

Abstract
Recombinant adeno-associated viruses (AAVs) are promising vectors for human gene therapy. However, current methods for evaluating AAV particle populations and vector purity are inefficient and low resolution. Here, we show that charge detection mass spectrometry (CDMS) can resolve capsids that contain the entire vector genome from those that contain partial genomes and from empty capsids. Measurements were performed for both single-stranded and self-complementary genomes. The self-complementary AAV vector preparation appears to contain particles with partially truncated genomes averaging at half the genome length. Comparison to results from electron microscopy with manual particle counting shows that CDMS has no significant mass discrimination in the relevant mass range (after a correction for the ion velocity is taken into account). Empty AAV capsids are intrinsically heterogeneous, and capsids from different sources have slightly different masses. However, the average masses of both the empty and full capsids are in close agreement with expected values. Mass differences between the empty and full capsids for both single-stranded and self-complementary AAV vectors indicate that the genomes are largely packaged without counterions.
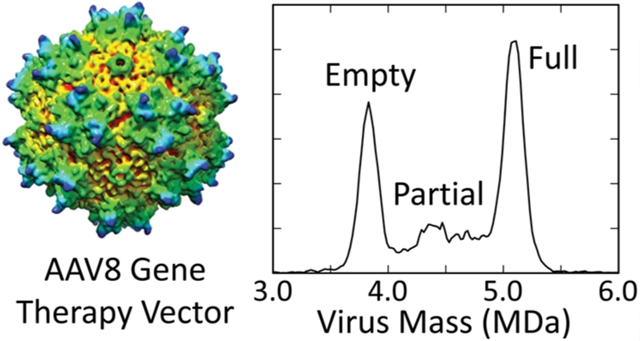
Graphical Abstract
There has been growing interest in using native mass spectrometry to investigate protein complexes and other assemblies including viruses, with masses into the MDa range.1–4 However, there are challenges associated with the mass analysis of such large objects. The main issue is that the peaks in the m/z spectrum broaden and shift due to mass heterogeneity, either intrinsic or due to adduct formation. Poorly resolved peaks in the m/z spectrum prevent charge state assignment and subsequent mass deduction. In particular, viruses have a proclivity for being heterogeneous in mass because they have the ability to encapsidate varying amounts of genetic material. Earlier studies demonstrated the feasibility of using time-of-flight mass spectrometry to measure the m/z spectrum of the ~2.5 MDa bacteriophage MS2 capsid, albeit without sufficient charge state resolution to calculate an accurate mass.5 More recently, high resolution m/z spectra of empty hepatitis B virus (HBV) capsids assembled from proteins lacking the C-terminal RNA-binding domain have been reported.6 However, the m/z spectrum for HBV assembled from the full-length capsid protein lacked charge state resolution due to heterogeneity in the amount of packaged RNA.7 For human rhino virus, incipient charge state resolution was obtained for the empty HRV-A2 capsid allowing a mass (5,210 kDa) to be assigned. On the other hand, for wild type HRV-A2 (with RNA), charge state resolution was absent, so its mass could not be determined from the m/z spectrum.8
Charge detection mass spectrometry (CDMS) bypasses the need to resolve charge states. CDMS is a single particle technique, where the m/z and z of individual ions are measured concurrently, thereby allowing direct determination of the mass of each ion.9–14 CDMS can analyze heterogeneous mixtures of protein complexes and other large assemblies that are intractable by conventional MS methods.15–17 In the studies reported here we have used CDMS to determine the DNA content of recombinant adeno-associated viral (AAV) vectors.
AAV vectors have emerged at the forefront of gene therapy due to their lack of pathogenicity, relatively low immunogenicity and persistent gene expression in different tissue types.18 From a structural perspective, this helper-dependent parvovirus has a nonenveloped, icosahedral capsid ~25 nm in diameter that packages a single-stranded DNA (ssDNA) genome ~4.7kb in length.19 Despite promising outcomes, a recurring concern noted in hemophilia gene therapy clinical trials is the potential for vector dose-related immunotoxicity in patients.20 Although resolvable by administration of anti-inflammatory steroids such as methyl prednisolone, several studies have indicated that the composition of clinical AAV vector preparations can influence these outcomes.21,22 In this regard, it is well-known that recombinant AAV vector preparations can contain different levels of full or partial genome-containing particles as well as empty capsids.23,24 Such particle diversity can be attributed to multiple factors such as genome packaging efficiency, production methods, downstream purification techniques and storage conditions.21,24
Though AAV packages ssDNA, the use of a self-complementary (sc) DNA genome bypasses the rate-limiting second-strand synthesis process and leads to more efficient and rapid onset of transgene expression.25–27 scDNA is formed by joining two sequence-inverted vectors by a hairpin, so the effective length of the unique transgene sequence is halved. scDNA is packaged in a single-stranded form, but upon release into the host cell, the two sequence-inverted regions can undergo intramolecular base pairing and anneal into a base-paired form. Though scAAV vectors show promise in the clinic, characterization of their heterogeneous packaging behavior remains a challenge due to the potential for contamination from vectors packaging fragmented or truncated genomes.28
Currently, electron microcopy (EM) and qPCR are used in combination to characterize the ultrastructural composition of AAV vector preparations. While EM can easily distinguish empty capsids from particles containing a full genome, it is much more challenging to characterize particles containing partial or truncated genomes. In particular, EM cannot accurately determine the size of partial genomes. It is also important to note that current quantitative PCR-based methods, although robust and accurate, cannot help distinguish partial or truncated vector genomes from fully packaged genomes.29,30 Thus, the development of cutting edge methods that can help analyze ultrastructural heterogeneity in recombinant AAV vector preparations at high resolution is an unmet need in the gene therapy field. Other techniques, such as fluorescence-based thermal shift assays and dynamic light scattering, as well as analytical ultracentrifugation, are being explored as low-resolution techniques for the characterization of recombinant AAV vectors.28 Here, we report proof of principle studies showing that charge detection mass spectrometry can be used to analyze AAV vectors (packaging both single-stranded and self-complementary genomes). We show that CDMS can quantitatively characterize diverse AAV particle populations including particles packaging the complete genome, empty particles, particles packing partial genomes, and particles with impurities. A number of different naturally occurring AAV serotypes have been described.31–33 They differ primarily in the surface properties of the capsid and show tropism for different tissue types. The studies described here were performed with AAV serotype 8 (AAV8).
