

Abstract
In this article we discuss a streamlined, scalable, laboratory approach that enables medium-to-large dataset analysis. The presented approach combines data management, artificial intelligence, containerization, cluster orchestration and quality control in a unified analytic pipeline. The unique combination of these individual building blocks creates a new and powerful analysis approach that can readily be applied to medium-to-large datasets by researchers to accelerate the pace of research. We have applied the proposed framework to a project that counts the number of plasmonic nanoparticles bound to peripheral blood mononuclear cells in dark-field microscopy images. By using the techniques presented in this article, we automatically process the images overnight, without user interaction, streamlining the path from experiment to conclusions.
Keywords: optics, data processing, automation, image analysis
Graphical Abstract
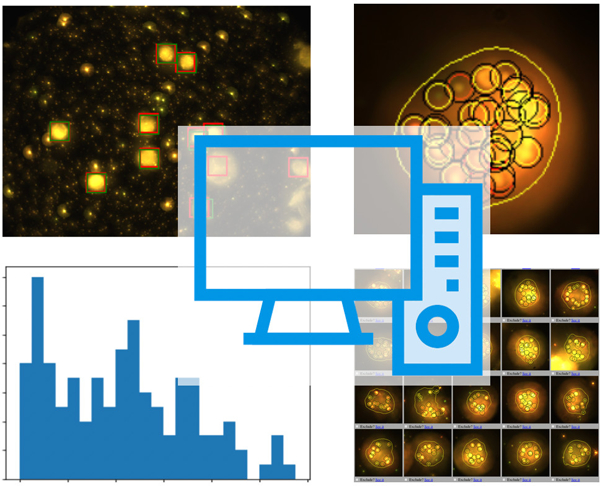
High throughput acquisition devices leave researchers with a deluge of data. Automated processing is often a challenging task. We propose a data processing framework based on deep learning, containerization and orchestration for the analysis of medium to large datasets. The framework is applied to microscopy images of cells.
1. Introduction
Advances in computer technology, data acquisition hardware, laser technology, and automated imaging platforms have transformed the field of biomedical research. Hardware systems that once required manual intervention can now be programed to run continuously for days or even weeks. High content-screening systems enable the simultaneous testing of several experimental hypotheses automatically.[1],[2],[3],[4],[5] Precision mechanical advances enable optical systems that can scan and re-scale entire centimeters of samples at sub-cellular resolution.[6] High-bandwidth communications networks enable large multi-site scientific datasets.[7] Data storage technology has evolved substantially to the point where the units of data measurements are terabytes, enabling the exploration of increasingly complex scientific questions.
These technologies have had an impact on biomedical research. Where it was once adequate to acquire a few data points to images to address a hypothesis, today’s tools enable considerably greater capabilities drive the exploration of increasingly complex scientific questions.
Archiving and indexing large datasets is a non-trivial, but possible task, as demonstrated by the plethora of database repositories currently in use for biology research, [8],[9],[10],[11],[12],[13],[14],[15],[16],[17],[18] electron microscopy, [19] microarray analysis, [20] radiology [21],[22],[23],[24] or multidiscipline research.[25],[26]
While these tools allow us to acquire substantially more data, it is still incumbent on the scientist to transform data into useful and actionable information. Manual image processing is unfeasible for those datasets and automated data analysis techniques and methods are continuously being developed, [27],[28],[29],[30],[31],[32],[33] even more now with the advent of machine learning tools, [34],[35],[36],[37],[38],[39],[40],[39] that often perform on par with human observers. Open-source artificial intelligence software libraries such as Keras (https://keras.io, accessed on 13.03.2019) [41] and Tensorflow (https://www.tensorflow.org, accessed on 13.03.2019), enable the power of neural networks and AI for the analysis of the datasets. Data inspection interfaces are built to inspect the results of such automated methods.[42],[36]
Beyond image processing, data visualization algorithms are being used to extract information from high dimensional datasets.[43],[44],[45],[46],[47] Reusable and reproducible software built over open source software such as python (www.python.org, accessed on 13.03.2019) and R (https://www.r-project.org, accessed on 13.03.2019), making the development of software in repositories based on Git and Github commonplace.[48] Python notebooks, for example, enable data analysis sharing [49] and containerization software such as Docker (http://www.docker.com, accessed on 13.03.2019), enables the running of such software across computers and operating systems with minimal configuration overhead [50] Examples exist in the medical imaging community, to find image-based signatures of cancer,[51] and in the optical coherence tomography (OCT) community,[52] where high-resolution volumetric images of vessel, esophagus or colon are routinely acquired.
All these tools can be daunting, especially to those who have developed many of their skills using more traditional, stand-alone computational analysis methods. This is the method predominantly taught to most scientists and engineers during their undergraduate and post-graduate training. While this approach is perfect for initial development, small datasets and for exploring some outcomes, it generally suffers from a lack of scalability, making the transition to large datasets challenging.
In this article, we present data management, automated analysis and software development methods and practices that are designed to enable the management and processing of medium-to-large datasets in a reliable and reproducible manner. Key aspects presented here are: a data policy to maintain order through projects that spans throughout years; a data processing pipeline based in artificial intelligence to automatically process imaging data; software techniques that ease the transition from prototyping to deployment; clustering techniques that can ease data analysis; and finally, data inspection techniques to monitor the validity of automation-generated analyses. We propose to make a distinct split among such elements of the data cycle, even if they are performed in the same machine or by the same researchers. This division enables distinct processing by different entities and establishes quality control procedures for each step (Fig 1). We present a concrete example of the presented concepts: a novel image processing pipeline that, from 3D microscopy image stacks, extract cells and cell contours using deep convolutional neural networks and then analyzes individuals cell using computer vision image processing techniques. The pipeline is designed to be run overnight in a small cluster of computers. Further, a quality-control web-based interface is used to validate the results of the automated pipeline. This approach will be useful to researchers that seek to acquire and process significant amounts of data all within the laboratory environment.
Figure 1.
