

Abstract
GoxA is a cysteine tryptophylquinone (CTQ)-dependent glycine oxidase that is a member of a family of LodA-like proteins. The electrochemical midpoint potential (Em) values for the quinone/semiquinone couple and semiquinone/quinol couple were determined to be +111 and +21, respectively. The Em value for the overall two-electron quinone/quinol couple was similar to those of CTQ and tryptophan tryptophylquinone (TTQ) bearing dehydrogenases. However, for the well-studied TTQ-dependent methylamine dehydrogenase, the quinone/semiquinone couple is more negative than the semiquinone/quinol couple; the opposite of what was determined for GoxA. The change in Em value for the two-electron quinone/quinol couple of CTQ in GoxA with pH indicates that the overall two-electron transfer process is associated with the transfer of one proton. Thus, the quinol is anionic. The data reported herein further suggest that in GoxA the CTQ semiquinone is neutral, in contrast to the TTQ-dependent dehydrogenases where it is an anionic TTQ semiquinone. These results are discussed in the context of the structure and function of this glycine oxidase, compared to that of the tryptophylquinone-dependent dehydrogenases.
Graphical Abstract
INTRODUCTION
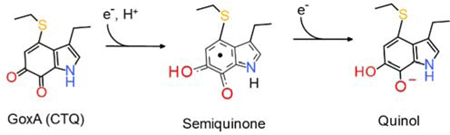
Tryptophylquinone enzymes possess protein-derived cofactors formed by posttranslational modifications of Trp residues.1–3 Two such cofactors have been characterized, tryptophan tryptophylquinone (TTQ)4 and cysteine tryptophylquinone (CTQ).5 In each of these, two oxygen atoms have been inserted into the indole ring of a Trp side-chain that is cross-linked to the side-chain of another amino acid residue. In TTQ, it is another Trp side-chain. In CTQ, it is the sulfur of a Cys side chain. All known tryptophylquinone enzymes catalyze the oxidative de-amination of a primary amino group on a substrate.3 TTQ and CTQ are redox-active and can potentially exhibit three redox states, the fully oxidized quinone, the one-electron reduced semiquinone and the two-electron reduced quinol. All TTQ-bearing enzymes are dehydrogenases. CTQ has been found in both dehydrogenases and oxidases. Quinohemoprotein amine dehydrogenase (QHNDH) contains CTQ, as well as two hemes.5 The enzyme in this study, GoxA, is a glycine oxidase with CTQ as the sole cofactor.6–8 GoxA is a member of a family of CTQ-bearing oxidases that are referred to as LodA-like proteins.9 These are named after the lysine ε-oxidase, LodA.10 The reductive half-reactions of the transformations that are catalyzed by TTQ-bearing11, 12 and CTQ-bearing13 dehydrogenases are similar to that of GoxA, but with a significant difference. The initial step in the reaction of each of these is the formation of a covalent Schiff-base adduct, which results from the substrate amine nitrogen displacing a quinone oxygen. This is followed by deprotonation of the α-carbon of the amine substrate concomitant with two-electron reduction of the quinone to yield a product Schiff base (Figure 1). However, during the reaction of the dehydrogenases the product Schiff base is immediately hydrolyzed to yield an iminoquinol, whereas for GoxA the product Schiff-base generated anaerobically is stable and not hydrolyzed until it is exposed to air.14 Furthermore, the oxidative-half reactions between these dehydrogenases and the CTQ-bearing oxidases are different. The reduced dehydrogenases transfer electrons to either the Cu2+ of a cupredoxin or the Fe3+ of a hemoprotein.15 In contrast, the reduced LodA and GoxA are each re-oxidized by O2.
Figure 1.
Reaction mechanisms of the TTQ-dependent methylamine dehydrogenase (top) and CTQ-dependent GoxA (bottom).
The redox properties of TTQ in methylamine dehydrogenase (MADH) and aromatic amine dehydrogenase (AADH) have been characterized.16, 17 For MADH and AADH the pHdependence of the electrochemical midpoint potential (Em) value for the two-electron quinone/quinol couple was determined and found to be consistent with a two electron, one proton transformation.17 It was not possible to characterize the redox properties of CTQ in QHNDH because of interference from the two hemes that are also present in this enzyme. However, it was possible to determine the Em value for the two-electron quinone/quinol couple of CTQ-bearing subunit of this enzyme after separation from the subunit bearing the hemes.18 The semiquinone species was not observed γ during the reductive titrations of MADH, AADH or the QHNDH subunit. For MADH it was possible to calculate the Em values for the oxidizedsemiquinone and semiquinone-reduced couples from the experimentally-determined ∆G° values for the forward and reverse rates of the electron transfer reactions between the MADH semiquinone and its electron acceptor, amicyanin, and between fully reduced MADH and amicyanin.19
The results presented herein are the first detailed description of the redox properties of CTQ within the native state of its host protein and the first description of the redox properties of a tryptophylquinone cofactor that functions as an oxidase. For GoxA, it was possible to observe the semiquinone during the reductive titration and determine the Em values of each of the two one-electron oxidized/semiquinone and semiquinone/reduced couples. The dependence on pH of the two-electron redox couple was also determined. Furthermore, it is shown that the absorption spectrum of the dithionite-reduced GoxA is quite different from that of the previously reported glycine-reduced GoxA, consistent with that species being a reduced CTQ-product Schiff base intermediate.14 These overall results reveal interesting similarities and distinctions between the redox properties of CTQ in GoxA and the previously studied tryptophylquinone cofactors of the dehydrogenases. This may be related to the function as an oxidase rather than a dehydrogenase.
