

SUMMARY
Mycobacterium tuberculosis, the causative agent of tuberculosis, withstands diverse environmental stresses in the host. The periplasmic protease HtrA is required only to survive extreme conditions in most bacteria but is predicted to be essential for normal growth in mycobacteria. We confirm that HtrA is indeed essential in Mycobacterium smegmatis and interacts with another essential protein of unknown function, LppZ. However, the loss of any of three unlinked genes, including those encoding Ami3, a peptidoglycan muramidase, and Pmt, a mannosyltransferase, suppresses the essentiality of both HtrA and LppZ, indicating the functional relevance of these genes’ protein products. Our data indicate that HtrA-LppZ is required to counteract the accumulation of active Ami3, which is toxic under the stabilizing influence of Pmt-based mannosylation. This suggests that HtrA-LppZ blocks the toxicity of a cell wall enzyme to maintain mycobacterial homeostasis.
In Brief
Wu et al. show that in Mycobacterium smegmatis, the putative cell wall amidase Ami3 can accumulate to toxicity under the stabilizing influence of Pmt mannosylation. To control Ami3 levels, an essential complex between the periplasmic serine protease HtrA and the lipoprotein LppZ regulates Ami3 levels, maintaining cellular integrity.
Graphical Abstract
INTRODUCTION
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis, remains the greatest infectious killer in human history, claiming 1.5 million lives each year (World Health Organization, 2017). Despite this enormous public health burden, efforts to curb Mtb’s spread and prevent rising rates of drug resistance have stagnated in recent decades. To combat the extremes of host defenses, Mtb deploys a tightly controlled array of stress response systems, including a large number of proteases. Recently mycobacterial proteases have emerged as appealing therapeutic targets because several of the most conserved homologs, including Clp, FtsH, HtrA, and the proteasome, are essential for the growth or virulence of Mtb (Raju et al., 2012a).
Despite their integral role in Mtb cell biology, mycobacterial proteases remain understudied. Recent work in our lab has implicated the mycobacterial Clp protease in the turnover of the essential transcriptional repressor WhiBI (Raju et al., 2012b, 2014). Similarly, transposon insertion sequencing data have indicated that the periplasmic serine protease HtrA (MSMEG_5070, Rv1223) is essential in mycobacteria (Griffin et al., 2011; Zhang et al., 2012; DeJesus and loerger, 2013). Despite this, the reasons for HtrA’s essentiality have remained unclear.
In E. coli and other organisms, HtrA is characterized as a nonessential, periplasmic protease with secondary chaperone function (Clausen et al., 2011). Although dispensable for normal growth, HtrA is crucial for the virulence of several intracellular pathogens, including Shigella, Listeria, and Salmonella (Ingmer and Brondsted, 2009). In these species, HtrA is required to tolerate a common set of stressful conditions, including high temperature, oxidative stress, and macrophage survival. The well-characterized E. coli HtrA homolog DegP is induced in conditions of membrane stress and becomes essential during heat shock (Ingmer and Brondsted, 2009). DegP contains a protease domain with a conserved Ser-His-Asp catalytic triad and two PDZ domains (Figure 1A) that regulate substrate binding and access to the proteolytic chamber. Although a handful of substrates have been identified in E. coli (Clausen et al., 2002), DegP appears to be mostly indiscriminate in its specificity, preferring denatured, unfolded substrates with hydrophobic C termini.
Figure 1. HtrA-LppZ Are Essential Interacting Proteins in Mycobacterium smegmatis.

(A) DegP/HtrA homolog domain architecture. E. coli DegP contains a protease domain and two C-terminal PDZ domains. In contrast, mycobacterial HtrA is anchored in the inner membrane and contains a single PDZ domain. Additionally, mycobacterial HtrA has a cytoplasmic domain with no homology to any known protein.
(B) HtrA and LppZ are essential by L5 swap. Top: a schematic of the L5 essentiality swap. Placing a second copy of htrA, along with a nourseothricin resistance cassette, at the L5 phage integration site allows replacement of endogenous htrA with a hygromycin resistance cassette. This copy of htrA can be swapped for another copy of htrA with a different antibiotic resistance marker but not for a functionally unrelated gene such as gfp. Bottom: quantification of htrA and gfp swaps. A total of 200 transformants were tested for antibiotic resistance. An equivalent swap was performed for IppZ and enumerated in the same manner.
(C) Cells depleted of HtrA grow at a slower rate. When regulated by an aTc-repressible promoter, htrA can be transcriptionally depleted from cells. Two strains were constructed: htrAHIGH, which used a strong promoter susceptible to aTc-based repression, and htrALOW, a weak promoter susceptible to aTc-based repression. These strains were grown, with or without aTc, in each of four conditions: rich media at 37°C, high temperature (42°C), carbon-nitrogen starvation (PBS-Tween 20), and oxidative stress (0.02% tert-butyl hydroperoxide). Error bars represent SD of the mean.
(D) HtrA and LppZ interact. HtrA-Strep and LppZ-FLAG were individually immunoprecipitated using anti-Strep and anti-FLAG magnetic beads, respectively, and the following fractions were analyzed using western blotting: L, lysate; FT, flow through; W1, wash 1; W2, wash 2; W3, wash 3; and E, elution. Control immunoprecipitations lacking the respective bait were performed simultaneously and lysate, flow through, and elution were analyzed.
(E) LppZ is not a substrate of HtrA. Strains expressing htrA or htrA(S354A) under htrA’s native promoter, htrAHIGH, or htrALOW were grown to log phase, and cell lysate was analyzed using western blotting to detect levels of HtrA-Strep and LppZ-FLAG. In the case of htrAHIGH and htrALOW, strains were also grown with or without 100 ng/mL aTc.
(F) Cells lacking HtrA or LppZ are highly sensitive to cell wall-targeting antibiotics. htrALOW and lppZLOW, strains expressing the respective essential gene under a weak promoter susceptible to aTc-based repression, were grown in teicoplanin and vancomycin, antibiotics targeting D-ala-D-ala crosslinking in the mycobacterial cell wall, and isoniazid, an antibiotic that inhibits mycolic acid synthesis.
(G) Depletion of HtrA or LppZ induces morphological defects. htrALOW and lppZ~OW were grown to log phase in aTc and observed under the microscope. Two representative images for each strain are shown. At least 100 cells were quantified in each condition. Dotted black lines indicate median values. Western blot images were cropped, but display all relevant lanes and reactive bands. ****p < 0.0001.