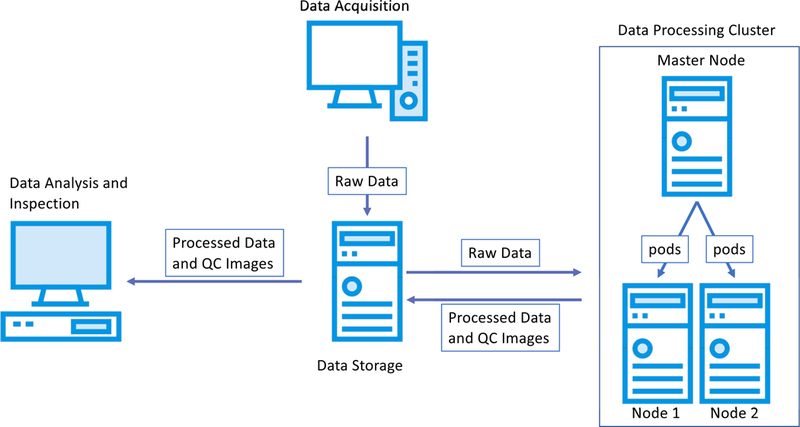
Flow of data for an experiment. The data is acquired with an acquisition system and is archived in a data storage system. A processing cluster, with one or more nodes, retrieves the acquired data and process it through an analysis pipeline generating processed data and intermediate quality control images. Internally, the processing cluster can split tasks among their nodes using cluster orchestration software. Finally, the processed data and quality control images are inspected by the researcher to draw the conclusions of the experiment.
2. Standardized data enable automated processing
Data is the cornerstone of research. Strong efforts are being made towards data interoperability[53] and standardized data and metadata formats.[54],[55],[56] Guidelines for data storage and management are actively being published with a focus on open standards.[57],[58],[59],[30],[60],[61] In this article we will detail methods to manage medium-to-large acquired datasets within the laboratory, with a focus on storing data for automated analysis.
Data in biomedical research is often acquired by devices that enable high throughput data acquisition such as microscopes, OCT devices, or MRI scanners. The data may be initially stored on the same computer that is attached to the physical acquisition device. After the acquisition session is ended, the data should be immediately archived to a data storage system. This can be automated via free and open source software such as rsync (https://rsync.damba.org, accessed on 13.03.2019) to ensure that acquired data is safely stored.
To maximize efficiency, there should always be an inherent hierarchy associated with the data. It should be defined before the start of the project and imposed through its duration. In our laboratory, for example, we keep the following hierarchy: Project / Data type (raw, processed, ...) / Date of experiments/ Subject / Experiment. The hierarchy can be reflected in the directory structure of the file server system of the laboratory, or through the imposition of relational databases. In practice, we favor the former solution, for its ease of use. When using home-built programs and data acquisition software, data storage structure can and should be built directly into the code. For example, in one of our projects, a home-built confocal microscopy software package was designed to automatically create a file structure based on user-defined conditions for each experiment. This automatically generated structure included the project name, the date of data acquisition, the type of data being stored, and a timestamp that accompanied each collected file. This automatic storage structure mechanism made the collected data instantly traceable and straightforward to analyze en masse post-acquisition.
For automated data processing, we make use of centralized data storage systems, such as network attached storage (NAS) devices. This approach allows the different members of the team to “mount” and externally access data on their networked desktop machine for inspection. Centralized data storage systems with a large HD capacity (around 50 TB) are available for only a few thousand dollars. The market of NAS systems has blossomed, especially with the addition of easy-to-use web interfaces, computing capabilities, and containerization to these servers. In our laboratory, for example, we have centralized the data for a single project into a 10-disk 40Tb NAS server, to which more HDs could be added as need-be.
Alternatively, the data can be stored on the cloud and cloud computing can be used for its analysis. Cloud services refer to online, on-demand computational services, such as file storage, that are managed by a cloud provider such as Google. Universities and hospitals often make these services available directly to users. Recently, services such as Dropbox, Google Cloud and Amazon Web Services can even be made HIPAA (the US Health Insurance Portability and Accountability Act) compliant if the right care is taken in their configuration. Cloud services can offer many advantages at the cost of a larger set-up time. Their use should be considered based on the amount of data to be collected, the time required to upload the data to cloud services, as well as the processing power needed to analyze the data. For example, Dropbox and other companies offer cloud storage tiers as large as tens of petabytes, storage sizes that are currently unavailable to many scientists. While setting up servers for remote computing and storage on a cloud provider can be slower than deploying on a locally-accessed machine, the availability of virtualized computing power for some applications can more than make up for this. For example, Google offers the Google Cloud Platform for data analysis where a user can log in to request a virtual computer with specific characteristics for data analysis, such as number of CPUs, RAM size, and number and type of graphics processing cards. Processing data on such cloud systems can make use of computing time on hardware, billed by the minute, that may otherwise be too expensive or exotic to be normally accessible. At the same time, cloud data storage and processing can be expensive; terabytes of cloud data storage can lead to prohibitively large costs if not planned or managed correctly. Table 1 shows a price comparison of data storage and computing possibilities between local and cloud-based sollutions.
Table 1.
Comparison of data storage and computing alternatives, advantages and disadvantages. Prices estimated on March 2019.
| Data Storage | |||||
|---|---|---|---|---|---|
| Brand | Type | Capacity | Price | Pros | Cons |
| QNAP | NAS | 50Tb | 4,000$. 80$/Tb |
Transmission speed. Data availability. Unlimited users. |
Local setup and management. Limited capacity. |
| Dropbox | Cloud sync | ∞ | 180$/user year - min of 540$ | Wide adoption. | Manual selection of files to sync. |
| Box | Cloud sync | ∞ | 162$/user year – min of 486$ | Data available locally through a cache system. | If many files are accessed at the same time, speed on the internet connection. |
| Amazon S3 | Cloud storage | ∞ | 264$/Tb year | ∞ | Pricing. Access through key-value pairs. |
| Cloud storage | ∞ | 240$/Tb year | ∞ | Pricing. Access through key-value pairs. | |
| Computing | |||||
| Server | Local | 1 GPU, 4CPU, 32Gb RAM | 3200$ | Always available. Multi-purpose. |
Local Management. |
| Amazon | Cloud | 4600$/year $0.75/hour | Pay per hour. Scalability. |
Data needs to be in the cloud in key/value. | |
| Cloud | 4294$/year $0.49/hour | Pay per hour. Scalability. |
Data needs to be in the cloud in key/value. | ||
3. Automated pipelines turn raw data into actionable insights
Data processing can be abstracted in the concept of a pipeline - a set of processes applied to the data to synthesize conclusions. Often, at each step of the pipeline, the data is reduced by eliminating information that is irrelevant for the project. Depending on the specific project and analytical methods, the steps of the pipeline can be automated or require manual input. For many of our projects, the ultimate goal of automated data processing is to let the scientist draw conclusions from the acquired data without any manual input. This push towards full automation removes the burden of step-by-step manual data analysis, freeing the scientist to focus on experiment design, data acquisition and interpretation.