METHODS
AAV Vector preparations.
All recombinant AAV vectors were generated at the UNC Vector Core Facility. AAV8 vectors packaging an ssDNA genome with a chicken beta-actin (CBA) promoter driving a firefly luciferase transgene (Luc) or a scDNA genome with a hybrid chicken beta-actin (CBh) promoter driving a green fluorescent protein (GFP) transgene flanked by AAV2 inverted terminal repeats (ITRs) from 3 to 4 different production runs were individually purified using a discontinuous iodixanol gradient, followed by ion exchange chromatography.34,35 Titers were obtained by qPCR with primers specific for the Luc transgene. Separate fractions of purified empty and genome-containing capsids were further dialyzed into 100 mM ammonium acetate for CDMS analysis.
Charge Detection Mass Spectrometry.
Mass analysis was performed using a home-built charge detection mass spectrometer that has been described previously.36–40 AAV ions were generated by nanoelectrospray using an automated TriVersa NanoMate system (Advion Inc., Ithaca, NY) and transported into the vacuum chamber through a heated stainless-steel capillary. The ions were separated from the background gas by passing them through three differentially pumped regions containing an RF ion funnel, an RF hexapole, and an RF quadrupole, respectively. They are thermalized by collisional cooling in the hexapole region. A DC voltage on the hexapole reaccelerates them to ~100 eV per charge (z) before they enter a fourth differentially pumped region where they were focused into the entrance of a dual hemispherical deflection analyzer (HDA). The dual HDA passes a narrow band of ion kinetic energies (centered on 100 eV/z) into the fifth differentially pumped region that contains an electrostatic ion trap with a cylindrical charge detection tube. With each oscillation in the trap, the ion passes through the detector tube. The induced charge is detected by a cryogenically cooled JFET (2SK152) and then amplified with a charge-sensitive preamplifier (Amptek A250). The periodic signals resulting from ion oscillation are digitized and sent to a computer for offline analysis using fast Fourier transforms. The oscillation frequency of the ions is related to the m/z and the magnitude of the fundamental is proportional to the ion charge. Charge and m/z of individual ions are multiplied to give m. Only ions that remain trapped for the entire trapping event (94 ms in this study) are compiled and binned to create a mass histogram.
The uncertainty in the charge measurement in CDMS is a function of the trapping time and oscillation frequency (and hence the m/z). Here, we used a trapping time of 94 ms, which leads to a charge uncertainty (RMSD) of ~1.2 e for an AAV capsid with an average m/z of ~24 000 Da. For an average charge of ~155 e the relative uncertainty in the charge is ~0.8%. The relative uncertainty in the m/z measurement (~0.8%) depends primarily on the ions’ kinetic energy distribution. Typically, masses for 2–3 thousand ions are required to characterize a sample and these measurements can usually be performed in around 2 h with the nanomolar particle concentrations employed here.
Electron Microscopy.
300-μm mesh carbon-coated copper transmission electron microscopy grids were prepared by spotting ~3 uL of AAV8 (1012 vg/mL) on the entire grid area for ~1 min. Grids were washed with HPLC-grade water and subsequently stained with 2% uranyl acetate for 1 min. After drying, grids were imaged using a JEOL 1010 transmission electron microscope. At least 2 images for each grid were taken for qualitative analysis and up to 12 images of each grid were taken for quantitative analysis.
RESULTS AND DISCUSSION
The AAV capsid has icosahedral symmetry and it is assembled from 60 copies of the capsid viral protein (VP). There are three different VPs: VP1, VP2, and VP3. VP1 is the longest, and for AAV8 it consists of 738 residues. The VP2 sequence is identical to VP1 except that 137 residues are truncated from the N-terminus. In a similar way, the VP3 sequence is identical to VP2 except that 66 residues are truncated from the N-terminus. The numbers of copies of VP1, 2, and 3 in each capsid are stochastic with an average ratio of around 1:1:10 (i.e., the numbers of VP1, 2, and 3 are random with an average of five copies of VP1, five copies of VP2, and 50 copies of VP3, and a total of 60 VPs altogether).41–46 The masses of AAV8 VP1, 2, and 3, determined from their sequences, are 81,624, 66,649, and 59 762 Da,47–49 respectively, so the average mass of the empty capsid is expected to be 3.729 MDa. Note that there will be a distribution of masses due to variations in the numbers of VP1, 2, and 3 in each capsid. We consider this distribution further below.
In the preceding paragraph we determined the expected mass for the empty AAV8 capsid, we now compare this to the measured mass distribution for empty capsids. Figure 1a shows a typical CDMS mass spectrum for empty capsids separated from AAV8 vectors with a CBA-Luc genome. There is a large peak centered on ~3.8 MDa and a small high mass tail. CDMS is a single particle technique and so the charge and mass can be correlated. The points that overlay the main peak in Figure 1a are a scatter plot of the charge and mass of each ion. The charge distribution for the main peak shows a single component centered on ~155 elementary charges (e).