Like E. coli and many other organisms, virulent mycobacteria express three orthologs of HtrA. In Mtb, these are htrA (Rv1223), htrA2/pepD (Rv0983), and htrA3/pepA (Rv0125); of these, only HtrA is predicted to be essential. However, this essentiality appears to be conserved across all mycobacteria, regardless of pathogenicity, including in the fast growing Mycobacterium smegmatis (Msm), which encodes both htrA (MSMEG_5070) and the non-essential pepD (MSMEG_5486) (Lew et al., 2011). Nevertheless, htrA’s synteny in mycobacteria indicates that its transcription may be stress responsive, as it lies in an operon with sigE, a stress-responsive alternative sigma factor (Manganelli et al., 2004). Additionally, its high degree of sequence conservation with homologs in other species indicates that HtrA may be capable of recapitulating some stress-responsive or virulence functions. Although HtrA’s role in mediating the mycobacterial stress response may partially overlap with its homologs in other species, its essentiality indicates it must be involved in a unique regulatory pathway that supports mycobacterial growth and survival.
Here, we present evidence that HtrA serves a previously undescribed and essential role in the regulation of the growth of Msm by degrading a putative cell wall muramidase. HtrA engages in a periplasmic complex with another essential protein, LppZ, to control levels of the lethal hyperactivity of Ami3, an amidase stabilized by Pmt-mediated O-mannosylation. Loss of either ami3 or pmt is sufficient to relieve the essentiality of both htrA and lppZ. These data expand upon a growing body of literature illustrating the complex ways in which bacteria regulate growth and division.
RESULTS
HtrA-LppZ Forms an Essential Complex in the Mycobacterial Periplasm
Previous transposon insertion-based (Tn-seq) screens have predicted the essentiality of htrA in both Msm and Mtb (Griffin et al., 2011; Zhang et al., 2012; DeJesus and loerger, 2013). To confirm htrA essentiality, we attempted to delete these genes from the genome of M. smegmatis through mycobacterial recombineering and homologous recombination (Figure 1B, top). Briefly, a second copy of htrA was transformed into the L5 phage integration site with a nourseothricin resistance cassette, allowing the replacement of the endogenous copy of htrA with a hygromycin resistance cassette. We then attempted to transform htrA or an empty vector with a kanamycin resistance cassette into the L5 integration site (an “L5 swap”). Three outcomes can result from these experiments. First, integrations resulting in nonviable cells will prevent the growth of transformants on antibiotic selection. Second, successful “swaps” are those that acquire kanamycin resistance at the expense of nourseothricin resistance, indicating the replacement of the original integrated vector with the transformed vector. Third, the second vector may recombine in alongside the original vector, creating a double integrant that retains resistance to both antibiotics. In these experiments, htrA swapped in at a rate of 63.5%, while the empty vector yielded only double integrants (Figure 1B, bottom), indicating that HtrA is essential.
To assess the phenotype of htrA hypomorphs, we constructed depletion strains in which the only copies of htrA were under the control of anhydrotetracycline (aTc)-repressible promoters of different strengths (htrALOW and htrAHIGH). With the addition of aTc, HtrALOW cells clumped in liquid culture and exhibited a slower growth rate (Figure 1C) but remained viable, suggesting that a minimal amount of HtrA is sufficient to sustain growth. As HtrA homologs in other bacterial species are associated with protection against environmental stress, we tested the ability of depleted htrALOW cells to survive extreme conditions, including high temperature, oxidative stress, and carbonnitrogen starvation (Figure 1C). All tested conditions impaired the growth of or killed depleted htrALOW cells, indicating that higher amounts of HtrA are required to sustain growth under environmental stress.
We reasoned that identifying binding partners could help define HtrA’s function. To And interactors, we immunoprecipitated Strep-tagged HtrA and identified eluted proteins using mass spectrometry. The most abundant protein, LppZ (MSMEG_2369, Rv3006), had no known function. LppZ is also predicted to be essential in both M. smegmatis and M. tuberculosis and is annotated as a putative secreted lipoprotein (Griffin et al., 2011; Zhang et al., 2012; DeJesus and loerger, 2013). We confirmed LppZ essentiality with an L5 swap (Figure 1B) and detected a stable interaction between HtrA and LppZ by co-immunoprecipitation of tagged proteins (Figure 1D).
On the basis of this interaction, LppZ could be an HtrA substrate or adaptor. To distinguish between these possibilities, we tagged LppZ with a FLAG sequence and monitored its stability during depletion of htrA. LppZ levels were completely insensitive to alterations in HtrA (Figure 1E). This suggests that LppZ may be an adaptor of HtrA proteolytic activity, analogous to interactions in E. coli between the periplasmic protease Prc and the lipoprotein Nlpl (Singh et al., 2015; Tadokoro et al., 2004) and between DegP and recombinant mutants of the lipoprotein Lpp (Park et al., 2017).
If LppZ is an HtrA adaptor, LppZ mutants should phenocopy HtrA mutants. Indeed, both htrALOW and lppZLOW depletion strains exhibited increased sensitivity to antibiotics targeting cell wall crosslinking (but not other antibiotics such as isoniazid) (Figure 1F) and severe morphological defects (Figure 1G). Cells bulged and shortened upon htrALOW or lppZLOW depletion; however, a small subpopulation branched in only the htrALOW depletion. Both these morphologies are reminiscent of previously characterized strains in which dysregulation of cell wall machinery induces swelling and branching (Chao et al., 2013; Kieser et al., 2015). These data suggest that HtrA and LppZ act cooperatively as a complex in the same step of an essential genetic pathway that engages the cell wall.
Loss-of-Function Mutations in ami3, pmt, or mprB Suppress htrA and lppZ Essentiality
Unlike E. coli DegP, mycobacterial HtrA is predicted to be membrane anchored and have only one PDZ domain. Furthermore, it has a unique cytoplasmic domain at its N terminus that bears no homology to any known protein (Kapopoulou et al., 2011). To understand structure-function relationships in HtrA, we made a series of domain deletions and tested their viability using L5 swaps. We found that both the cytoplasmic and PDZ domains are essential for function (Figure S1A) but do not affect HtrA’s interaction with LppZ (Figure S1B). However, these experiments yielded a small number of survivors that grew despite loss of functional HtrA. We hypothesized that these might be due to mutations elsewhere in the chromosome and performed whole-genome sequencing of these strains to identify potential extragenic suppressors of htrA essentiality.