Recent advances in computer vision and artificial intelligence have brought automated image analysis algorithms to a level that could be often considered on-par with inter-reader agreement. Deep convolutional neural networks excel at object localization, image segmentation and object tracking in video sequences. These findings have been adopted for biomedical image analysis. As an example, In Fig. 2 we present an automated data processing pipeline for microscopy. Raw images are first acquired. Cells are then identified in each field of view using an object detector based on a deep convolutional neural network (SSD).[62] Regions of interest around the cells are then extracted from the raw data. From each of them we extract the contour of the cell, including bound PNPs, with a segmentation deep convolutional neural network (U-Net).[63] Finally, we count -- using a standard image processing technique (blob-detection) -- the number of gold nanoparticles attached to the cell. This tabular data is then ready for analysis via R or Python scripts. In this project, the raw data collected can be quite large. Each experiment consists of roughly 20 fields of view, each field of view consisting of an uncompressed 3D color image 650 MB in size. If four experiments are carried daily, this makes the total data collection nearly 52 GB per day.
Figure 2.
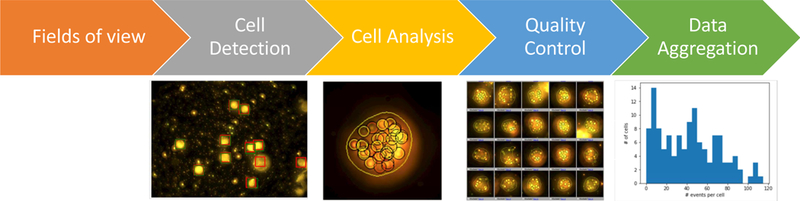
Example of data flow in an image processing pipeline. From left to right: the raw image of the field of view is loaded and pre-processed. Cells are extracted from such field of view and are then analyzed to find the number specific features within. A quality control image is generated to inspect if the cells are properly located and if the bright locations are properly identified. All cell counts within the fields of view of the experiment are aggregated into a CSV file and plotted in a histogram.
It is worth noting that while the analytic tools developed here, including deep-learning based methods, are used for post-acquisition data analysis, such approaches can be implemented in the future to improve data acquisition. Neural networks and computer vision techniques could play a potential role in real-time processing of a live image feed to provide guidance and feedback a scientist or operator. For example, images acquired on a camera system could be processed to guide the selection of optimal image fields, or to ensure that the cells of interest are at the center of the microscope focus. Similarly, neural networks could be used to process image data during acquisition in a quality control step: real-time analysis of the image could provide a user information on the signal-to-noise, signal-to-background, viability of the cells, lineage of the cells, and so on. These automated feedback methods may be of importance when technology of this kind is commercialized to ensure quality data acquisition by minimally trained users or even enable fully automated, computer-guided image acquisition.
3.1. Deep learning as a pipeline building block
Training and development of deep learning neural networks is still subject of research. However, for many applications, recently developed methods are readily applicable. In the processing pipeline we use deep learning to automate two tasks: finding cells in a field of view and outlining the contour of the cells for subsequent analysis. Each task is solved by a different network structure. For instance, in cell detection networks, the input space is an image and the output is a list of boxes representing the location and size of the cells. For cell contouring, the input space is an image and the output is another image where pixels belonging to the cell are marked as ‘1’, while pixels belonging to the background are marked as ‘0’. A key aspect of training deep learning algorithms is the definition of a high-quality training database that spans all of the expected image variabilities.
3.1.1. Finding cells in fields of view with deep learning object detection networks
Finding objects in natural images has been an intense are of research in computer vision and artificial intelligence. Open databases, such as ImageNet [64] have been used to benchmark the performance of different algorithms, such as Fast R-CNN [65], SSD[62] or YOLO.[66] The choice of which algorithm to implement should be done on the basis of the performance of the network on images and the availability of open-source implementations that can be integrated with the rest of the pipeline. In our case there is only one type of object to be detected: cells. While this may seem a simplistic target at first glance, the cells in our experiments are extracted from blood, span a range of sizes and compositions, and are often accompanied by spurious image features such as dead cells and cellular debris. We have chosen to implement a SSD network due to the availability of a python-based implementation, its good performance on ImageNet and the underlying simplicity of the network structure.
Briefly, we reduce each input image stack of 2496 pixels x 3328 pixels x 40 layers x 3 RGB channels into a maximum intensity projection down-sampled image of 624 pixels x 832 pixels x 3 RGB channels. Each cell occupies roughly a window of 20 × 20 pixels on such down-sampled image. We define the output of the network as a 39 × 52 grid with 4 channels, where the first channel denotes the likelihood of the pixel of belonging to the background, the second one the likelihood of the pixel belonging to the cell, and the third and fourth channels model the displacement in normalized units in the x and y directions. The reference standard is composed of similar grids where background pixels have a 1 in the first channel and 0 on the second. In this scenario, pixels that have cells have a 1 in the second channel and a 0 in the first channel and the third and fourth channel encode their normalized displacement in the original image.
The network is trained using the adaptive momentum optimizer and a hybrid cost function, where the first term corresponds to the normalized cross entropy between the first two channels of the reference standard and the network’s output. The second term is defined only on pixels that have cells in the reference standard and represents the mean error between computed and reference displacements. There is a lambda factor between two terms to modulate the weight of correct localization with respect to detection, which is fixed experimentally to 0.1.
The training database is formed of 7097 fields of view (fov) manually annotated by image analysis experts by placing a dot in the central point of the cells. We use 4000 fovs for training, 1000 for validation and 2097 for testing. A custom non-maxima suppression method is applied to eliminate close-by detections. The Pearson correlation coefficient between the number of detected cells using this SSD-based method and the number of cells obtained by an expert is of 0.827. The resulting average error in the test set between the number of cells found in a field of view and the number of cells present is of 1.7 cells/fov. Such results were deemed of good quality by the researchers that are using the system. Most of the errors happened in close-by cells, or cells that were partially in the field of view. Improvements on the metrics would probably arise from a cleaner dataset. Examples of the fovs can be found in Figure 3. The network process the 2097 testing images in 73 seconds, at a rate of 35 ms/image using an Nvidia GTX1080Ti graphics card.
Figure 3.