Figure 1.

a) Shows the CDMS mass histogram measured for empty capsids separated from AAV8 vectors with a CBA-Luc genome. The bin width is 20 kDa. The inset shows an expanded view of the measured peak (black line). The red line in the inset shows the calculated histogram for homogeneous capsids consisting of only VP3 (60 copies). The width of this peak is due to the experimental uncertainty in the m/z and charge measurements (see text). The blue line in the inset shows the calculated histogram for capsids with a stochastic distribution of VP1, 2, and 3 in the expected 1:1:10 ratio. The black points in a) are a scatter plot of each ion’s charge and mass for ions with masses greater than 3.4 MDa. There is a single charge distribution centered around 155 elementary charges (e). b) Shows the masses and abundances that result for a stochastic distribution of VP1, 2, and 3 in the expected 1:1:10 ratio. The separation between the peaks in this spectrum is too small for the peaks to be resolved in the measured histogram in a).
The inset in Figure 1a shows an expanded view of the measured peak (black line). The red line shows a simulation of the peak expected for a homogeneous sample of capsids consisting of only VP3 (the lightest VP). This illustrates the peak shape expected for a capsid with a single mass. The width of the peak, ~ 97 kDa full width at half-maximum (fwhm), results from the uncertainties in the m/z and z measurements, which are well characterized (see Methods section).36–40 Combining the relative uncertainties from the charge and m/z leads to an overall relative uncertainty in the mass of ~1.1%. Note that this is the uncertainty associated with a single mass measurement. For many independent mass measurements the uncertainty leads to the peak width. The average mass (the center of the peak) can be defined more accurately than the uncertainty associated with a single mass measurement.
The blue line in the inset in Figure 1a shows the simulated peak for a stochastic mixture of VP1, 2, and 3 in the expected 1:1:10 ratio. To calculate this peak, we first determined the masses and abundances of all possible VP1, 2, and 3 compositions from a multinomial distribution. The resulting mass distribution is shown in Figure 1b. The peaks in Figure 1b are too close in mass to be resolved by CDMS with the conditions employed here. When we account for the uncertainties in the m/z and z measurements we obtain the blue Gaussian peak in the inset of Figure 1a. The peak is centered on 3.729 MDa, and it is around 152 kDa wide (fwhm). It is considerably broader than the peak for the homogeneous sample (red line) because of the distribution of VP1, 2, and 3 compositions that are present.
The heterogeneous mass distribution in Figure 1b is a challenge to analyze by conventional mass spectrometry because each peak in the mass distribution would lead to a number of overlapping peaks in the m/z spectrum (due to ions in different charge states). However, Heck and co-workers have made some progress in analyzing the m/z spectrum of AAV1 albeit with a sample that is less heterogeneous than the expected 1:1:10 ratio and depleted in VP1.50 It is likely that different expression systems yield AAV particles with different compositions. For instance, some AAV capsid types with reduced levels of VP1 have been reported in first generation insect cell production systems.51–53 Newer generation systems have altered the composition of AAV capsids to include increased VP1 content, further supporting the notion that the heterogeneity of AAV capsids can be modulated.53 However, even for the sample with the reduced heterogeneity, overlapping charge states hindered the analysis of the m/z spectrum, and it was only possible to identify a few capsid compositions.
The measured peak in the empty AAV8 CDMS mass distribution (the black line in Figure 1a) is broader than the simulated peak for the 1:1:10 ratio and shifted to a higher mass. From four independent measurements performed on different days, the average mass is 3.819 ± 0.016 MDa (versus 3.729 MDa for the simulated peak) and the average width is 197 ± 8 kDa (versus 152 kDa for the simulated peak). The expected and measured masses and peak widths are collected together in Table 1. The difference between the measured mass and the expected mass (2.4%) is too large to be attributed to a systematic error in the mass measurement. A plausible explanations for the shift and the increased peak width is heterogeneity from salt adducts or incomplete desolvation. However, in our previous studies of hepatitis B virus capsids15 and woodchuck hepatitis virus capsids17 the peak widths were close to the values expected for a homogeneous sample, which suggests that this explanation is not the main cause of the discrepancies here. The shift and increased width could be due to a sample that is enriched in VP1 and VP2 or more likely due to the incorporation of small genomic DNA fragments which are known to be present during assembly.54
Table 1.
Compilation of the Expected and Measured Masses and Peak Full Width at Half Maximum (fwhm) (See Text)a
| expected mass (MDa) | measured mass (MDa) | difference (kDa) | expected fwhm (kDa) | measured fwhm (kDa) | |
|---|---|---|---|---|---|
| 60 copies AAV8 VP3 | 3.586b | – | – | 97 | – |
| AAV8 Capsidc | 3.729b | 3.819 ± 16 | +90 | 152 | 195 ± 8 |
| CBA-Luc genome | 1.269d | 1.261 ± 21 | −8 | – | – |
| AAV8/CBA-Lucc | 4.998 | 5.080 ± 13 | +82 | 174 | 208 ± 7 |
| AAV8 Capsidc | 3.729b | 3.704 ± 13 | −25 | 152 | 180 ± 5 155 ± 11e |
| scGFP Genome | 1.389d | 1.392 ± 13 | +3 | – | – |
| AAV8/scGFPc | 5.118 | 5.095 ± 25 | −23 | 177 | 172 ± 15 |
Empty AAV8 capsids were measured after separation from CBA-Luc-containing capsids and from a sample which packaged scGFP. The measurements for these two samples of empty capsids are grouped together with the appropriate genome in the table.
See text for masses of VP1, VP2, and VP3.
For a stochastic mixture of VP1, VP2, and VP3 in an average ratio of 1:1:10.