We sequenced nine strains in total and compared their genomes with that of the parental strains. Each swapped strain carried at least one of the following mutations (Figure S1C): one of five different frameshift mutations in ami3 (MSMEG_6406, or Rv8311 in Mtb), a putative N-acetylmuramoyl-L-alanine amidase; a frameshift mutation in pmt (MSMEG_5447, or Rv1002c in Mtb), a dolichyl-phosphate-mannose-protein man- nosyltransferase; or two SNPs in mprB (MSMEG_5487, or Rv0982 in Mtb), the sensor histidine kinase of a two-component regulatory system that also includes mprA (MSMEG_5488, or Rv0981 in Mtb). All of these genes are non-essential in Mtb and Msm (Griffin et al., 2011; Zhang et al., 2012; DeJesus and loerger, 2013). To verify these suppressors, we generated double-deletion strains in fresh backgrounds. The Δami3ΔhtrA and ΔpmtΔhtrA double-deletion strains grew robustly under all growth conditions tested, including those in which the htrALOW depletion strain failed to grow or died (Figures 2A and 2B). However, the ΔmprBΔhtrA double-deletion strain, although viable, grew slowly even under optimal conditions and remained moderately susceptible to environmental stress (Figure 2C). Importantly, loss of ami3, pmt, or mprB also allowed disruption of the otherwise essential lppZ (Figure S2). Given that mprB only partially suppresses the essentiality of htrA, and the previously characterized, wide-reaching transcriptional effects of deleting mprB (He et al., 2006), we focused our subsequent experiments on ami3 and pmt.
Figure 2. Loss-of-Function Mutations in ami3, pmt, or mprB Suppress htrA Essentiality.

(A-C) Double-mutant phenotypes of htrA suppressor strains: ami3 (A), pmt (B), and mprB (C). Wild-type Mycobacterium smegmatis (Msm), single suppressor gene deletions, suppressor deletions with htrA deletions, and htrA complemented strains (see Figures S1 and S6) were grown under conditions identical to those in Figure 1, including growth in rich media at 37°C, high temperature (42°C), carbon-nitrogen starvation (PBS-Tween 20), and oxidative stress (0.02% tert-butyl hydroperoxide). Error bars represent SD of the mean.
(D) htrA suppressor mutants exhibit reduced cell length. Single suppressor deletion strains (blue), htrA double-deletion strains (red), and htrA complemented strains (green) were grown to log phase and analyzed for total cell length (see Figure S2). At least 70 cells were quantified in each condition. Dotted black lines indicate median values. ****p < 0.0001.
Double-deletion strains allowed us to study the growth and morphology of strains that lacked htrA and lppZ. The suppressed strains grew at near normal rates (Figures 2A–2D), unlike the htrA depletion strain. Additionally, Δami3ΔhtrA and Δami3ΔlppZ were less sensitive to teicoplanin and vancomycin than the htrA and lppZ depletions (Figure S2C). However, double-deletion strains in all backgrounds exhibited reduced cell length (Figures 2D, S2A, and S2B), much like the htrA and lppZ depletions, indicating that the HtrA-LppZ complex may have an additional, non-essential function that affects cell division or elongation.
Ami3 Is Toxic when Overexpressed
Because deletions of ami3 and pmt could independently suppress the essentiality of both htrA and lppZ, we reasoned that these genes may operate within the same pathway. In E. coli, the peptidoglycan endopeptidase MepS has been shown to be negatively regulated by the periplasmic protease Prc in conjunction with a lipoprotein adaptor, Nlpl (Schwechheimer et al., 2015; Singh et al., 2015). Because the htrA and lppZ depletion phenotypes (Figures 1F and 1G) mimic cell wall dysregulation (Chao et al., 2013; Kieser et al., 2015), we hypothesized that the putative cell wall enzyme Ami3 might be a substrate of HtrA-LppZ. Indeed, when expressed under an aTc-inducible promoter (Figure S3A), ami3 overexpression quickly led to cell death (Figure 3A, top). We found the aTc minimum inhibitory concentration (MIC) of this strain to be about 12 ng/mL. This toxicity was exacerbated at high temperature (Figure 3A, bottom), mirroring the sensitivity of htrALOW depletion (Figure 1C). Furthermore, cells overexpressing ami3 exhibited reduced cell length and large polar bulges (Figure 3B), phenocopying htrA and lppZ depletion (Figure 1G). When C-terminally tagged with mRFP, Ami3 localized to the poles, mid-cell, and bulges, all points of cell growth and division (Figure S3B).
Figure 3. Ami3 Hyperactivity Is Toxic.

(A) Overexpression of Ami3 is lethal. Left: strains expressing wild-type ami3 or its catalytic mutants regulated by an aTc-inducible promoter were grown in the presence or absence of 100 ng/mL aTc (see Figure S3). Aliquots were taken at the indicated time points and analyzed for colony-forming units (CFUs). The uninduced wild-type ami3 strain is representative of all uninduced strains. Error bars represent SD of the mean. Right: aTcMIC of wild-type ami3 overexpression at different temperatures.
(B) Overexpression of different variants of Ami3 yields divergent phenotypes. Strains expressing wild-type ami3 or its catalytic mutants regulated by an aTc-inducible promoter were grown in the presence or absence of100 ng/mL aTc and observed under the microscope. At least 50 cells were quantified in each condition. Dotted black lines indicate data median. ***p < 0.001 and ****p < 0.0001.
(C) Ablation of Ami3 catalytic activity suppresses the essentiality of htrA and IppZ. Top: the endogenous copies of ami3 and htrA were replaced with zeocin and hygromycin resistance cassettes, respectively, and a copy of htrA was integrated at the L5 phage integration site. ami3 or a catalytically inactive variant of ami3 (ami3[H226A], ami3[H362A], and ami3[C370S]), was transformed into this background. ami3* indicates the respective ami3 allele. Full swaps that acquire kanamycin resistance at the expense of nourseothricin resistance render strains devoid of htrA (or IppZ and must thus carry a suppressor mutation. Bottom: quantification of ami3 and ami3* swaps. A total of 68 transformants were tested for antibiotic resistance. An equivalent swap was performed for IppZ and enumerated in the same manner.