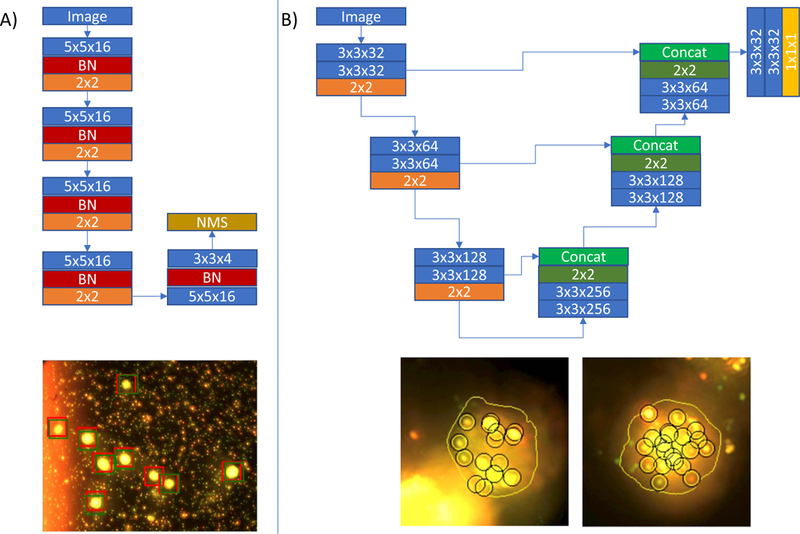
Deep learning networks. A) Network used to find cells in a field of view and an example of its performance. Red boxes in the fov stand for the reference standard. Green boxes stand for the detected cells. Please note the variable background. B) network used to find the contour of cells and examples of the contour on processed cells (with detected bound PNPs). Please note how the method is robust to background artifacts. In the network images, blue boxes correspond to convolution operations, red boxes to batch normalization, orange to max-pooling, dark green to upsampling operations, concat stands for concatenate, NMS for non-maxima-suppression and the yellow box stands for a convolution with a sigmoid activation function. Each layer has its associated parameters on the box.
It is important to note that, using current AI libraries, this network can be written in 300 lines of code, as shown in the github repository of the project (https://github.com/evansgroup/BioEssays2018 ).
3.1.2. Deep learning finds the contours of cells with segmentation networks
Encoder-decoder network structures with skip connections, such as the U-Net,[63] outperform most specialized segmentation methods in a wide variety of tasks. The U-Net has, since its creation, been subject to modifications and improvements, with the addition of more complex convolutional blocks[67] or squeeze-and-excite methods.[68] While the SSD network structure was well suited for object identification tasks, such as the automated identification of cells, the architecture of the U-net is particularly optimized for the partitioning of images based on image features such as shapes and edges. This makes the U-net a natural choice for image segmentation tasks.
In this work we have used a simplified version of the U-Net to segment the contour of the cells and bound PNPs due to its ease to code with current AI libraries. The network is composed of a 3-step encoder path, each of them with two convolution layers, each of 32, 64 or 128 filters according to the depth. An image of the network can be found in Fig. 3. The network is trained with a database of 1704 manually segmented maximum intensity projections of 200 × 200 pixel cell images. 800 images were used for training, 200 for validation and 704 were used for testing. The network was optimized using the DICE coefficient between the resulting segmentation and the reference standard. The DICE coefficient measures the similarity between two segmentation masks by taking twice the area of the product of such images and dividing it by the sum of the areas of the two segmentation masks. The DICE coefficient will approach 1 when the masks are equal and 0 when the masks do not share common pixels. Such metric has been proven to have better properties than a per-pixel classification method.[69] The network is trained with the ADAM optimizer, with a learning rate of 1e-5 and default parameters. The average DICE coefficient on the test set was of 0.935. Examples of the contours over the cells can be found in Fig. 3. Once the data is loaded, cell contouring provides a mask for a cell in 1ms using a Nvidia GTX1080Ti GPU.
3.2. Automated algorithms need to be quality controlled
While a given image analysis routine may seem to be performing well, it is critical to ensure that data is being processed correctly and reproducibly. We configure our code to generate Intermediate quality control (QC) images that can be readily checked so that researchers can trust the data generated via automated processes. QC intermediates, whether they be images, tables, or spreadsheets, allow for rapid inspection to make sure that the data and associated analysis is working correctly. A QC inspection step can save considerable time and effort and ensures that the data meets standards consistently long before a paper, presentation, or publication is written. QC is especially important in large-scale experiments, since often it is impossible to analyze each intermediate step of the pipeline. Semi-automated methods for the identification of outliers are valuable tools to control large-size experiments.[70],[71],[72],[73] QC-inspection interfaces can be put into place. They are especially important when the conclusions of the experiments challenge the experimental setup or the data acquisition method. Current web-frameworks, such as Django, (https://www.djangoproject.com, accessed on 13.03.2019) enable the creation of software that expose the contents of a folder structure and create links among their items with few lines of code, enabling data analysis to all members of the team. An example of an interface developed to inspect the results acquired with the pipeline of Fig. 2 can be seen in Fig. 4. Such interface has the capacity to mark cells for exclusion for analysis, for example, in case the image processing pipeline fails.
Figure 4.
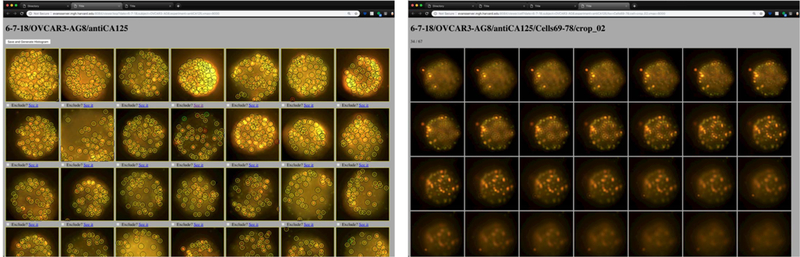
Quality control interface. The interface, based on web-technologies, allow to easily inspect an experiment, consisting on approximately 150 cells and select which ones to exclude from further analysis from a maximum intensity projection of the cell with the bound PNPs (left image). Clicking on any cell allows for a display of each of the z-layers of the cell (right image).
The analysis step typically follows automated image analysis to validate the hypotheses of the experiment or infer if more data needs to be acquired. The analysis is often performed at the researcher’s computer since a graphical visualization of the extracted data and statistical methods are generally needed. Any data analysis software, from python and R to Matlab or SAS, can be used to perform such exploration. Both Jupyter and R Studio additionally provide outstanding notebook environments that allow for data analysis, exploration, and documentation. When analysis output is either too large to easily transport between computers or too complex to analyze on a researcher’s computer, Jupyter and R Studio server software provide a browser-based means of remote graphical processing that process data in-place. For example, we have deployed multiprocessor and GPU-equipped data processing servers running Jupyter to enable GPU-accelerated data analysis routines that would not be feasible on a personal computer.
4. Deploying experimental software for routine use
Software is often developed on personal computer, validated in a subset of the data, and then encapsulated for deployment in the whole dataset. This process is often iterative as improvements or adaptations are needed when new data arises. For good practices on software development we refer the reader to Wilson et al.[74] Software version control systems, such as git, should be used to track changes in the developed software. General guidelines on their use can be found in Blischak et al.[48] Git provides a powerful set of tools for writing code both individually or as a team. Rolling back to an earlier code version is straightforward and git is scalable to large teams all writing code together.