EXPERIMENTAL PROCEDURES
Expression and Purification of GoxA.
Recombinant mature GoxA from Pseudoalteromonas luteoviolaceae was produced in E. coli in which the goxA and goxB genes were co-expressed and purified as described previously.6
Spectrochemical Redox Titrations.
Em values were determined by spectrochemical titration in which the ambient potential and absorption spectrum of GoxA are monitored simultaneously. The titrations were performed under anaerobic conditions in solutions that had been subjected to alternating cycles of degassing under vacuum and purging with argon. Absorbance spectra were recorded with an HP4852A diode array spectrophotometer. The ambient potential was measured using a Microelectrodes Micro-ORP Electrode that was calibrated using quinhydrone (a 1:1 mixture of hydroquinone and benzoquinone) as a standard. Em values are reported versus the normal hydrogen electrode. Titrations were performed in 50 mM potassium phosphate buffer at the indicated pH at 25 °C. The following mediators were included in the solution to facilitate the electron transfer between GoxA and electrode: phenazine methosulfate (2 µm) (Acros Organics), Safranin T (2 µm) (Acros Organics), and 2,6dichlorophenolindophenol (1 µm) (Fluka). The oxidized GoxA was titrated by addition of incremental amounts of dithionite, which was used to decrease the ambient potential. The reverse oxidative titration was performed by addition of incremental amounts of air.
Data analysis.
Data from the spectroscopic changes that occurred during the glycine titration of oxidized GoxA were fit to eq 1, which describes allosteric binding. The fraction of GoxA with glycine bound was determined from the ΔA/ΔAmax at each point in the titration.
| [1] |
To determine of Em values from the spectrochemical reductive titrations of GoxA, the data were fit by a form of the Nernst equation that has previously been used to simultaneously determine the Em values associated with oxidized/semiquinone and semiquinone/reduced couples of flavoproteins (eq 2).20, 21 A is the total absorbance at each applied potential (E) and a, b and c are the component absorbance values contributed by the quinone, semiquinone and quinol states, respectively, at the measured wavelength. Eox/sq and Esq/red are the midpoint potentials for the one-electron quinone/semiquinone and semiquinone/quinol couples, respectively. R is the universal gas constant, T is the temperature in Kelvin, F is the Faraday constant, and n is the number of electrons transferred.
| [2] |
For the oxidative titrations during which no semiquinone was observed, data were fit to eq 3 where Eox/red is the midpoint potential for the two-electron quinone/quinol couple. The fraction of the quinol ([CTQred]/[CTQtotal]) was determined from the ∆A/∆Amax at 412 nm at each applied potential divided.
| [3] |
RESULTS
Reductive titration of GoxA with glycine.
It was previously noted that addition of a large excess of glycine was required to fully reduce GoxA under anaerobic conditions. It was subsequently shown that the absorbance spectrum of the glycine-reduced GoxA was that of the reduced CTQ product Schiff base adduct.14 The reduction of GoxA with glycine was repeated using incremental additions of glycine (Figure 2A) under anaerobic conditions and unexpected results were obtained. As noted before,6 it was not possible to reduce GoxA stoichiometrically with glycine. However, during the incremental titration there was an apparent lack of reaction at lower glycine concentrations. This was followed by changes in absorbance that did not show a linear dependence on glycine concentration. It was previously reported that the steady-state kinetics of GoxA exhibited non-Michaelis-Menton, allosteric behavior.6 As such, the changes in absorbance with increasing glycine concentration were fit to an equation that describes allosteric binding (eq 1). Similar results were obtained from fitting the changes in absorbance at 600 nm and 364 nm. The fit of the increase in absorbance at 600 nm yielded values of h=3.7 ± 0.4, Kd =103 ± 3 µM and an R2 = 0.991 (Figure 2B). The fit of the decrease in absorbance at 364 nm yielded values of h=3.4 ± 0.3, Kd = 92 ± 3 µM and an R2 = 0.993 (Figure 2C). In the previously reported steady-state kinetic analysis, the dependence of the reaction rate on glycine concentration was also sigmoidal and yielded a smaller but significant h value of 1.8.6 The larger h values obtained in the current study indicate stronger cooperativity, and are quite consistent with the fact that this GoxA is a homotetramer.6 The final absorbance spectrum in this titration is that of the product Schiff base, which results from substrate binding followed by deprotonation of the bound substrate adduct. The observed strong cooperativity suggests that the concentrationdependence of the changes in absorbance describe the initial binding step and not the subsequent deprotonation (see Figure 1). This is why the data fit so well to eq 1. For this to be true, the rate of binding is slow relative to the subsequent deprotonation, such that what is actually being measured is the extent of specific binding as a function of glycine concentration. The weaker cooperativity observed in the steady-state analysis may be attributed to the fact that product formation was being measured in that assay, and that this rate is not solely dependent on the rate of glycine binding, but could also be influenced by catalytic step and reaction with O2.