Sequence mass calculated using http://www.bioinformatics.org/sms2/dna_mw.html.
For the higher charge cluster of empty capsids in Figure 5.
Above, we described mass measurements for empty AAV8 particles. We now turn to consider the more complex scenario of genome containing particles. Figure 2a shows an example of the normalized mass histogram recorded for recombinant AAV8 vectors that have packaged a single-stranded CBA-Luc genome. The mass histogram measured for empty capsids (blue line) is overlaid. The spectrum for the genome-containing particles contains a major peak at around 5.1 MDa as well as a broad distribution extending down to roughly 4.0 MDa. The average mass of the 5.1 MDa peak is 5.080 ± 0.013 MDa (see Table 1). The width of the peak is 208 ± 7 kDa. The expected width is 174 kDa, so while the measured peak is slightly broader than the measured peak for the empty capsids, it is slightly narrower when compared to the expected peak width (120% versus 130%).
Figure 2.

(a) Mass histograms of separated empty (blue) and genome-containing (black) AAV8 vectors packaging a ssDNA CBA-Luc cassette. The histograms have been intensity normalized for comparison. The bin widths used to generate the histograms are 20 kDa. (b and c) TEM images of the corresponding empty and full capsids, respectively (scale bars = 100 nm).
The difference between the measured masses of the empty particles and the genome containing particles is 1.261 ± 0.021 MDa. The expected mass of the CBA-Luc ssDNA genome with flanking inverted terminal repeats (ITRs) is 1.269 MDa (see Table 1). Thus, the 5.1 MDa peak is attributed to capsids that have packaged the full length vector genome. The broad distribution of ions below 5 MDa most likely highlights the presence of capsids that have packaged partial ssDNA genomes. Thus, CDMS easily resolves empty and genome-containing particles. In addition, CDMS can differentiate particles that have packaged partial lengths of DNA. These results are consistent with cryo-EM analysis of AAV1 particles, which revealed that as many as four distinct capsid structures representing empty, partial and full virions can coexist in a single AAV preparation.55
The results described above are also corroborated by transmission electron micrographs of the empty capsids and recombinant AAV8 vector preparations shown in parts b and c of Figure 2, respectively. In Figure 2b, the donut-like structures result from stain entering the empty capsids, while in Figure 2c the bright uniform structures result from capsids that have encapsidated the full genome while those with darkened interiors (bottom right-hand corner of Figure 2c) are attributable to capsids with a partial genome.
The difference between the measured masses of the empty particles and the genome-containing particles (1.261 MDa) is slightly less than the expected mass of the genome (1.269 MDa). The average mass of the empty particles is larger than expected and if the excess mass is partly due to the incorporation of small DNA fragments, then they would be expected to be expelled when the genome is packaged, leading to a smaller difference between the measured masses of the empty particles and the genome-containing ones. The fact that the mass distribution for the full particles is slightly less heterogeneous than the empty ones is consistent with this expulsion.
The molar mass of the genome was calculated assuming that the backbone phosphates are un-ionized. In solution, the phosphates are expected to ionize and neutrality to be retained through counterions. The phosphate groups in the DNA backbone are known to have a strong affinity for Na+. If all the H+ ions associated with the backbone phosphates were replaced by Na+ ions, the mass of the CBA-Luc genome would increase by 90 kDa or 7.1%. The fact that the measured mass is close to the mass expected for the genome with un-ionized backbone phosphates suggests that the degree of incorporation of Na+, or any other counterion, is small. Although we electrosprayed from a low sodium buffer, the particles were not assembled in a low sodium environment. Replacement of Na+ ions by H+ seems unlikely to occur once the particles are assembled. It is also possible that some of the charge on the phosphate groups is compensated by interactions with basic residues on the capsid’s interior surface. However, there are not enough basic residues on the interior surface to interact with all the phosphate groups on the DNA. Estimating the number of basic residues on the capsid interior is complicated by the fact that only residues in VP3, and the C-termini of VP1 and VP2 that are shared with VP3, were resolved in the crystal structure of the AAV8 capsid.56 Assuming that the unresolved N-termini of VP1 and VP2 are completely inside the capsid, there would still only be 720 basic residues available (according to the Φ–Ψ Explorer in VIPERdb). Even with this most favorable count, the number of basic residues is less than one-fifth of the number needed to compensate for the backbone phosphates. Thus, these interactions cannot explain the absence of counterions.
To examine how well CDMS can detect compositional heterogeneity in AAV samples, stock solutions of empty and genome-containing capsids were mixed in a number of volume ratios and CDMS spectra recorded. Typical results for mixtures with empty/genome containing ratios of 1:0, 1:1, 1:15, and 1:100 are shown in Figure 3a. Figure 3b shows the fraction of empty particles determined from the CDMS spectra plotted against the volume fraction of empty capsid solution used in the mixture. The relationship between the fraction of empties determined by CDMS, FCDMS, and the volume fraction, FV, is
| (1) |
where A is the concentration ratio of the solutions of empty and full capsids. Note that if the solutions had equal concentrations, A = 1 and the equation simplifies to the linear form, FCDMS = FV. The blue line is the fit to the data where A = 5.84, indicating that the empty solution is 5.84 times more concentrated than the full solution.
Figure 3.

Empty and genome containing capsid solutions mixed in ratios of 1:0, 1:1, 1:15, and 1:100 v/v and analyzed by CDMS. Individual CDMS histograms are stacked in panel a. The fraction of empty capsids determined by CDMS is plotted against the volume fraction of the empty capsid solution in panel b. The blue line (see text) is the best fit to the data using eq 1 (see text). A representative micrograph of the 1:15 v/v empty/genome containing mixture is shown in panel c. The image shows empty capsids (blue arrow), full capsids (red arrow), and partially filled capsids (white arrow) (scale bar =100 nm).