When co-developing software, it is important that the team uses the same libraries on their different development machines. The python programming language, for example, provides tools to list all software dependencies installed. New users can use such lists to re-generate the same precise development environment across different computers and operating systems. Package managers for languages such as python and R provide a simple means of identifying and installing all the required libraries for analysis. Examples of such lists are the requirements.txt files generated by the pip (Pip Installs Python) package manager. All one has to do to make their code portable to other users is to generate a requirements.txt file and include it with the packaged code. When a user downloads the code repository, pip can read the file and set up a “virtual” environment that houses all of the correct versions of required libraries. Similarly, the conda python distribution also can manage python packages and generate virtual environment description files to accomplish the same purpose.
Running the same software in different machines under different operating systems can be challenging, making multi-platform development cumbersome. Even when using the same operating system, different installed versions of computational libraries can lead to incompatibilities that render software unreliable. A first abstraction to deal with such issue was the use of the virtual machine: a guest operating system that runs within the host operating system in virtualized hardware. This model has recently been extended with the concept of operating system virtualization, also called containerization. Each container holds a stripped-down version of the operating system that encapsulates only the libraries required to run specific code. Containers have the advantage of being relatively lightweight, can be created as needed and replicated on-demand to run processes in parallel. Today, the most popular containerization software is Docker (www.docker.com, accessed on 13.03.2019). Docker containers can be run locally using standard Linux tools and can be configured run at scheduled times using cron. This approach is appropriate for cases when there is only one server processing the data. Importantly, Docker is available for Linux, Windows, and MacOS, meaning that once a container is made, it can be run anywhere and on any machine.
Containers are now widely in use for development and web services and are finding new life in the burgeoning area of data science. In a scientific research environment, the allure of containers is clear: a program and all the software libraries necessary for its execution can simply be placed in a container and distributed. This ensures that the program will run consistently across all computers and servers in the laboratory, whether it is on a student’s Windows laptop or a departmental Linux server. This can be ideal for research environments, as it becomes simple to distribute both data and its associated analysis as readily traceable and easily documented bundles. This approach is immediately attractive for translational research, where bench-side routines need to be transformed into analyses that can be run by clinical collaborators. Distributing containers can ease this process and ensure that the code and environment running on a clinical computer system matches the needs of the project. Containers are also a potential means by which scientists can address some concerns regarding data and analysis repeatability. Analysis routines written in one lab can be easily transferred to another and be guaranteed to work out-of-the box. When used along with proper data storage approaches and version control, containerization can be a powerful way to ensure standardized analysis.
5. Parallelizing the effort: running the code automatically in several computers simultaneously
Let’s imagine we have already collected the data, generated manual annotations, trained artificial intelligence models, generated a Docker infrastructure to guarantee that our code runs in any computer, and that we acquire 60Gb of data from a day of experiments. How do we process that data to generate actionable information? In our laboratory we process the data using the Linux utility called cron (https://linux.org/docs/man8/crond), that runs a background process at a given time. Every evening at a fixed time, a script inspects the directory structure for unprocessed data, and in the case that it exists, triggers the image processing pipeline on it. This works well for normal, routine data collection in the laboratory, as the size of any new data is amenable for analysis within a few hours. In some cases, when the data processing is slow or the amount of new data is large, overnight processing in a single device might not be enough. In these cases, the pipeline is run for several days until complete. In some scenarios, however, this approach may result in days or weeks of analysis time, making parallel data analysis methods preferred.
Running the code on several computers is a complex topic covered by the field of clustering and cluster orchestration. In our case, clustering operations are simple, since each image is independent of the others. This situation, often called ‘embarrassingly easily parallelizable’, only requires of infrastructure of a master node that allocates individual image analyses to the processing nodes according to their capacities. The parallelization will bring benefits to computation time when the image processing time is large in comparison with the image loading time.
While several cluster management and orchestration methods exist, we advocate for the use of Kubernetes (https://kubernetes.io, accessed on 13.03.2019), an open-source platform, due to its availability and tight bounds with Docker. This enables the use of heterogeneous nodes, as might be the case in small-scale laboratories. Kubernetes requires a Kubernetes cluster. A computer is assigned the role of master node, to which the rest of the computers are registered. The master node dispatches pods (running processes) to the nodes according to their capacities. A job is the set of pods that are created to perform a set of operations on independent data (Figure 5).
Figure 5.
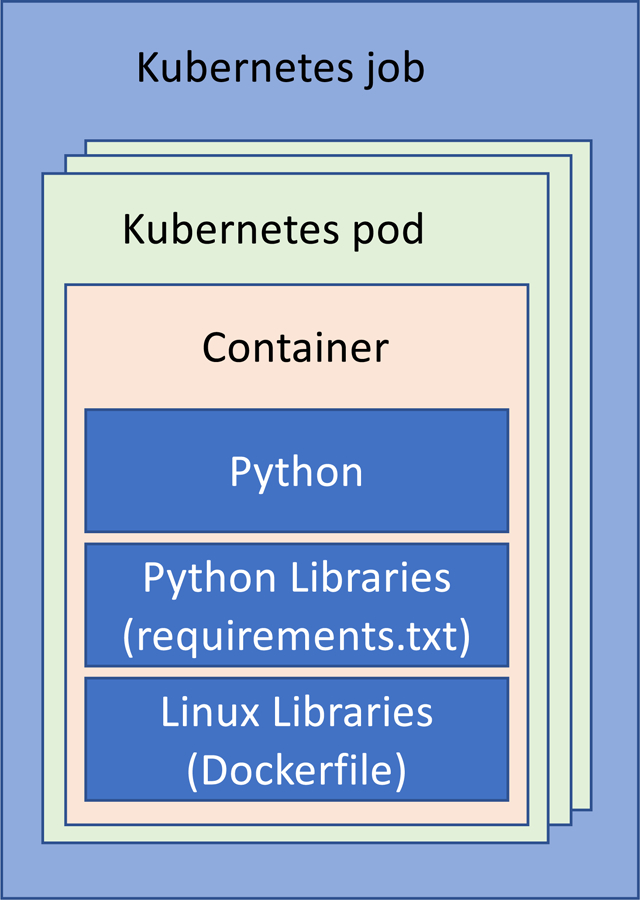
Abstract layers of software development using python as an example. The python environment configuration is specified through the use of a requirements.txt file. The operating system where the python code runs is specified through the use of a Dockerfile. Such OS is encapsulated within a container and the container within a Kubernetes pod. The cluster manager generates the set of pods that consist a job (for example, running cell extraction routine of the pipeline of Fig. 2 on all acquired images). The job is then executed on the processing cluster.