Figure 2.
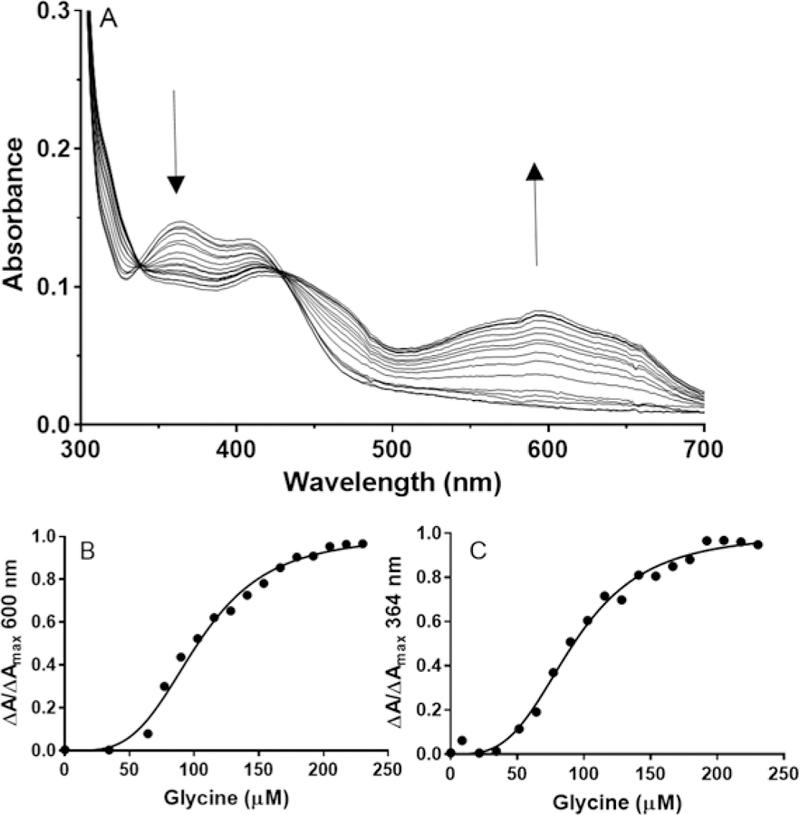
Titration of changes in absorbance of GoxA during the reduction by glycine and analysis of glycine binding. A. GoxA (20 µM) was present in 50 mM potassium phosphate buffer, pH 6.5, under anaerobic conditions. Spectra were recorded after incremental additions of glycine to final concentrations from 0–250 µM. The directions of the spectral changes are indicated with arrows. B. The fraction of GoxA with glycine bound as determined from the fraction of the total increase in absorbance at 600 nm versus glycine concentration is shown and the line is a fit of the data by eq 1. C. The fraction of GoxA with glycine bound as determined from the fraction of the total decrease in absorbance at 364 nm versus glycine concentration is shown and the line is a fit of the data by eq 1.
Spectrochemical reductive titrations of GoxA.
It was previously shown that GoxA could be reduced to what appeared to be the semiquinone state by addition of a large excess of dithionite. However, the reaction was slow and the semiquinone could not be further reduced. In the current study, redox mediators are present with GoxA under anaerobic conditions and dithionite is incrementally added to the solution to decrease the ambient potential, rather than to directly reduce GoxA. In this case, it was possible to completely reduce CTQ to the quinol. When the titration was performed at pH 6.5, it clearly proceeded through the semiquinone intermediate state (Figure 3), which allowed determination of the Em values for each of the two one-electron couples. It is difficult to ascertain the fraction of semiquinone present at any given time during the titration because the absorbance features of the oxidized, semiquinone and reduced forms of CTQ overlap. Figures 3A and B display two portions of the overall titration, which highlight the direction of the changes in absorbance in different parts of the spectra during the conversion from oxidized to reduced CTQ. The absorbance at 376 nm, the absorbance maximum of the semiquinone, with potential were fit to a two-component version of the Nernst equation (eq 2) to determine the Em values associated with both one-electron reductions. This analysis makes no assumption as to the exact midpoint in the titration or fraction of semiquinone present at any given time. The fit of the data in this biphasic plot (Figure 3C) yielded Em values of +111 ± 6 mV for the quinone/semiquinone couple and +21 ± 2 mV for the semiquinone/quinol couple. The R2 value for the fit is 0.999. At higher pH values the accumulation of semiquinone during the titration was much less (discussed later). As such, it was not possible to determine the Em values for each of the two one-electron couples from the analysis of those data. For this reason, only the results of the titration at pH 6.5 are presented here.