Ami3 exhibits some structural homology to E. coli AmiD, a zinc metalloprotease in the amidase_2 domain family of cell wall hydrolyzing amidases (Kelley et al., 2015; Senzani et al., 2017). Threading the Ami3 sequence into the AmiD structure using Phyre2 allowed us to identify a potential triad of zinc-coordinating residues in Ami3 consisting of two histidines (H226, H362) and a cysteine (C370). To test the contribution of Ami3 enzymatic activity to HtrA essentiality, we constructed mutants carrying amino acid substitutions in each of these residues and attempted to swap enzymatically inactive versions of ami3 into a strain lacking the endogenous copies of both ami3 and htrA and carrying a copy of htrA at the L5 integration site. We found that that the inactive alleles could exchange with htrA and, in an analogous experiment, lppZ (Figure 3C). Thus, HtrA and LppZ are only essential in the presence of Ami3 catalytic activity.
If Ami3 hyperactivity leads to cell death, mutating the catalytic residues of Ami3 might relieve toxicity. Indeed, overexpression of ami3(H226A), ami3(H362A), or ami3(C370S) was no longer lethal to cells (Figure 3A). However, full wild-type viability was not restored, indicating that even inactive ami3 still retains some toxicity despite being expressed at levels broadly similar to the wild-type allele (Figures 3C and S3C). Cells that overexpressed the mutant protein were still abnormal, albeit with a morphology distinct from that of wild-type Ami3 overexpression: overaccumulation of the catalytically inactive mutants of Ami3 resulted in long, branching cells (Figure 3B). These cells resemble a subpopulation of cells with increased length and branching observed upon htrA depletion (Figure 1G).
HtrA-LppZ Regulates Ami3
On the basis of the correlation in phenotypes between ami3 overexpression and htrA and lppZ depletion, we hypothesized that HtrA-LppZ negatively regulates Ami3. To test this, we constructed strains in which the only copies of htrA and lppZ were expressed only in the presence of inducer and monitored levels of Ami3.
As expected, Ami3 levels decreased with HtrA induction (Figure 4A). We next mutated the catalytic serine of htrA, creating htrA(S354A). In PepD, another mycobacterial HtrA homolog, this mutation ablates up to 90% of proteolytic activity (White et al., 2010, 2011). As expected, Ami3 levels were less affected by HtrA(S354A) induction; this is not due to a difference in stability between wild-type and catalytically inactive HtrA (Figure 1E). Increasing HtrA(S354A) expression further decreased Ami3 levels, but not to the levels achieved by wild-type HtrA (Figure 4B). Accordingly, strains expressing only HtrA(S354A) were more sensitive to Ami3 overexpression (Figure S3D). A similar but milder decrease in Ami3 protein levels was observed upon LppZ induction (Figure 4A), indicating that LppZ makes a less substantial contribution to Ami3 regulation.
Figure 4. Induction of HtrA-LppZ Decreases Cellular Ami3 Levels.

(A) HtrA and LppZ expression decrease Ami3 levelsto varying degrees. Strains in which the only copy of htrA or lppZ was under an aTc-inducible promoter were constructed and grown with or without 100 ng/mL aTc for 12 hr. HtrA(S354A) is an allele of htrA presumed to exhibit reduced catalytic activity in HtrA homologs due to a mutation in its catalytic serine (see Figure S3). Whole-cell lysate was analyzed using western blotting using anti-Strep and anti-RpoB as a loading control.
(B) Higher expression of HtrA(S354A) further decreases Ami3. A second copy of aTc-inducible htrA(S354A) was transformed into the mutant strain described in (A), and all strains were grown with or without 100 ng/mL aTc for 8 hr. Whole-cell lysate was analyzed using western blotting using anti-Strep and anti-HtrA and anti-RpoB as a loading control. Western blot images were cropped but display all relevant lanes and reactive bands.
Finally, we tested the dependency of the HtrA-LppZ interaction on the presence of its potential substrate Ami3. We found that HtrA and LppZ interacted even in a Δami3 background (Figure S1D). We conclude that the HtrA-LppZ complex negatively regulates Ami3, with HtrA likely serving as the limiting factor and catalytic agent.
Pmt Stabilizes Ami3 through Mannosylation
Because loss of pmt suppressed the essentiality of htrA and IppZ (Figures 2B and 2D), we hypothesized that it might be a positive regulator of Ami3 toxicity by driving its accumulation. Pmt has been previously characterized as a periplasmic mannosyltrans- ferase with a catalytic aspartic acid at residue 68 (D68) (VanderVen et al., 2005; Liu et al., 2013); however, few of its targets have been identified. We tested the importance of this activity by mutating the catalytic aspartic acid D68 to an alanine, which decreases mannosyltransferase activity by more than 50% (VanderVen et al., 2005). Using an L5 swap, we found that pmt(D68A), but not wild-type pmt, permitted growth of a strain missing either htrA or lppZ (Figure 5A).
Figure 5. Pmt Mannosylation Stabilizes Ami3.

(A) Mutating the catalytic activity of Pmt suppresses the essentiality of htrA and IppZ. Top: the endogenous copies of pmt and htrA were replaced with zeocin and hygromycin resistance cassettes, respectively, and a copy of htrA was integrated at the L5 phage integration site. pmt or pmt(D68A), a catalytically inactive variant of pmt, was transformed into this background. Full swaps that acquire kanamycin resistance at the expense of nourseothricin resistance render strains devoid of htrA (or IppZ) and must thus carry a suppressor mutation. Bottom: quantification of pmt and pmt(D68A) swaps. A total of 60 transformants were tested for antibiotic resistance. An equivalent swap was performed for IppZ and enumerated in the same manner.
(B) Removing the mannosylation residues of Ami3 suppresses the essentiality of htrA and IppZ. Top: the endogenous copies of ami3 and htrA were replaced with zeocin and hygromycin resistance cassettes, respectively, and a copy of htrA was integrated at the L5 phage integration site. ami3 or ami3(T122A/T130A/T138A) (ami3***), an allele of ami3 in which all mannosylation sites have been mutated (see Figure S4), was transformed into this background. Full swaps that acquire kanamycin resistance at the expense of nourseothricin resistance render strains devoid of htrA (or lppZ) and must thus carry a suppressor mutation. Bottom: quantification of ami3 and ami3*** swaps. A total of 120 transformants were tested for antibiotic resistance. An equivalent swap was performed for lppZ and enumerated in the same manner; a total of 60 transformants were tested for antibiotic resistance.
