH; Mah S; Han SH; Lee H; Paik SG; Lee JO Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 2009, 31, 873–884. [DOI] [PubMed] [Google Scholar]
- (28).Wang J; Hou T; Xu X Recent advances in free energy calculations with a combination of molecular mechanics and continuum models. Curr. Comput.-Aided Drug Des. 2006, 2, 287–306. [Google Scholar]
- (29).Hou T; Wang J; Li Y; Wang W Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hou T; Wang J; Li Y; Wang W Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Minor W; Cymborowski M; Otwinowski Z; Chruszcz M HKL-3000: the integration of data reduction and structure solution-from diffraction images to an initial model in minutes. Acta Crystallogr.,Sect. D: Biol. Crystallogr. 2006, 62, 859–866. [DOI] [PubMed] [Google Scholar]
- (32).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read R J. Phaser crystallographic software. J. Appl Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Murshudov GN; Vagin AA; Dodson EJ Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- (35).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive Python-based system for macro-molecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kollman PA; Massova I; Reyes C; Kuhn B; Huo S; Chong L; Lee M; Lee T; Duan Y; Wang W; Donini O; Cieplak P; Srinivasan J; Case DA; Cheatham TE 3rd Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [DOI] [PubMed] [Google Scholar]
- (37).Wang J; Morin P; Wang W; Kollman PA Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J. Am. Chem. Soc. 2001, 123, 5221–5230. [DOI] [PubMed] [Google Scholar]
- (38).Wang J; Kang X; Kuntz ID; Kollman PA Hierarchical database screenings for HIV-1 reverse transcriptase using a pharmacophore model, rigid docking, solvation docking, and MM-PB/SA. J. Med. Chem. 2005, 48, 2432–2444. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.