Figure 3.
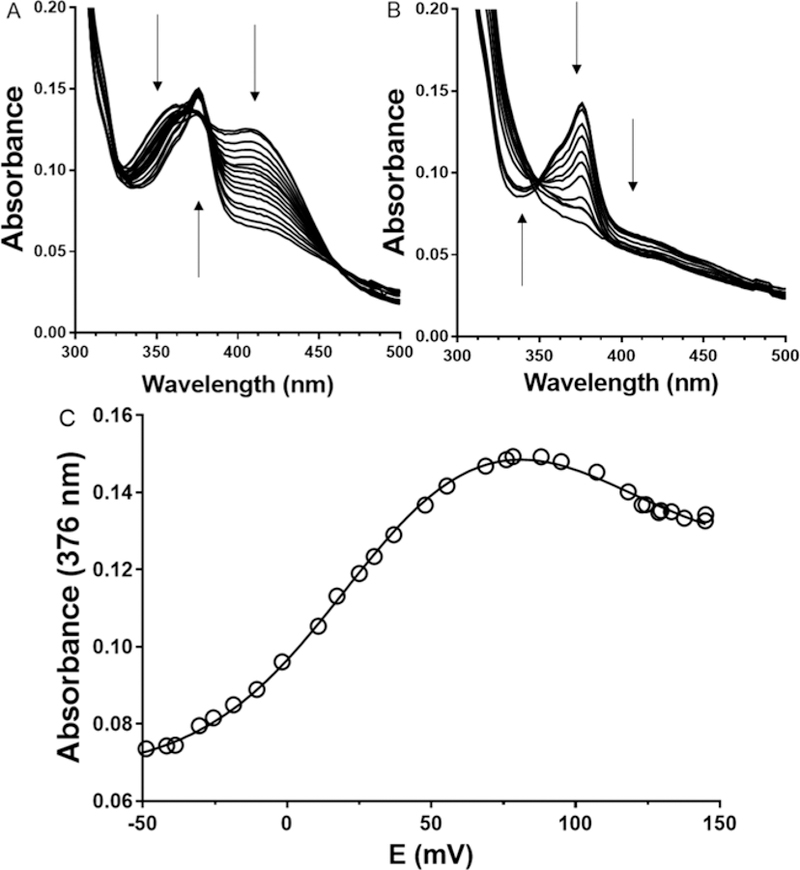
Reductive spectrochemical titration of GoxA. Absorbance spectra were recorded at several values of ambient potential and the direction of changes in absorbance are indicated by arrows. A. First portion of the titration of oxidized GoxA. B. Second portion of the titration to yield reduced GoxA. C. Determination of the two Em values associated with the reduction of oxidized GoxA via the semiquinone intermediate. The absorbance at 376 nm is plotted against the measured ambient potential. The line is a fit of the data by eq 2.
Oxidative titrations of quinol GoxA.
While it was possible to perform the reverse oxidative titration using ferricyanide as an oxidant, the absorbance of ferricyanide overlaps that of CTQ making it difficult to accurately measure the spectral changes. Alternatively, it was determined that the ambient potential could be gradually increased by incremental addition of air into the system, again in the presence of redox mediators. During the oxidative titration, no significant accumulation of the semiquinone species was observed (Figure 4). The oxidative titration was performed immediately after the completion of the reductive titration. As this is a lengthy process, there was a small amount of precipitation of the protein, which accounts for the background absorbance in the spectrum. In order to better visualize the relevant spectral changes difference spectra which eliminated this background interference are presented in the inset.
Figure 4.
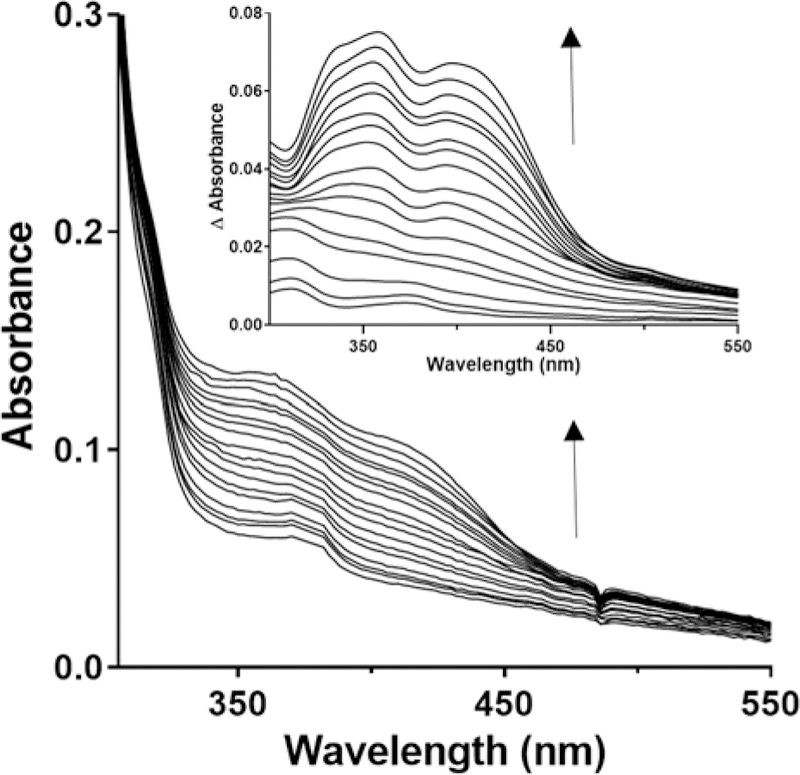
Oxidative titration of reduced GoxA with air at pH 6.5. The fully reduced GoxA was generated as described in Figure 3. Spectra were recorded after equilibration of the ambient potential after incremental additions of air. Inset. The difference spectra for the oxidation of reduced GoxA. For clarity, the initial spectrum of the reduced GoxA that was the product of the reductive titration was subtracted from the spectra obtained after each addition of air.