(C) Ami3 levels decrease in cells missing Pmt. Strains expressing Ami3-Strep or Ami3*** in a wild-type, Δpmt, Δpmt+pmt, or Δpmt+pmt(D68A) background were grown to log phase. Whole-cell lysate was analyzed using western blotting using anti-Strep and anti-RpoB as a loading control.
(D) Pmt mannosylation increases the stability of Ami3 protein. At indicated time points after adding chloramphenicol, aliquots of cells were lysed, and levels of Ami3-Strep were monitored using western blot. Results are representative of three independent experiments.
(E) Quantification of Ami3 stability. Independent values of RpoB-normalized levels of Ami3 from (D) quantified using densitometry analysis. Western blot images were cropped but display all relevant lanes and reactive bands. *p < 0.05 and **p < 0.01. Error bars represent SD of the mean.
To test if Ami3 is itself mannosylated by Pmt, we immunoprecipitated epitope-tagged Ami3, analyzed it using mass spectrometry, and found hexose modifications on three threonines: T122, T130, and T138 (Figures S4A and S4B; Table S1). These modifications were no longer detected when an Ami3 sample was isolated from a Δpmt strain (Figure S4A; Table S1). Given that diverse hexose modifications emerge at similar molecular weights by mass spectrometry, we were not able to chemically confirm mannosylation; however, given our genetic data, we proceeded under the assumption that we had found a putative mannosylation. An allele substituting alanines for these residues (Ami3[T122A/T130A/T138A] or Ami3***) allowed growth of a strain lacking htrA (Figure 5B). We assessed the contribution of each mannosylated residue to Ami3 toxicity and found that T122 and T130 were critical, while T138 did not affect Ami3 toxicity (Figure S4C). To test whether mannosylation contributed to Ami3 stability, we monitored Ami3 levels by western blot in a Δpmt strain (Figure 5C, left). Adding wildtype pmt, but not pmt(D68A), restored wild-type levels of Ami3. In contrast, Ami3*** was insensitive to the presence of Pmt (Figure 5C, right).
The simplest explanation for the effect of mannosylation on protein quantity is that modification affects protein stability. To test this, we assayed the half-life of Ami3 in wild-type and Δpmt strains after inhibiting translation with chloramphenicol. Ami3 levels decreased significantly faster in the Δpmt strain compared with wild-type cells (Figures 5D and 5E) demonstrating that mannosylation stabilizes Ami3.
Because Pmt increases Ami3 stability, we reasoned that overexpression of Ami3 in cells lacking pmt may be less toxic. Indeed, Ami3 overexpression in a Δpmt background and Ami3*** overexpression exhibit less severe morphological defects and 5-fold increased survival compared with wild-type Ami3 overexpression (Figures 6A–6C and S5) To determine whether abundance and catalytic activity make independent contributions to toxicity, we monitored the growth of strains overexpressing Ami3(H362A), the catalytic mutant of Ami3 with protein levels most closely mimicking those of wild-type Ami3 (Figure S3B) in the Δpmt background. As expected, growth of a strain overexpressing Ami3(H362A) was further rescued (Figure 6C) and its morphological defects were lessened (Figures 6A and 6B) by knocking out pmt. These results are consistent with a model in which Ami3 activity and stability are independent, and altering both in combination has an additive effect on relieving the toxicity of overexpression.
Figure 6. Removal of Mannosylation Partially Rescues Morphology and Survival of Ami3 Overexpression.

(A) Loss of pmt relieves morphological defects in Ami3 overexpression. Wild-type ami3, ami3(H362A), or ami3*** was overexpressed in either Δami3 or Δami3Δpmt cells (see Figure S5).
(B) Quantification of cellular morphology of Ami3 overexpression. Cells were observed by microscopy and measured for total cell length, maximum cell width, and the proportion of the cell bulging (length of bulge over total cell length). At least 80 cells were quantified in each condition. Dotted black lines indicate median values.
(C) Survival and morphological quantification ofAmi3 overexpression. Strains expressing wild-type ami3, ami3(H362A), or ami3*** under an aTc-inducible promoter were grown in the presence of absence or 100 ng/mL aTc. Aliquots were taken at the indicated time points for CFU analysis. The uninduced wildtype Ami3 strain is representative of all uninduced strains. The growth of various strains was compared, and demarcated by asterisks (comparing the overexpression of ami3 in a wild-type or Δpmt background), stars (comparing the overexpression of wild-type ami3 or ami3***), or X’s (comparing the overexpression of ami3(H362A) in a wild-type or Δpmt background).
*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Error bars represent SD of the mean.
Finally, mannosylation does not appear to affect the interaction of HtrA and LppZ (Figure S1D). Additionally, HtrA still reduces Ami3 levels in a Δpmt strain (Figure S4D), demonstrating that mannosylation is not required to elicit the activity of HtrA-LppZ against Ami3.
Amidases Have Redundant Functions in Msm
Although Ami3 has the potential to accumulate to toxicity, it is not an essential gene, and loss of ami3 is not associated with any obvious phenotype (Figures S6A and S6B). However, Ami3 is not the only putative N-acetylmuramoyl-L-alanine amidase in Msm; Ami1 and Ami4 are also members of this family (Machow- ski et al., 2014), and several studies have presented evidence of Amil’s influence on normal cell division (Senzani et al., 2017; Li et al., 2018). In E. coli, amidases have redundant functions and are not individually essential for growth, and mutants missing multiple amidases remain viable (Heidrich et al., 2001; van Heijenoort, 2011). To test for similar redundancy in Msm, we constructed strains lacking individual and multiple amidases. We found that all of these strains had normal growth kinetics and morphologies (Figures S6A and S6B) but were altered in their accumulation of the fluorescent molecule calcein (Figure S6C). In accordance with previous studies (Rego et al., 2017), we found rifampin susceptibility in these mutants in a manner that correlated with calcein permeability (Figure S6D). However, loss of either ami1 or ami4 was unable to suppress htrA essentiality (Figure S6E), highlighting Ami3’s unique contribution to necessitating the activity of HtrA-LppZ.