To examine how well CDMS can quantify the different types of AAV particles, a 1:15 mixture of the stock solutions of empty and genome-containing capsids was spotted onto EM grids for analysis by image counting. A representative portion of a transmission electron micrograph of the 1:15 mixture is shown in Figure 3c. Because of the ambiguity in assigning capsid types, eight subjects each counted over 700 particles from 10 different images. Subjects were asked to classify the particles as empty (donut-like appearance), full (bright, uniform appearance), or ambiguous (lower contrast or differential staining pattern). As shown in Table 2, 43% of the counted particles were deemed to contain full cargo, 30% empty capsids, and 27% ambiguous.
Table 2.
Comparison of the AAV Particle Types Determined by Electron Microscopy with Manual Image Counting (EM) and Charge Detection Mass Spectrometry (CDMS)a
| full | ambiguous/partial | empty | |
|---|---|---|---|
| EM | 0.43 (±0.16) | 0.27 (±0.15) | 0.30 (±0.16) |
| CDMS | 0.421 (±0.004) | 0.288 (±0.006) | 0.291 (±0.002) |
The numbers indicate the fractions of the different particle types in the 1:15 v/v mixed sample (see text). Numbers in parentheses are standard deviations for multiple analyses of the same data set.
The assignment of a particle to a particular group depends on the incorporation of stain. To be assigned “empty”, the particle must acquire enough stain to attain a donut appearance. Empty particles that acquire less stain are classified as ambiguous or even full. Thus, the number of empty particles may be underestimated. The ambiguous particles could be understained empty particles. On the other hand, the CDMS measurements show that there are a significant number of partially filled particles, and so the ambiguous particles could also be stained partially filled particles. A bright uniform appearance suggests a full particle; however, this appearance could also be due to particles that are partially full (or empty) that have not picked up enough stain. Thus, the number of full particles may be overestimated.
For comparison, a single CDMS histogram of the same empty/full capsid mixture used for EM analysis was fit with a series of Gaussians with widths corresponding to that of the empty peak. The areas underneath the Gaussians were used to determine the relative abundances of each particle type. Any Gaussian not associated with the empty or full capsids were assigned as ambiguous or to capsids packaging partial genomes. As shown in Table 2, 42% of the detected ions had a mass that corresponded to the capsid plus the full-length genome; 29% of ions had a mass between that of an empty and full particle; and 29% of ions had a mass corresponding to an empty capsid.
It is evident from Table 2 that the relative abundances determined by CDMS are in good agreement with those obtained from EM particle counting. The values in parentheses in Table 2 are the standard deviation from multiple analyses of the same data set. The standard deviations for the EM-derived data are large and reflect the high degree of subjectivity associated with manually classifying particle types. On the other hand, CDMS is able to unambiguously differentiate between empty particles, particles containing a partial genome, and particles containing the full genome. The measurement time for CDMS was around 2 h, which is longer than it takes to spot grids and measure EM images. On the other hand, it takes a few minutes to generate a mass histogram from the CDMS data, while the manual particle counting of multiple EM images we performed to generate the data given in Table 2 took more than an hour per person. However, the processing of the EM images could be automated, and this would dramatically reduce the time needed to process the images.
There are two main sources of discrimination associated with ion detection in CDMS. First, for ions that carry a small charge there is the possibility that the signal is lost in the noise. However, with the instrument and data analysis scheme employed here the detection efficiency is expected to be 100% for trapped ions with more than 10 charges.39 Therefore, low detection efficiency is not expected to be an issue here, where the ions usually have more than 100 charges. The second source of discrimination is that the probability of being trapped depends on the ion velocity. Faster ions spend less time in the trap and hence they have a lower probability of being trapped. Because the ion energy is known and constant, it is straightforward to correct for mass discrimination resulting from different ion velocities, the ions are weighted by the square root of their m/z (which is directly measured for each ion). This correction has been included in Table 2. The good agreement between the particle counting and CDMS values in Table 2 points to a lack of significant mass discrimination in the relevant mass range, after the correction for the ion velocity has been taken into account. Since the ion velocity is inversely proportional to the square root of the m/z, the detection efficiency for light ions is less than for heavy ions in the continuous trapping mode used here. This is usually beneficial because light ions are generally much more abundant than heavy.
For comparison with the results presented above for the single-stranded genome we used CDMS to analyze AAV particles containing a self-complementary genome, in this case scGFP, which is 4.174 kb long. Figure 4a shows a representative CDMS mass histogram. There are four main features: two narrow, intense peaks at ~3.7 and ~5.1 MDa; a comparably intense but broader distribution between 4 and 5 MDa; and a small peak at ~5.6 MDa. The peak at ~3.7 MDa (average mass 3.704 ± 0.013 MDa) is close to the mass expected for the empty capsid (3.729 MDa, see Table 1). In this case, the average mass is slightly less than the measured mass. The difference is small and could be due to the amount of VP1 and VP2 in this sample being slightly less than in the expected 1:1:10 ratio. The average mass of the capsid separated from the CBA-Luc-containing capsids was 3.819 MDa, so these results confirm that AAV capsids from different sources can have slightly different masses.
Figure 4.

(a) Mass histogram for AAV8 packaging an scGFP genome and (b) electron micrograph of the same sample (scale bar = 200 nm).