It was possible to determine the Em value for the two-electron quinone/quinol couple from the data obtained in the oxidative titration. The Em value at pH 6.5 was 77 ± 2 mV. It was also possible to perform the oxidative titrations at higher pH to determine the effect of pH on the Em value for the two-electron quinone/quinol couple (Figure 5). Going from pH 6.5 to 7.5, the Em value decreased to 53 ± 1 mV. It did not decrease further at higher pH as the value was also 53 ± 1 at pH 9.0. The R2 values for the fits at pH 6.5, 7.5, and 9.0 were 0.983, 0.994 and 0.995, respectively.
Figure 5.
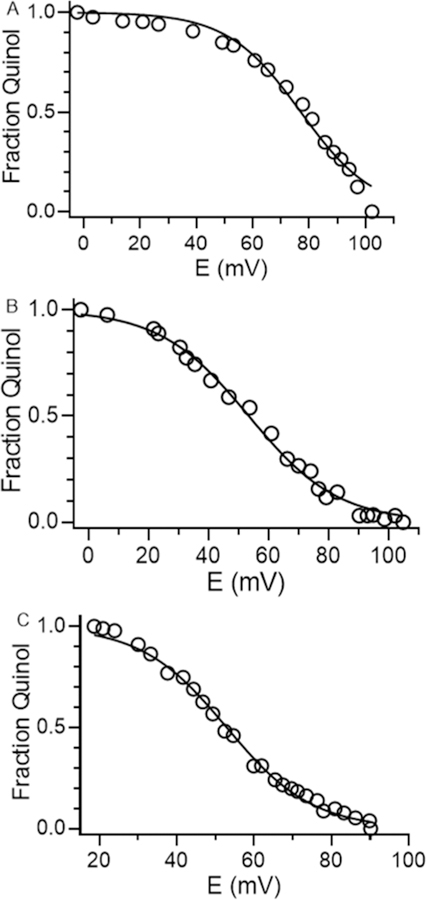
Oxidative titrations of reduced GoxA. Titrations were performed at pH 6.5 (A), 7.5 (B) and 9.0 (C). The line in each plot is a fit of the data by eq 3.
It should be noted that it was not possible to perform the oxidative titration with the substrate-reduced GoxA. As shown in Figure 1, in contrast to other quinoproteins that oxidize amines, the reduced species with only an amino group bound is not a stable intermediate. In the absence of O2, the reduced form of GoxA is a product Schiff base adduct. Hydrolysis of the Schiff base only occurs in the presence of O2, and this is linked to the oxidative half-reaction, yielding the oxidized amino-quinone.
If the two-electron interconversion of the quinol and quinone was accompanied by the transfer of two protons, as is the case for a neutral quinol with two OH groups, then the change in Em value would be approximately 59 mV per pH unit. For GoxA the change in Em value between pH 6.5 and 7.5 is 24 ± 2 mV. This value is more consistent with the process involving the transfer of two electrons and only one proton, for which a value of 29.5 would be expected. This indicates that only one of the quinol oxygens is protonated in the fully reduced state. An explanation for this observation may be inferred from the crystal structure of GoxA (Figure 6). Only the quinone oxygen at the C6 position is exposed to solvent in the active site. In fact, it is hydrogen bonded to a water molecule. The other oxygen at the C7 position is shielded from solvent in a hydrophobic pocket. This oxygen forms a hydrogen bond with a backbone amide hydrogen of residue His583. Thus, in the reduced state this quinone oxygen will be negatively charged while coordinated to this non-exchangeable hydrogen.
Figure 6.