Following with the example of Fig. 5, a pod processes a field of view to extract cells, and for each of them find their contour and count the number of bound PNPs to it. The job would be to process all fields of view. Each pod will be dispatched among the available servers according to their capacities. Several pods can be placed at the same time in the same node. In our laboratory, using 3 nodes, we have reduced the average processing time for each field of view from 1 minute to 12 seconds. This 5x improvement comes from the parallelization between the nodes and within the nodes.
We run the jobs in the Kubernetes cluster every night, using a script triggered by the Linux cron utility. Such scripts inspect the data structure, finds unprocessed images and generates a Kubernetes pod description file for such image. Then the master node generates and orchestrates all pods, that will be consequently executed.
This use of Kubernetes demonstrates that these analysis pipelines can be parallelized and accelerated. A future challenge, especially if these types of analysis move out of the laboratory and into diagnostic settings, will be the need for far faster computing for on-demand or even real-time analysis. This so-called “edge computing” challenge may be well addressed by the emergence of specially designed neural network processors designed to efficiently carry out machine learning tasks. While the training of neural network models largely requires GPU-powered workstations, the actual step of calculating inference can be carried out on these special purpose chips, very much like how machine learning currently augments the cameras in many smartphones. These chips are being combined with system-on-a-chip devices (SoCs) in a new wave of single-board computers, such as the Google Coral. The low cost (~$150) and small form factor of these edge computing devices may enable the construction of data analysis systems composed to dozens of boards capable of providing efficient and fast parallel computation for the pipeline approach described here.
6. Conclusions and outlook
The analysis of large image datasets is an everyday research task for biomedical engineers. A concise data policy and automated image processing can help the everyday work of laboratories. Deep convolutional neural network methods show high level of performance and can automate daunting tasks. Recent computer science techniques developed for cloud computing, data sharing, operating-system virtualization, and cluster orchestration provide abstractions of data and computation management applicable to biomedical engineering and translational research. While these technologies take some time to set up, from our experience, they can pay off greatly for medium to large research studies.
When dealing with medium-to-large datasets, we propose the following recommendations: a) centralize the data, b) establish a data storage policy, c) establish a data processing pipeline, d) process overnight automatically, e) use the same code for prototyping and processing, f) compartmentalize using container software, g) deploy using clustering management software if needed and h) perform quality control images of the results. Such recommendations have been adopted in our laboratory for the last 3 years and have enabled us to deal with multi-terabyte research projects.
While the tools described here are applied to imaging data, they are not unique to images and can certainly be applied to other types of data, such as mass spectral data and DNA sequences.
We are now in the process of expanding the discussed analysis approaches into all current and future studies to eliminate analysis bottlenecks and accelerate the pace of translation. To provide those interested in further examples, a code repository has been set up via the Evans Laboratory Github page (https://github.com/evansgroup/BioEssays2018, accessed on 13.03.2019).
Acknowledgements
We would like to thank Petra Krauledat and Peter Hansen of PnP Research Corporation for their mentorship, expertise, and constant drive to push the state-of-the-art in research. None of the work described here would be possible without their continued and valued collaboration. We would like to thank members of the Evans laboratory with their help in preparing the manuscript, in particular Sinyoung Jeong, Haley Marks, John Nguyen, Sam Osseiran, and Emmanouil Rousakis. We also would like to acknowledge Marcelo Periera de Silva for his help in network and server debugging.
Footnotes
Disclosures
Germán González is partner at PNP Research Corp. and founder and CEO of Sierra Research S.L., an imaging processing consulting company. He has no conflicts of interests with respect to the technologies mentioned in this work from either company. Conor L. Evans is an Assistant Professor at the Wellman Center. He has no conflicts of interest with respect to the technologies discussed here.
Bibliography
- [1].Zhang X, Boutros M, BMC Bioinformatics 2013, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Singh S, Carpenter AE, Genovesio A, Journal of Biomolecular Screening 2014, 19, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kang J, Hsu CH, Wu Q, Liu S, Coster AD, Posner BA, Altschuler SJ, Wu LF, Nature Biotechnology 2016, 34, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bougen-Zhukov N, Loh SY, Lee HK, Loo LH, Cytometry Part A 2017, 91, 115. [DOI] [PubMed] [Google Scholar]
- [5].Rajwa B, Assay and Drug Development Technologies 2017, 15, 15. [DOI] [PubMed] [Google Scholar]
- [6].Ouyang W, Zimmer C, Current Opinion in Systems Biology 2017, 4, 105. [Google Scholar]