DISCUSSION
Peptidoglycan hydrolases must be carefully regulated in a spatiotemporal manner to coordinate the complex processes of cell expansion and division. Dysregulation of cell wall enzymes has been shown to be toxic in species from E. coli to the various mycobacteria (van Heijenoort, 2011; Chao et al., 2013). The rapid turnover of cell wall requires an equally fast disposal of unwanted enzymes, and in the extracellular compartment, fewer mechanisms are available than in the cytoplasm. Proteolysis is one of the more rapid ways to effect change in the periplasm (Mukherjee et al., 2009; Festa et al., 2010). Protein processing and degradation have already been shown to play important roles in the fine-tuning of cell division proteins. For instance, the hydrolase RipA must be cleaved by the serine protease MarP to process peptidoglycan (Botella et al., 2017). Cleavage can also efficiently halt the activity of enzymes: PBPB is cleaved by the metallopro- tease Rv2869 in Mtb during oxidative stress (Mukherjee et al., 2009). Finally, several factors involved in cell wall assembly have been shown to be targeted for pupylation and subsequent proteasomal degradation, including MurA, KasB, and MtrA (Festa et al., 2010). However, none of these proteolytic or processing systems are essential for viability and only take on critical roles under environmental stress.
We propose a model in which HtrA and LppZ are essential to degrade Ami3, thereby controlling its toxicity in Msm (Figure 7). Over the course of the cell cycle, preexisting cell wall material must be cleaved to make room for newly synthesized precursors. Cell wall hydrolases such as Ami3 are double-edged swords: although they are important for proper growth and septation, overactivity can destabilize the cell wall and lead to autolysis. Without spatiotemporal regulation, sustainable cellular growth cannot occur, thus necessitating the activity of a periplasmic protease such as HtrA that can regulate a highly active enzyme like Ami3 in Msm.
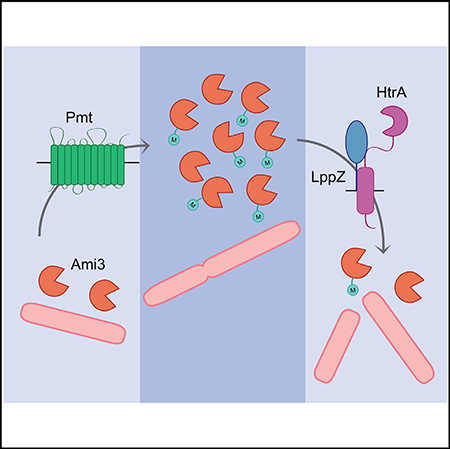
Figure 7. Model of HtrA-LppZ-Mediated Regulation of Ami3.

The muramidase Ami3, under the stabilizing influence of Pmt, potentially accumulates to toxic levels over the course of the cell cycle. The HtrA-LppZ complex restores normal Ami3 levels to abrogate this lethality and is thus essential for viability.
Notably, because Ami3 is still not completely stable even upon htrA depletion (Figures 5D and 5E), other proteases may have a more minor effect on Ami3 levels and will be the subject of future work.
The finding that HtrA requires a second protein is consistent with other observations of periplasmic proteases that rely on partners for fine-tuned regulation. Lipoproteins have been shown to be allosteric modulators of proteases in a variety of contexts (Tadokoro et al., 2004; Singh et al., 2015; Park et al., 2017) and have also been shown to directly activate the activity of cell wall enzymes (Paradis-Bleau et al., 2010; Typas et al., 2010; Egan et al., 2014; Lupoli et al., 2014). It is possible that LppZ facilitates the interaction between HtrA and Ami3, enhances the activity of HtrA, primes Ami3 for regulation by HtrA, or some combination of the above.
Although we were unable to measure amidase activity directly, Ami3 is likely a bona fide amidase on the basis of the phenotype of overexpression and requirement of catalytic residues for full toxicity, which mirror prior work in this field (Heidrich et al., 2001; Chao et al., 2013). In our work, all three N-acetylmuramoyl-L-alanine amidases in Msm are largely dispensable, though the importance of different amidases may vary in different Msm strains (Bavesh Kana, personal communication). Several alternative enzymes able to cleave peptidoglycan crosslinks likely operate in our strain under the growth conditions that we assayed, compensating for the loss of this family of amidases. Moreover, we simply may not know the growth condition in which these amidases play a critical role.
If Ami3 is indeed an active amidase, it is perhaps surprising that even a catalytically inactive variant of Ami3 can arrest cell growth. There are multiple possibilities to explain our observations. First, these mutants might retain a small amount of activity that stunts growth when enough protein is produced. Second, amidase activity may account for only a portion of ami3 toxicity, which can still impair growth when expressed at high levels, perhaps by aggregating to levels the cell cannot tolerate. Third, Ami3 may engage with another cell wall enzyme that depends on Ami3 catalytic activity to execute its function. Enzymatically inactive Ami3 may thus lock its binding partner in a futile interaction that results in a failure to divide when the complex is sequestered or it fails to dissociate from peptidoglycan (Figure 3B). These models are not mutually exclusive.
In this context, mannosylation could alter protein folding or recognition by proteolytic enzymes. Post-translational modifications play an important role in the activation or stabilization of cell wall enzymes in mycobacteria. For instance, the peptidoglycan synthase PonAI and MurA regulator CwlM must be phosphorylated to be active (Kieser et al., 2015; Boutte et al., 2016). Although this could also be a possible consequence of Ami3 mannosylation, it is unlikely given that ami3 overexpression in a Δpmt background does not phenocopy overexpression of catalytically inactive ami3. Additionally, pmt mannosylation does not appear to affect localization, as Ami3-mRFP is recruited to the same locales regardless of background (Figure S3B).
Furthermore, Pmt might play a broad role in cell wall maintenance, as it contributes to the virulence, immunogenicity, permeability, and antibiotic susceptibility of Mtb and M. abscessus (Becker et al., 2017; Liu et al., 2013; Deng et al., 2016; Harriff et al., 2017). In fact, a Δpmt mutant in M. abscessus is more susceptible to several antibiotics including rifampin (Becker et al., 2017).