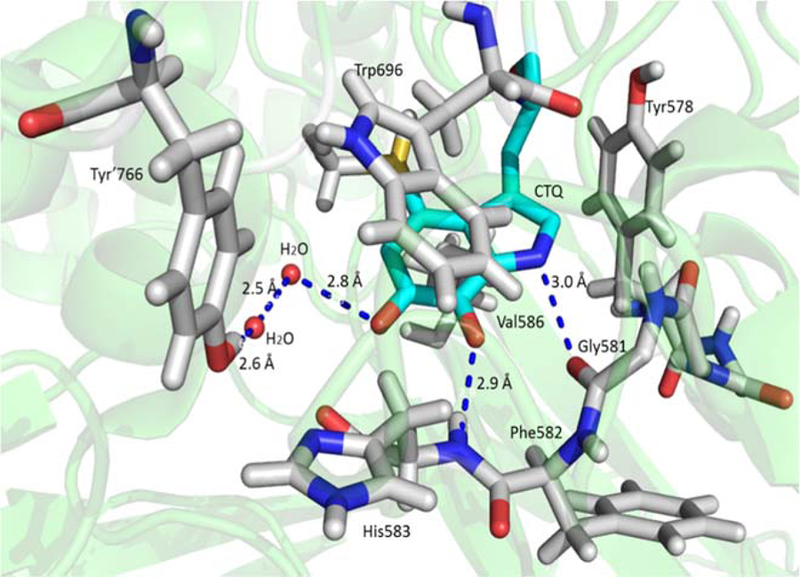
CTQ and surrounding residues in the crystal structure of GoxA (PDB entry 6BYW). Most of CTQ is surrounded by a hydrophobic pocket formed by Trp696, Tyr578 Val586 and His583, except for the quinone oxygen at the C6 position, which is exposed to the solvent. The other quinone oxygen at the C7 position forms a hydrogen bond with backbone amide hydrogen of His583.
DISCUSSION
The redox properties of CTQ in GoxA are distinct from those of TTQ and CTQ in dehydrogenases. With the TTQ-dependent MADH it was possible to generate a stable semiquinone by stoichiometric addition of one electron-equivalents of dithionite. The semiquinone was resistant to oxidation by O2, and subsequent addition of a second-electron equivalent completely reduced the cofactor to the quinol.16 However, no formation of the semiquinone was observed during the spectrochemical reductive titration of MADH, which yielded a direct two-electron reduction of the quinone to the quinol.17 A similar result was obtained for the redox titration of TTQ in AADH.17 In stark contrast to what occurred during the spectrochemical titration of TTQ in MADH, the titration of CTQ in GoxA proceeded via an observable semiquinone. This distinction is likely related to the relative differences in the Em values of the oxidized-semiquinone and semiquinone-reduced couples. For MADH the oxidizedsemiquinone couple is more negative than the semiquinone-reduced couple (+14 mV versus +190 mV),19 whereas for CTQ in GoxA the oxidized-semiquinone couple is more positive than the semiquinone-reduced couple (+111 mV versus +21 mV). Thus, with MADH any semiquinone formed during the reductive titration will be immediately reduced to the quinol. In contrast, during the reductive titration of GoxA reduction to the semiquinone will occur prior to the applied potential decreasing to the point at which further reduction to the quinol occurs. Another distinction is that it was possible to fully reduce MADH by addition of a stoichiometric amount of the methylamine substrate. This was not possible with GoxA where titration with glycine starting at zero concentration yielded a sigmoidal curve consistent with the previously observed cooperativity exhibited by GoxA in steady-state kinetic studies.6
Reduction potentials of the two-electron oxidized-reduced couples of TTQ in MADH and CTQ in GoxA were each dependent on pH. In each case, the data were consistent with the twoelectron reduction being accompanied by a single proton. In MADH and GoxA, this may be explained by the fact the O7 oxygens in TTQ in MADH17 and CTQ in GoxA6 are each shielded from solvent in their respective protein structures. Thus, in each case only the O6 oxygen that is situated at the end of the solvent-accessible channel can be protonated. In the TTQ-dependent dehydrogenases it was concluded that the single proton transfer occurred during the semiquinone reduction to the quinol, indicating that the one-electron reduced species was an anionic semiquinone.17 In contrast, the data presented herein for GoxA are more consistent with the proton transfer occurring during the reduction of the quinone to the semiquinone indicating that it is present as a neutral semiquinone (Figure 7). The reasoning is as follows. For the oneelectron transfer that is accompanied by a proton transfer, the Em value will decrease by 59 mV going from pH 6.5 to7.5. For the other one-electron transfer that is not accompanied by a proton transfer, the Em value will not change with pH. If it is the formation of the semiquinone that is accompanied by proton transfer, then it will be a protonated neutral semiquinone. In this case, the difference in Em values for the two one-electron transfers will become smaller as pH increases. This will mean that less semiquinone will accumulate during the titration at higher pH, which is what was observed. If instead, the proton transfer accompanies the semiquinone reduction to the quinol, then the difference in Em values for the two one-electron transfers will be greater as pH increase and one would expect it to be easier to observe the semiquinone at the higher pH. In this scenario, the semiquinone would be anionic, but this is not observed. This explains why the semiquinone form of GoxA did not fully form during the reductive titrations at higher pH. It is because the gap between the Em values of the two redox couples decreased such the semiquinone is further reduced to the quinol before it is completely formed.
Figure 7.