- [7].Regan E. a., Hokanson JE, Murphy JR, Lynch D. a., Beaty TH, Curran-everett D, Silverman EK, Crapo JD, C. Investigators, Epidemiology 2011, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sarkans U, Gostev M, Athar A, Behrangi E, Melnichuk O, Ali A, Minguet J, Rada JC, Snow C, Tikhonov A, Brazma A, McEntyre J, Nucleic Acids Research 2018, 46, 1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Orloff DN, Iwasa JH, Martone ME, Ellisman MH, Kane CM, Nucleic Acids Research 2013, 41, 1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bray MA, Gustafsdottir SM, Rohban MH, Singh S, Ljosa V, Sokolnicki KL, Bittker JA, Bodycombe NE, Dan?Ìk V, Hasaka TP, Hon CS, Kemp MM, Li K, Walpita D, Wawer MJ, Golub TR, Schreiber SL, Clemons PA, Shamji AF, Carpenter AE, GigaScience 2017, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cook CE, Bergman MT, Cochrane G, Apweiler R, Birney E, Nucleic Acids Research 2018, 46, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Antal B. l., Chessel A, Carazo Salas RE, Genome Biology 2015, 16, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Allan C, Burel JM, Moore J, Blackburn C, Linkert M, Loynton S, MacDonald D, Moore WJ, Neves C, Patterson A, Porter M, Tarkowska A, Loranger B, Avondo J, Lagerstedt I, Lianas L, Leo S, Hands K, Hay RT, Patwardhan A, Best C, Kleywegt GJ, Zanetti G, Swedlow JR, Nature Methods 2012, 9, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Burel JM, Besson S, Blackburn C, Carroll M, Ferguson RK, Flynn H, Gillen K, Leigh R, Li S, Lindner D, Linkert M, Moore WJ, Ramalingam B, Rozbicki E, Tarkowska A, Walczysko P, Allan C, Moore J, Swedlow JR, Mammalian Genome 2015, 26, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li S, Besson S, Blackburn C, Carroll M, Ferguson RK, Flynn H, Gillen K, Leigh R, Lindner D, Linkert M, Moore WJ, Ramalingam B, Rozbicki E, Rustici G, Tarkowska A, Walczysko P, Williams E, Allan C, Burel JM, Moore J, Swedlow JR, Methods 2016, 96, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bauch A, Adamczyk I, Buczek P, Elmer F-J, Enimanev K, Glyzewski P, Kohler M, Pylak T, Quandt A, Ramakrishnan C, Beisel C, Malmstrom L, Aebersold R, Rinn B, BMC Bioinformatics 2011, 12, 468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Adebayo S, McLeod K, Tudose I, Osumi-Sutherland D, Burdett T, Baldock R, Burger A, Parkinson H, Journal of Animal Science and Biotechnology 2016, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Martel AL, Hosseinzadeh D, Senaras C, Zhou Y, Yazdanpanah A, Shojaii R, Patterson ES, Madabhushi A, Gurcan MN, Cancer Research 2017, 77, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Iudin A, Korir PK, Salavert-Torres J, Kleywegt GJ, Patwardhan A, Nature Methods 2016, 13, 387. [DOI] [PubMed] [Google Scholar]
- [20].Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarahan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Rylstov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J, Microarray Technologies 2003, 34, 374. [DOI] [PubMed] [Google Scholar]
- [21].Crawford KL, Neu SC, Toga AW, Neuroimage 2016, 124, 1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gorgolewski KJ, Varoquaux G, Rivera G, Schwarz Y, Ghosh SS, Maumet C, Sochat VV, Nichols TE, Poldrack RA, Poline J-B, Yarkoni T, Margulies DS, Frontiers in Neuroinformatics 2015, 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kennedy DN, Haselgrove C, Riehl J, Preuss N, Buccigrossi R, Neuroimage 2016, 124, 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Erickson BJ, Fajnwaks P, Langer SG, Perry J, Translational Oncology 2014, 7, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Muehlboeck J-S, Westman E, Simmons A, Frontiers in Neuroinformatics 2014, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Williams E, Moore J, Li SW, Rustici G, Tarkowska A, Chessel A, Leo S, Antal B. l., Ferguson RK, Sarkans U, Brazma A, Carazo Salas RE, Swedlow JR, Nature Methods 2017, 14, 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pincus Z, Theriot JA, Journal of Microscopy 2007, 227, 140. [DOI] [PubMed] [Google Scholar]
- [28].R‰mˆ P, Sacher R, Snijder B, Begemann B, Pelkmans L, Bioinformatics 2009, 25, 3028. [DOI] [PubMed] [Google Scholar]
- [29].Rajaram S, Pavie B, Wu LF, Altschuler SJ, Nature Methods 2012, 9, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Caicedo JC, Cooper S, Heigwer F, Warchal S, Qiu P, Molnar C, Vasilevich AS, Barry JD, Bansal HS, Kraus O, Wawer M, Paavolainen L, Herrmann MD, Rohban M, Hung J, Hennig H, Concannon J, Smith I, Clemons PA, Singh S, Rees P, Horvath P, Linington RG, Carpenter AE, Nature Methods 2017, 14, 849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fetz V, Prochnow H, Brˆnstrup M, Sasse F, Nat. Prod. Rep 2016, 33, 655. [DOI] [PubMed] [Google Scholar]
- [32].Gordonov S, Hwang MK, Wells A, Gertler FB, Lauffenburger DA, Bathe M, Integr. Biol 2016, 8, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pennisi E, Science 2016, 352, 877. [DOI] [PubMed] [Google Scholar]
- [34].Jones TR, Carpenter AE, Lamprecht MR, Moffat J, Silver SJ, Grenier JK, Castoreno AB, Eggert US, Root DE, Golland P, Sabatini DM, Proceedings of the National Academy of Sciences 2009, 106, 1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Horvath P, Wild T, Kutay U, Csucs G, Journal of Biomolecular Screening 2011, 16, 1059. [DOI] [PubMed] [Google Scholar]
- [36].Dao D, Fraser AN, Hung J, Ljosa V, Singh S, Carpenter AE, 2016, 32, 3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Grys BT, Lo DS, Sahin N, Kraus OZ, Morris Q, Boone C, Andrews BJ, Journal of Cell Biology 2016, 216, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kraus OZ, Ba JL, Frey BJ, Bioinformatics 2016, 32, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kraus OZ, Grys BT, Ba J, Chong Y, Frey BJ, Boone C, Andrews BJ, Molecular Systems Biology 2017, 13, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Godinez WJ, Hossain I, Lazic SE, Davies JW, Zhang X, Bioinformatics 2017, 33, 2010. [DOI] [PubMed] [Google Scholar]
- [41].Keras; Chollet F, Deep learning with python, Manning Publications Co., 2017. [Google Scholar]
- [42].Jones TR, Kang IH, Wheeler DB, Lindquist RA, Papallo A, Sabatini DM, Golland P, Carpenter AE, BMC Bioinformatics 2008, 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Van Der Maaten LJP, Hinton GE, Journal of Machine Learning Research 2008, 9, 2579. [Google Scholar]
- [44].Zare H, Shooshtari P, Gupta A, Brinkman RR, BMC Bioinformatics 2010, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Qiu P, Simonds EF, Bendall SC, Gibbs KD, Bruggner RV, Linderman MD, Sachs K, Nolan GP, Plevritis SK, Nature Biotechnology 2011, 29, 886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Amir EAD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe?Er D, Nature Biotechnology 2013, 31, 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Anchang B, Hart TDP, Bendall SC, Qiu P, Bjornson Z, Linderman M, Nolan GP, Plevritis SK, Nature Protocols 2016, 11, 1264. [DOI] [PubMed] [Google Scholar]