The average mass difference between the two major peaks in Figure 4a is 1.392 ± 0.013 MDa. This is in good agreement with the sequence mass of the scDNA (1.389 MDa), so the peak near 5.1 MDa is assigned to the capsid with the full sc genome. A representative micrograph (Figure 4b) is shown below the mass histogram. The image confirms the presence of empty and full particles. The sequence mass for the scDNA given above was calculated for un-ionized DNA so the close agreement with the measured genome mass (the differences between the masses of the empty and full particles) indicates that the scDNA is mainly packaged without counterions. We reached a similar conclusion for the ssDNA genome discussed above.
The broad distribution between 4 and 5 MDa in Figure 4a is attributed to partially filled capsids. The peak in this distribution occurs roughly halfway between the peaks due to the empty and full capsids indicating a propensity to package truncated genomes averaging approximately half of the full genome length. This indicates a preference for cleaving the genome near the hairpin that joins the two self-complementary segments. The peak attributed to packaging of a partial genome is broad indicating that there is some variability in where the genome is cleaved relative to the hairpin. In contrast, for single-stranded CBA-Luc (Figure 2a), the distribution of partially packaged genomes is broader and does not show a preference for packaging half the genome.
In Figure 4a, approximately 35% of the capsids are empty, 23% packaged the full genome, and nearly 42% of the capsids have masses that lie between the empty and full capsids. We found that the amount of monomeric and self-complementary genomes packaged can vary significantly for different samples with as little as 5% of the capsids containing the full genome in one case.
A scatter plot of z vs m for the scGFP-filled capsids is shown in Figure 5a. Each point represents an ion. There are clusters of ions associated with empty (~3.7 MDa), partially full (4–5 MDa), and full capsids (~5.1 MDa). There are two clusters for the empty particles: the main cluster centered at ~155 e and a more-diffuse cluster centered at ~135 e. Note that the lower-charge cluster is slightly heavier, on average, than the higher-charge cluster. Similar lower-charge clusters of ions are absent for the partially filled and full capsids. The higher-charge cluster of empty particles and the clusters due to the partially full and the full capsids have similar average charges that increase slightly with mass (from ~155 to to ~160 e). Finally, the small cluster of ions near 5.6 MDa has a higher average charge of ~195 e.
Figure 5.

(a) Charge versus mass scatter plot for AAV8 packaging an scDNA genome. The red diagonal lines are lines of constant m/z and (b) m/z histogram for AAV8 packaging an scDNA genome.
The mechanism by which ions are produced by electrospray depends on their size. The large ions studied here are expected to be generated by the charge residue mechanism.57,58 Here, the water droplet containing the analyte evaporates away, leaving the ion with a charge close to the Rayleigh limit for a water droplet with the same radius as the analyte. Inspection of Figure 5 shows that the main clusters of ions associated with the empty, partial, and full capsids have similar charges indicating that the genome and partial genome are packaged inside the capsid. If some of the genome extended outside the capsid the charges would be higher. Indeed this might provide an explanation for the tail of highly charged ions that extend from the cluster due to the full particles (see Figure 5a).
There are two distinct charge state distributions for the empty capsids in Figure 5. This behavior has been observed for other empty capsids by CDMS where it was attributed to the compaction of some of the capsids during the electrospray process.17 The absence of low charge clusters for the partial and full capsids in Figure 5 can be attributed to the presence of the genome, which presumably helps the capsids resist compaction.59 Evidently, even a partial genome is enough to prevent compaction. A low charge cluster of ions was also not observed in the charge versus mass scatter plot for the empty particles separated from AAV8 vectors with a CBA-Luc genome (Figure 1). The scatter plot for these ions shows a single cluster centered at ~155 e. However, the masses of the separated AAV8 particles are significantly larger than expected for empty particles (see Table 1), leading to the suggestion that they may contain some small DNA fragments.
The small peak at ~5.55 MDa in Figure 4a is probably due to an impurity. If it is attached to the full capsid then its mass is around 416 ± 25 kDa. The fact that the ions in this peak have significantly higher average charge than the full particles (see Figure 5) suggests that the impurity is outside the capsid where it increases the average radius of the ion. Note that there is no evidence for the same impurity attached to the outside of the empty capsid. Such a low abundance impurity attached to the empty capsid would be difficult to detect in the mass distribution shown in Figure 4a; however, it would be easy to detect in the charge versus mass scatter plot.
Since the average charges on the empty, partial, and full particles are similar, it is possible that useful information on their relative abundances could be obtained from the m/z distribution that is accessible from conventional mass spectrometry methods, even without charge state resolution. The red diagonal lines in the charge versus mass plot (Figure 5a) show lines of constant m/z. The m/z histogram is shown in Figure 5b. There are peaks at 24, 28, and 32 kDa. Inspection of Figure 5a shows that these are due to empty, partial, and full capsids. Therefore, the m/z spectrum alone can reveal some information about the composition. However, the components are more poorly resolved and their relative abundances are different from in the mass distribution. Inspection of Figure 5a reveals the origin of the difference. The low charge component of the empty capsids has m/z values (~28 kDa) similar the partially filled particles, as does the high mass component at ~5.55 MDa. Thus, the m/z distribution underestimates the empty capsids, overestimates the partial, and cannot detect the high mass impurity.
In conclusion, CDMS can rapidly establish the purity of AAV vector preparations by resolving the particles into empty, partial, and full subpopulations. The sc AAV vector preparation appears to contain particles with partially truncated genomes averaging at half of the genome length. The close agreement between CDMS results and electron microscopy with manual particle counting confirms expectations that mass discrimination is small in the relevant mass range (after correcting for the ion velocity). The single particle resolution of AAV vector preparations by CDMS will enable rapid screening and quality testing of clinical samples. Because the empty, partial, and full capsids have similar charges (as expected from the charge residue model) it is possible to obtain some abundance information from the m/z spectra alone, even without charge state resolution. However, CDMS provides much more reliable information, and low abundance species can be detected by dispersing the ions in two dimensions (charge and mass). The differences between the masses of the empty and full particles, for both the ss and sc genomes, indicate that the DNA is packaged without counterions.