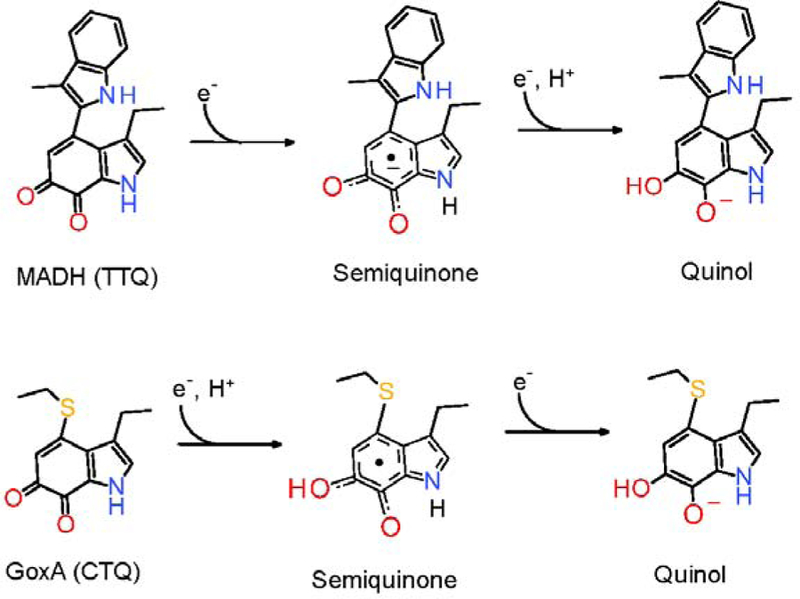
Distinctions between the two-step reductions of TTQ in MADH and CTQ in GoxA.
There is not an obvious structural basis for the observation that the oxidized-semiquinone couple is more negative than the semiquinone-reduced couple in MADH, while the opposite is the case for GoxA. It should be noted that the reduction titration of CTQ in the isolated γ subunit of QHNDH did not proceed via an observable semiquinone and only the Em value for the twoelectron redox couple was determined.18 This result is similar to what was observed for TTQ in MADH and suggests that the crosslink to a Cys rather than a Trp does not account for the differences described above. However, it is difficult to make a direct comparison since the titration of CTQ in QHNDH was on an isolated subunit and not in the native protein environment. In fact, while the semiquinone was not observed during the redox titration of the isolated γ unit,18 the semiquinone form of CTQ was observed by EPR spectroscopy of the intact QHNDH on partial reduction with dithionite.22
Some of the other observed differences in the redox properties of the TTQ-dependent dehydrogenases and GoxA may be explained by structural features as well as the enzyme function. The substrate channel of MADH is open to the surface and allows easy access to the substrate. The substrate channel of GoxA is obscured by another subunit of the heterotetramer and becomes more accessible as successive substrate molecules bind, as evidenced by the observed cooperativity. It is also noteworthy that the TTQ-dependent dehydrogenases react with other redox proteins during their catalytic reactions. As such, these are “designed” to efficiently transfer electrons to the protein surface from the reduced cofactor. This is consistent with the ability to stoichiometrically reduce the protein with an external electron donor such as dithionite. Additional small molecule redox mediators are required to efficiently reduce GoxA. As GoxA is an oxidase, it is re-oxidized by O2 and must therefore be “designed” to minimized electron transfer between the cofactor and the protein surface to avoid non-specific re-oxidation of the reduced cofactor that would short-circuit the catalytic reaction.
ACKNOWLEDGMENT
The authors thank Yu Tang for providing technical assistance.
Funding
This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R37GM41574 and R35GM130173 (VLD).
ABBREVIATIONS
- AADH
aromatic amine dehydrogenase
- CTQ
cysteine tryptophylquinone
- Em
electrochemical midpoint potential
- MADH
methylamine dehydrogenase
- QHNDH
quinohemoproteine amine dehydrogenase
- TTQ
tryptophan tryptophylquinone
Footnotes
ACCESSION CODES
UniProt KB entry A0A161XU12.
The authors declare no competing financial interest.