Proteases are attractive drug candidates, and in fact, an inhibitor of mycobacterial MarP and HtrA has been described (Zhao et al., 2015). However, at least for Msm, our results suggest that blocking HtrA could lead to high rates of resistance, as there are multiple mechanisms to escape drug-mediated killing. This might be less true for Mtb, especially during infection, when it is possible that Ami3 plays a larger role to maintain cellular integrity or HtrA, LppZ, and Pmt contribute to the pathogen’s virulence in additional ways (Liu et al., 2013; Deng et al., 2016; Harriff etal., 2017). Notably, because Mtb and Msm differ in many ways, including the existence of a third htrA homolog, pepA, in Mtb only, our findings may not be entirely generalizable. However, given the retention of all three HtrA family proteins in multiple pathogenic mycobacteria (Lew et al., 2011), it is likely that these proteases serve critical roles worthwhile of future study. In this light, targeting of the essential HtrA-LppZ pathway remains possible.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eric J. Rubin (erubin@hsph.harvard.edu).
EXPERIMENTAL MODEL DETAILS
Msm mc2155 was grown in liquid media containing 7H9 salts (Becton Dickinson) supplemented with 5 g/L albumin, 2 g/L dextrose, 0.85 g/L NaCl, 0.003 g/L catalase, 0.2% glycerol, and 0.05% Tween80, or plated on LB agar. For all oxidative stress experiments, strains were grown in Hartmans-de Bont (HdB) media, which was made as described (Hartmans and De Bont, 1992) with 0.05% Tween80. E. coli TOP10 was used for cloning. Antibiotic selection concentrations for M. smegmatis were as follows: 25 μg/mL kanamycin, 50 μg/mL hygromycin, 20 μg/mL zeocin, 20 μg/mL nourseothricin, and 5 μg/mL gentamicin. Antibiotic concentrations for E. coli were as follows: 50 μg/mL kanamycin, 100 μg/mL hygromycin, 50 μg/mL zeocin, and 40 μg/mL nourseothricin. Anhydrotetracyline was used at 100 ng/mL for gene induction or repression. All strains were grown at 37°C unless otherwise indicated.
METHOD DETAILS
Strain construction
Deletions of htrA and lppZ were generated by first transforming in a second copy of the gene at the L5 and Tweety phage integration sites (Lewis and Hatfull 2003; Pham et al. 2007), then using mycobacterial recombineering as previously described (van Kessel and Hatfull 2008) to delete the endogenous copy. Where indicated, htrA and lppZ were cloned into vectors with a tetON repressor to make their expression tetracycline inducible, or into vectors with a tetOFF repressor to make their expression tetracycline repressible.
All other deletions (ami3, pmt, and mprB) were generated by recombineering. To make the inducible overexpression strain, ami3 was sub-cloned into a multi-copy episomal vector carrying both the tet operator and the tet repressor.
L5 swaps were performed as previously described (Pashley and Parish 2003); this technique was used to 1) test essentiality and suppression where indicated and/or 2) generate different allelic variants of htrA or IppZ. When testing essentiality or suppression, 60–200 colonies were patched for kanamycin and nourseothricin resistance.
A full list of strain details, including genotypes, plasmids, and primers, is available in Table S2.
Microscopy and image analysis
Still images were taken of cells immobilized on agar pads on a Nikon Ti inverted widefield epifluorescence microscope with a Photometrics coolSNAP CCD monochrome camera and a Plan Apo 100X objective with a numerical aperture of 1.4. Images were processed using NIS Elements version 4.3 and ImageJ. All aTc-repressible and -inducible strains were depleted or induced by the addition of 100 ng/mL anhydrotetracycline (aTc). The red fluorescent 405 images were taken with a 528–553 excitation filter and a 590–650 emission filter.
Growth rate determination and kill curves
For growth curves, strains were grown to mid-log phase, diluted to OD600 0.005 in triplicate, and measured every 15 min in a Bioscreen growth curve machine (Growth Curves USA) at 37°C or 42°C, where indicated.
For environmental stress kill curves, strains were grown to mid-log phase, washed twice with PBS-Tween80, diluted to OD600 0.05, and rolled at 37°C in PBS-Tween80 or HdB + 0.02% tert-butyl hydroperoxide. At the indicated time points, 200 uL aliquots were removed, serially diluted in PBS-Tween80, and plated for CFUs.
For aTc-inducible overexpression kill curves, all steps were the same except cells were not washed before subculturing into 7H9 with kanamycin to maintain the episomal plasmid, with or without 100 ng/mL aTc.
Tert-butyl hydroperoxide and aTc were added once at the beginning of each experiment. Each kill curve was performed at least thrice, in triplicate, with similar results.
Immunoprecipitation and western blotting
To identify potential interactors of HtrA, 200 mL mid-log phase cultures of a strain expressing HtrA-Strep and an untagged control were spun down and resuspended in 2 mL of Buffer W (100 mM Tris, 150 mM NaCl, 1 mM EDTA) with an EDTA-free protease inhibitor cocktail (Roche, Switzerland). The cells were lysed by bead beating and SDS was added to the lysate to a final concentration of 1%. The lysate was pre-cleared of endogenously biotinylated proteins using Pierce Avidin Agarose for 1 hour at room temperature. The cleared supernatant was then added to MagStrep “type3” XT beads (IBA Lifesciences) and incubated overnight at 4°C. The beads were then washed three times with Buffer Wand eluted using Buffer BXT (IBA Lifesciences). The eluted samples were separated on a 4%−12% NuPAGE Bis-Tris precast gel (Invitrogen Novex) and stained with Coomassie Blue. The entire lanes of eluted protein from the HtrA-Strep and untagged control immunoprecipitations were cut out and analyzed by the Harvard Taplin mass spectrometry facility. The unbiased immunoprecipitation was performed twice, both identifying LppZ as the top hit enriched in HtrA-Strep compared to the untagged control. To search for mannosylation residues in Ami3-Strep, all steps were performed as above except single bands were excised instead of entire lanes.
For co-immunoprecipitations, 100 mL cultures were split in two and all steps were performed as above for samples incubated with MagStrep beads. For immunoprecipitations using a-FLAG M2 Magnetic Beads (Sigma Aldrich), SDS was not added, preclearing with avidin agarose was skipped, and elution was carried out with FLAG peptide. Co-immunoprecipitations were analyzed by western blotting.