ACKNOWLEDGMENTS
We gratefully acknowledge the UNC Vector Core facility for their help with this study as well as Staci Anthony, Benjamin Draper, Corinne Lutomski, Kevin Bond, and Carmen Dunbar for their contributions to the microscopy studies. We would also like to acknowledge grant support from NSF (Award 1531823 to M.F.J) and NIH (Grants R01HL089221 and P01HL112761 to A.A).
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Ganem B; Li YT; Henion JD J. Am. Chem. Soc. 1991, 113, 6294–6296. [Google Scholar]
- (2).Loo JA Mass Spectrom. Rev 1997, 16, 1–23. [DOI] [PubMed] [Google Scholar]
- (3).van den Heuvel RHH; Heck AJR Curr. Opin. Chem. Biol 2004, 8, 519–526. [DOI] [PubMed] [Google Scholar]
- (4).Zhou M; Robinson CV Trends Biochem. Sci 2010, 35, 522–529. [DOI] [PubMed] [Google Scholar]
- (5).Tito MA; Tars K; Valegard K; Hajdu J; Robinson CV J. Am. Chem. Soc 2000, 122, 3550–3551. [Google Scholar]
- (6).Uetrecht C; Versluis C; Watts NR; Wingfield PT; Steven AC; Heck AJR Angew. Chem., Int. Ed 2008, 47, 6247–6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Uetrecht C; Versluis C; Watts NR; Roos WH; Wuite GJL; Wingfield PT; Steven AC; Heck AJR Proc. Natl. Acad. Sci. U. S. A 2008, 105, 9216–9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Weiss VU; Bereszcazk JZ; Havlik M; Kalinger P; Gösler I; Kumar M; Blaas D; Marchetti-Deschmann M; Heck AJR; Szymanski WW; Allmaier G Anal. Chem 2015, 87, 8709–8717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Fuerstenau SD; Benner WH Rapid Commun. Mass Spectrom 1995, 9, 1528–1538. [DOI] [PubMed] [Google Scholar]
- (10).Benner WH Anal. Chem 1997, 69, 4162–4168. [Google Scholar]
- (11).Schultz JC; Hack CA; Benner WH J. Am. Soc. Mass Spectrom 1998, 9, 305–313. [DOI] [PubMed] [Google Scholar]
- (12).Schultz JC; Hack CA; Benner WH Rapid Commun. Mass Spectrom 1999, 13, 15–20. [DOI] [PubMed] [Google Scholar]
- (13).Doussineau T; Kerleroux M; Dagany X; Clavier C; Barbaire M; Maurelli J; Antoine R; Dugourd P Rapid Commun. Mass Spectrom 2011, 25, 617–623. [DOI] [PubMed] [Google Scholar]
- (14).Doussineau T; Désert A; Lambert O; Taveau J-C; Lansalot M; Dugourd P; Bourgeat-Lami E; Ravaine S; Duguet E; Antoine R J. Phys. Chem. C 2015, 119, 10844–10849. [Google Scholar]
- (15).Pierson EE; Keifer DZ; Selzer L; Lee LS; Contino NC; Wang JC-Y; Zlotnick A; Jarrold MF J. Am. Chem. Soc 2014, 136, 3536–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Keifer DZ; Pierson EE; Hogan JA; Bedwell GJ; Prevelige PE; Jarrold MF Rapid Commun. Mass Spectrom 2014, 18, 483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Pierson EE; Keifer DZ; Kukreja AA; Wang JC-Y; Zlotnick A; Jarrold MF J. Mol. Biol 2016, 428, 292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hastie E; Samulski R J. Hum. Gene Ther. 2015, 26, 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Agbandje-McKenna M; Kleinschmidt J Methods Mol. Biol 2012, 807, 47–92. [DOI] [PubMed] [Google Scholar]
- (20).High KH; Nathwani A; Spencer T; Lillicrap D Haemophilia 2014, 20 (Suppl.4), 43–49. [DOI] [PubMed] [Google Scholar]
- (21).Dismuke DJ; Tenenbaum L; Samulski RJ Curr. Gene Ther 2014, 13, 434–452. [DOI] [PubMed] [Google Scholar]
- (22).Gao K; Li M; Zhong L; Su Q; Li J; Li S; He R; Zhang Y; Hendricks G; Wang J; Gao G Mol. Ther.–Methods Clin. Dev 2014, 1, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Allay JA; Sleep A; Long S; Tillman DM; Clark R; Carney G; Fagone P; McIntosh JH; Nienhuis AW; Davidoff AM; Nathwani AC; Gray JT Hum. Gene Ther 2011, 22, 595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Grieger JC; Samulski RJ Methods Enzymol 2012, 507, 229–254. [DOI] [PubMed] [Google Scholar]
- (25).McCarty DM; Fu H; Monahan PE; Toulson CE; Naik P; Samulski RJ Gene Ther. 2003, 10, 2112–2118. [DOI] [PubMed] [Google Scholar]
- (26).Wu J; Zhao W; Zhong L; Han Z; Li B; Ma W; Weigel-Kelley KA; Warrington KH; Srivastava A Hum. Gene Ther 2007, 18, 171–182. [DOI] [PubMed] [Google Scholar]
- (27).McCarty DM Mol. Ther 2008, 16, 1648–1656. [DOI] [PubMed] [Google Scholar]
- (28).Burnham B; Nass S; Kong E; Mattingly M; Woodcock D; Song A; Wadsworth S; Cheng SH; Scaria A; O’Riordan CR Hum. Gene Ther: Methods 2015, 26, 228–242. [DOI] [PubMed] [Google Scholar]