References
- [1].Davidson VL (2018) Protein-derived cofactors revisited: Empowering amino acid residues with new functions, Biochemistry 57, 3115–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Klinman JP, and Bonnot F (2014) Intrigues and intricacies of the biosynthetic pathways for the enzymatic quinocofactors: PQQ, TTQ, CTQ, TPQ, and LTQ, Chem Rev 114, 4343–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yukl ET, and Davidson VL (2018) Diversity of structures, catalytic mechanisms and processes of cofactor biosynthesis of tryptophylquinone-bearing enzymes, Arch Biochem Biophys 654, 40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].McIntire WS, Wemmer DE, Chistoserdov A, and Lidstrom ME (1991) A new cofactor in a prokaryotic enzyme: tryptophan tryptophylquinone as the redox prosthetic group in methylamine dehydrogenase, Science 252, 817–824. [DOI] [PubMed] [Google Scholar]
- [5].Datta S, Mori Y, Takagi K, Kawaguchi K, Chen ZW, Okajima T, Kuroda S, Ikeda T, Kano K, Tanizawa K, and Mathews FS (2001) Structure of a quinohemoprotein amine dehydrogenase with an uncommon redox cofactor and highly unusual crosslinking, Proc Natl Acad Sci U S A 98, 14268–14273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Andreo-Vidal A, Mamounis K, Sehanobish E, Avalos D, Campillo-Brocal JC, Sanchez-Amat A, Yukl ET, and Davidson VL (2018) Structure and enzymatic properties of an unusual cysteine tryptophylquinone-dependent glycine oxidase from Pseudoalteromonas luteoviolacea, Biochemistry 57, 1155–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sehanobish E, Campillo-Brocal JC, Williamson HR, Sanchez-Amat A, and Davidson VL (2016) Interaction of GoxA with its modifying enzyme and its subunit assembly are dependent on the extent of cysteine tryptophylquinone biosynthesis, Biochemistry 55, 2305–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sehanobish E, Williamson HR, and Davidson VL (2016) Roles of conserved residues of the glycine oxidase GoxA in controlling activity, cooperativity, subunit composition, and cysteine tryptophylquinone biosynthesis, J Biol Chem 291, 23199–23207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Campillo-Brocal JC, Chacon-Verdu MD, Lucas-Elio P, and Sanchez-Amat A (2015) Distribution in microbial genomes of genes similar to lodA and goxA which encode a novel family of quinoproteins with amino acid oxidase activity, BMC.Genomics 16, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gomez D, Lucas-Elio P, Sanchez-Amat A, and Solano F (2006) A novel type of lysine oxidase: L-lysine-epsilon-oxidase, Biochim Biophys Acta 1764, 1577–1585. [DOI] [PubMed] [Google Scholar]
- [11].Brooks HB, Jones LH, and Davidson VL (1993) Deuterium kinetic isotope effect and stopped-flow kinetic studies of the quinoprotein methylamine dehydrogenase, Biochemistry 32, 2725–2729. [DOI] [PubMed] [Google Scholar]
- [12].Hyun YL, and Davidson VL (1995) Mechanistic studies of aromatic amine dehydrogenase, a tryptophan tryptophylquinone enzyme, Biochemistry 34, 816–823. [DOI] [PubMed] [Google Scholar]
- [13].Sun D, Ono K, Okajima T, Tanizawa K, Uchida M, Yamamoto Y, Mathews FS, and Davidson VL (2003) Chemical and kinetic reaction mechanisms of quinohemoprotein amine dehydrogenase from Paracoccus denitrificans, Biochemistry 42, 10896–10903. [DOI] [PubMed] [Google Scholar]
- [14].Avalos D, Sabuncu S, Mamounis KJ, Davidson VL, Moenne-Loccoz P, and Yukl ET (2019) Structural and spectroscopic characterization of a product Schiff-base intermediate in the reaction of the quinoprotein glycine oxidase, GoxA, Biochemistry 10.1021/acs.biochem.8b01145. [DOI] [PMC free article] [PubMed]
- [15].Davidson VL (2004) Electron transfer in quinoproteins, Arch Biochem Biophys 428, 32–40. [DOI] [PubMed] [Google Scholar]
- [16].Husain M, Davidson VL, Gray KA, and Knaff DB (1987) Redox properties of the quinoprotein methylamine dehydrogenase from Paracoccus denitrificans, Biochemistry 26, 4139–4143. [DOI] [PubMed] [Google Scholar]
- [17].Zhu Z, and Davidson VL (1998) Redox properties of tryptophan tryptophylquinone enzymes. Correlation with structure and reactivity, J Biol Chem 273, 14254–14260. [DOI] [PubMed] [Google Scholar]
- [18].Fujieda N, Mori M, Kano K, and Ikeda T (2002) Spectroelectrochemical evaluation of redox potentials of cysteine tryptophylquinone and two hemes c in quinohemoprotein amine dehydrogenase from Paracoccus denitrificans, Biochemistry 41, 13736–13743. [DOI] [PubMed] [Google Scholar]
- [19].Brooks HB, and Davidson VL (1994) Free energy dependence of the electron transfer reaction between methylamine dehydrogenase and amicyanin, J. Am. Chem. Soc 116, 11202–11202. [Google Scholar]
- [20].Finn RD, Basran J, Roitel O, Wolf CR, Munro AW, Paine MJ, and Scrutton NS (2003) Determination of the redox potentials and electron transfer properties of the FAD- and FMN-binding domains of the human oxidoreductase NR1, Eur J Biochem 270, 1164–1175. [DOI] [PubMed] [Google Scholar]
- [21].Munro AW, Noble MA, Robledo L, Daff SN, and Chapman SK (2001) Determination of the redox properties of human NADPH-cytochrome P450 reductase, Biochemistry 40, 1956–1963. [DOI] [PubMed] [Google Scholar]
- [22].Takagi K, Torimura M, Kawaguchi K, Kano K, and Ikeda T (1999) Biochemical and electrochemical characterization of quinohemoprotein amine dehydrogenase from Paracoccus denitrificans, Biochemistry 38, 6935–6942. [DOI] [PubMed] [Google Scholar]