- [48].Blischak JD, Davenport ER, Wilson G, PLoS Computational Biology 2016, 12, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shen H, Nature 2014, 515, 151. [DOI] [PubMed] [Google Scholar]
- [50].Boettiger C, ACM SIGOPS Operating Systems Review 2015, 49, 71. [Google Scholar]
- [51].Gillies RJ, Kinahan PE, Hricak H, Radiology 2016, 278, 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Probst J, Koch P, Hüttmann G, “Real time 3D rendering of optical coherence tomography volumetric data”, presented at European Conference on Biomedical Optics, 2009. [Google Scholar]
- [53].Sansone SA, Rocca-Serra P, Field D, Maguire E, Taylor C, Hofmann O, Fang H, Neumann S, Tong W, Amaral-Zettler L, Begley K, Booth T, Bougueleret L, Burns G, Chapman B, Clark T, Coleman LA, Copeland J, Das S, De Daruvar A, De Matos P, Dix I, Edmunds S, Evelo CT, Forster MJ, Gaudet P, Gilbert J, Goble C, Griffin JL, Jacob D, Kleinjans J, Harland L, Haug K, Hermjakob H, Sui SJH, Laederach A, Liang S, Marshall S, McGrath A, Merrill E, Reilly D, Roux M, Shamu CE, Shang CA, Steinbeck C, Trefethen A, Williams-Jones B, Wolstencroft K, Xenarios I, Hide W, Nature Genetics 2012, 44, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Linkert M, Rueden CT, Allan C, Burel JM, Moore W, Patterson A, Loranger B, Moore J, Neves C, MacDonald D, Tarkowska A, Sticco C, Hill E, Rossner M, Eliceiri KW, Swedlow JR, Journal of Cell Biology 2010, 189, 777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Freedman LP, Inglese J, Cancer Research 2014, 74, 4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Vempati UD, Chung C, Mader C, Koleti A, Datar N, Vidovi? D. i., Wrobel D, Erickson S, Muhlich JL, Berriz G, Benes CH, Subramanian A, Pillai A, Shamu CE, Sch¸rer SC, Journal of Biomolecular Screening 2014, 19, 803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Michener WK, PLoS Computational Biology 2015, 11, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Auffray C, Balling R, Barroso I, Bencze L. s., Benson M, Bergeron J, Bernal-Delgado E, Blomberg N, Bock C, Conesa A, Del Signore S, Delogne C, Devilee P, Di Meglio A, Eijkemans M, Flicek P, Graf N, Grimm V, Guchelaar HJ, Guo YK, Gut IG, Hanbury A, Hanif S, Hilgers RD, Honrado n., Hose DR, Houwing-Duistermaat J, Hubbard T, Janacek SH, Karanikas H, Kievits T, Kohler M, Kremer A, Lanfear J, Lengauer T, Maes E, Meert T, M¸ller W, Nickel D, Oledzki P, Pedersen B, Petkovic M, Pliakos K, Rattray M, i M‡s JR, Schneider R, Sengstag T, Serra-Picamal X, Spek W, Vaas LAI, van Batenburg O, Vandelaer M, Varnai P, Villoslada P, VizcaÌno JA, Wubbe JPM, Zanetti G, Genome Medicine 2016, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wilkinson MD, Dumontier M, Aalbersberg IJ, Appleton G, Axton M, Baak A, Blomberg N, Boiten J-W, da Silva Santos LB, Bourne PE, Bouwman J, Brookes AJ, Clark T, Crosas M, Dillo I, Dumon O, Edmunds S, Evelo CT, Finkers R, Gonzalez-Beltran A, Gray AJG, Groth P, Goble C, Grethe JS, Heringa J, Hoen P. A. C. t., Hooft R, Kuhn T, Kok R, Kok J, Lusher SJ, Martone ME, Mons A, Packer AL, Persson B, Rocca-Serra P, Roos M, van Schaik R, Sansone S-A, Schultes E, Sengstag T, Slater T, Strawn G, Swertz M. a., Thompson M, van der Lei J, van Mulligen E, Velterop J, Waagmeester A, Wittenburg P, Wolstencroft K, Zhao J, Mons B, Scientific Data 2016, 3, 160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Leonelli S, Davey RP, Arnaud E, Parry G, Bastow R, Nature Plants 2017, 3, 1. [DOI] [PubMed] [Google Scholar]
- [61].Nichols T, Kriegeskorte N, Yeo BTT, Milham MP, Poldrack RA, Poline J.-b., E. Proal, 2017, 20, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Liu W, Anguelov D, Erhan D, Szegedy C, Reed S, Fu C-Y, Berg AC, “Ssd: Single shot multibox detector”, presented at European conference on computer vision, 2016. [Google Scholar]
- [63].Ronneberger O, Fischer P, Brox T, “U-net: Convolutional networks for biomedical image segmentation”, presented at International Conference on Medical image computing and computer-assisted intervention, 2015. [Google Scholar]
- [64].Russakovsky O, Deng J, Su H, Krause J, Sathesh S, Ma S, Huang X, Karpathy A, Kosia A, Bernstein M, Berg AC, Fei-Fei L, International Journal of Computer Vision 2015, 115, 211. [Google Scholar]
- [65].Ren S, He K, Girshick R, Sun J, IEEE Transactions on Pattern Analysis & Machine Intelligence 2017, 39, 1137. [DOI] [PubMed] [Google Scholar]
- [66].Redmon J, Divvala S, Girshick R, Farhadi A, “You only look once: Unified, real-time object detection”, presented at Proceedings of the IEEE conference on computer vision and pattern recognition, 2016. [Google Scholar]
- [67].Jégou S, Drozdzal M, Vazquez D, Romero A, Bengio Y, “The One Hundred Layers Tiramisu: Fully Convolutional DenseNets for Semantic Segmentation”, presented at 2017 IEEE Conference on Computer Vision and Pattern Recognition Workshops (CVPRW), 21–26 July 2017, 2017. [Google Scholar]
- [68].Roy AG, Navab N, Wachinger C, IEEE Transactions on Medical Imaging 2018, 1. [DOI] [PubMed] [Google Scholar]
- [69].Milletari F, Navab N, Ahmadi S, “V-Net: Fully Convolutional Neural Networks for Volumetric Medical Image Segmentation”, presented at 2016 Fourth International Conference on 3D Vision (3DV), 25–28 Oct. 2016, 2016. [Google Scholar]
- [70].Prastawa M, Bullitt E, Ho S, Gerig G, Medical Image Analysis 2004, 8, 275. [DOI] [PubMed] [Google Scholar]
- [71].Goode A, Sukthankar R, Mummert L, Chen M, Saltzman J, Ross D, Szymanski S, Tarachandani A, Satyanarayanan M, 2008 5th IEEE International Symposium on Biomedical Imaging: From Nano to Macro, Proceedings, ISBI 2008, 249. [Google Scholar]
- [72].Bray MA, Fraser AN, Hasaka TP, Carpenter AE, Journal of Biomolecular Screening 2012, 17, 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lou X, Fiaschi L, Koethe U, Hamprecht FA, Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics) 2012, 7588 LNCS, 176. [Google Scholar]
- [74].Wilson G, Bryan J, Cranston K, Kitzes J, Nederbragt L, Teal TK, PLoS Computational Biology 2016, 13, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]