- (29).Aurnhammer C; Haase M; Muether N; Hausl M; Rauschhuber C; Huber I; Nitschko H; Busch U; Sing A; Ehrhardt A; Baiker A Hum. Gene Ther: Methods 2012, 23, 18–28. [DOI] [PubMed] [Google Scholar]
- (30).Lock M; Alvira MR; Chen S-J; Wilson JM Hum. Gene Ther: Methods 2014, 25, 115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zincarelli C; Soltys S; Rengo G; Rabinowitz JE Mol. Ther 2008, 16, 1073–1080. [DOI] [PubMed] [Google Scholar]
- (32).Cearley CN; Wolfe JH Mol. Ther. 2006, 13, 528–537. [DOI] [PubMed] [Google Scholar]
- (33).Wu Z; Asokan A; Samulski RJ Mol. Ther 2006, 14, 316–327. [DOI] [PubMed] [Google Scholar]
- (34).Zolotukhin S; Byrne BJ; Mason E; Zolotukhin I; Potter M; Chesnut K; Summerford C; Samulski RJ; Muzyczka N Gene Ther. 1999, 6, 973–985. [DOI] [PubMed] [Google Scholar]
- (35).Lock M; Alvira M; Vandenberghe LH; Samanta A; Toelen J; Debyser Z; Wilson JM Hum. Gene Ther 2010, 21, 1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Contino NC; Jarrold MF Int. J. Mass Spectrom. 2013, 345-347, 153–159. [Google Scholar]
- (37).Contino NC; Pierson EE; Keifer DZ; Jarrold MF J. Am. Soc. Mass Spectrom 2013, 24, 101–108. [DOI] [PubMed] [Google Scholar]
- (38).Pierson EE; Keifer DZ; Contino NC; Jarrold MF Int. J. Mass Spectrom 2013, 337, 50–56. [DOI] [PubMed] [Google Scholar]
- (39).Pierson EE; Contino NC; Keifer DZ; Jarrold MF J. Am. Soc. Mass Spectrom 2015, 26, 1213–1220. [DOI] [PubMed] [Google Scholar]
- (40).Keifer DZ; Shinholt DL; Jarrold MF Anal. Chem 2015, 87, 10330–10337. [DOI] [PubMed] [Google Scholar]
- (41).Rose JA; Maizel JV; Inman JK; Shatkin AJ J. Virol 1971, 8, 766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Johnson FB; Ozer HL; Hoggan MD J. Virol 1971, 8, 860–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Buller RM; Rose JA J. Virol 1978, 25, 331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Trempe JP; Carter BJ J. Virol 1988, 62, 3356–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Cassinotti P; Weitz M; Tratschin JD Virology 1988, 167, 176–184. [PubMed] [Google Scholar]
- (46).Becerra SP; Koczot F; Fabisch P; Rose JA J. Virol 1988, 62, 2745–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).The masses were obtained from the sequences with the following assumptions: a) the N-terminus Tyr in VP2 is translated as Met; b) the N-terminus Met in VP1 and VP3 (where the sequences are MAA⋯) are both removed after transcription; the N-terminus Met in VP2 (where the sequence is MAP⋯) is not removed because removal is inhibited by the proline.
- (48).Gao GP; Alvira MR; Wang L; Calcedo R; Johnston J; Wilson JM Proc. Natl. Acad. Sci. U. S. A 2002, 99, 11854–11859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Shen X; Storm T; Kay MA Mol. Ther 2007, 15, 1955–1962. [DOI] [PubMed] [Google Scholar]
- (50).Snijder J; van de Waterbeemd M; Damoc E; Denisov E; Grinfeld D; Bennett A; Agbandje-McKenna M; Makarov A; Heck AJR J. Am. Chem. Soc. 2014, 136, 7295–7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Kohlbrenner E; Aslanidi G; Nash K; Shklyaev S; Campbell-Thompson M; Byrne BJ; Snyder RO; Muzyczka N; Warrington KH; Zolotukhin S Mol. Ther 2005, 12, 1217–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Urabe M; Nakakura T; Xin KQ; Obara Y; Mizukami H; Kume A; Kotin RM; Ozawa K J. Virol 2006, 80, 1874–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Mietzsch M; Grasse S; Zurawski C; Weger S; Bennett A; Agbandje-McKenna M; Muzyczka N; Zolotukhin S; Heilbronn R Hum. Gene Ther 2014, 25, 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Levy HC; Bowman VD; Govindasamy L; McKenna R; Nash K; Warrington K; Chen W; Muzyczka N; Yan X; Baker TS; Agbandje-McKenna M J. Struct. Biol 2009, 165, 146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Kronenberg S; Böttcher B; von der Lieth CW; Bleker S; Kleinschmidt JA J. Virol 2005, 79, 5296–5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Nam H-J; Lane MD; Padron E; Gurda B; McKenna R; Kohlbrenner E; Aslanidi G; Byrne B; Muzyczka N; Zolotukhin S; Agbandje-McKenna M J. Virol 2007, 81, 12260–12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Fernandez de la Mora J Anal. Chim. Acta 2000, 406, 93–104. [Google Scholar]
- (58).Konermann L; Ahadi E; Rodriguez AD; Vahidi S Anal. Chem 2013, 85, 2–9. [DOI] [PubMed] [Google Scholar]
- (59).Thomas JJ; Bothner B; Traina J; Benner WH; Siuzdak G Spectroscopy 2004, 18, 31–36. [Google Scholar]