For western blots, cultures were spun down, resuspended in IP buffer (10 mM Tris-HCl (pH 8), 100 mM NaCl, 1 mM EDTA), and lysed by bead beating. Supernatants were normalized by A280 protein concentration, diluted with 6X Laemmli buffer, and run on SDS-PAGE gels with 4%−12% NuPAGE Bis Tris precast gels (Life Technologies). Membranes were blotted with rabbit α-FLAG (Sigma Aldrich) at 1:1000 in TBST + 5% milk, rabbit α-Strep (Genscript) at 1:1000 in TBST + 3% BSA, mouse α-RpoB (ThermoFisher Scientific) at 1:1000 in TBST + 5% milk, and rabbit α-HtrA (a generous gift from Helene Botella and Sabine Ehrt) at 1:1000 in TBST + 5% milk. For co-immunoprecipitation blots, the relevant fractions were run on a single membrane, blotted first with one antibody, stripped with Restore Western Blot Stripping Buffer (ThermoFisher Scientific 21059), and reblotted with the inverse.
MIC determination
The MIC of M. smegmatis strains were performed using the Alamar Blue assay as previously described (Kieser et al. 2015).
Calcein staining and flow cytometry
Mid-log-phase cultures were stained with 0.5 μg/mL calcein for 1 hour at 37°C. These cells were then analyzed by flow cytometry (MACSQuant VYB excitation: 488 nm; emission filter: 525/50).
Total cell length, bulge length, maximum cell width, and mean fluorescence intensity (MFI) were measured manually where indicated.
Hexose modification analysis by LC-MS/MS
To identify potential hexose modifications on Ami3, a 200 mL mid-log phase cultures of strains expressing Ami3-Strep in a wild-type or Δpmt background were spun down and resuspended in 2 mLof Buffer W (100 mM Tris, 150 mM NaCl, 1 mM EDTA) with an EDTA-free protease inhibitor cocktail (Roche, Switzerland). The cells were lysed by bead beating and SDS was added to the lysate to a final concentration of 1%. The lysate was pre-cleared of endogenously biotinylated proteins using Pierce Avidin Agarose for 1 hour at room temperature. The cleared supernatant was then added to MagStrep “type3” XT beads (IBA Lifesciences) and incubated overnight at 4°C. The beads were then washed three times with Buffer W and eluted using Buffer BX (IBA Lifesciences). The eluted samples were separated on a 4%−12% NuPAGE Bis-Tris precast gel (Invitrogen Novex) and stained with Coomassie Blue. A single band running at the predicted molecular weight of Ami3-Strep was excised from each sample.
The samples were reduced with 1 mM DTT for 30 minutes at 60°C, then alkylated with 5mM iodoacetamidefor 15 minutes at room temperature in the dark. Samples were then subjected to a modified in-gel trypsin digestion procedure (Shevchenko et al. 1996). Gel pieces were washed and dehydrated with acetonitrile for 10 minutes, followed by removal of acetonitrile and complete drying in a speed-vac. Rehydration of the gel pieces was performed with an 50 mM ammonium bicarbonate solution containing 12.5 ng/μl modified sequencing-grade trypsin (Promega, Madison, WI) at 4°C. Samples were then incubated at 37°C overnight. Peptides were later extracted by removing the ammonium bicarbonate solution, followed by one wash with a solution containing 50% acetonitrile and 1% formic acid. The extracts were then dried in a speed-vac for about an hour. The samples were then stored at 4°C until further analysis, upon which samples were reconstituted in 5 to 10 μl of HPLC solvent A (2.5% acetonitrile, 0.1% formic acid). A nano-scale reverse-phase HPLC capillary column was created by packing 2.6 μm C18 spherical silica beads into a fused silica capillary (100 μm inner diameter × ~30 cm length) with a flame-drawn tip (Peng and Gygi 2001). After the column was equilibrated, each sample was loaded onto the column via a Famos auto sampler (LC Packings, San Francisco, CA). A gradient was formed and peptides were eluted with increasing concentrations of solvent B (97.5% acetonitrile, 0.1% formic acid).
As each peptide was eluted, it was subjected to electrospray ionization and then entered into an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher Scientific, San Jose, CA). Eluting peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences were determined by matching the acquired fragmentation pattern with the Ami3 amino acid sequence, using a software program, Sequest (ThermoFinnigan, San Jose, CA) (Eng et al. 1994). The modification of 180.156 mass units to serine, and threonine was included in the database searches to determine hexose-modified peptides. Modifications were scored from 0 to 1000. Scores over 19 were considered confidently assigned. Scores of 1000 were considered unequivocal. All databases include a reversed version of all the sequences and the data was filtered to between a one and two percent peptide false discovery rate.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of experiments can be found in the figure legends. All experiments were performed at least twice. Means were compared using a two-sided Student’s t test, and all error bars indicate standard deviation around the mean. p < 0.05 was considered significant. For most microscopy experiments, roughly 100–200 cells were analyzed and data medians are shown. No statistical methods were used to predetermine sample size, and the researchers were not blinded to sample identity.
DATA AND SOFTWARE AVAILABILITY
Full, uncropped Western blots for relevant figures have been deposited to the Mendeley database under the following DOI: https://doi.org/10.17632/r7mm9mtt3v.1#folder-26c6e562-7d8d-44c8-a65a-21fa0ae692eb.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Strep-tag II (NWSHPQFEK) | GenScript | Cat#89494–724 |
| Rabbit polyclonal anti-FLAG | Sigma Aldrich | Cat#F7425 |
| Rabbit polyclonal anti-HtrA | Helene Botella and Sabine Ehrt | N/A |
| Mouse polyclonal anti-RpoB | ThermoFisher Scientific | Cat#MA1–25425 |
| Bacterial Strains | ||
| Escherichia coli TOP10 | ThermoFisher Scientific (formerly Invitrogen) | Cat#404010 |
| Msm mc2 155 | Laboratory of Barry Bloom | N/A |
| Full list of strains in Table S2 | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MagStrep type3 XT Beads, 5% suspension | IBA Life Sciences | Cat#2–4090-002 |
| Anti-FLAG M2 Magnetic Beads | Sigma Aldrich | Cat#M8823 |
| FLAG peptide | Sigma Aldrich | Cat#F3290 |
| Concanavalin A from Canavalia ensiformis (Jack bean) peroxidase conjugate | Sigma Aldrich | Cat#L6397 |
| Pierce Avidin Agarose | ThermoFisher Scientific | Cat#20219 |
| 10X Buffer BXT | IBA Life Sciences | Cat#2–1042-025 |
| Deposited Data | ||
| Full, uncropped Western blots | This paper | https://doi.org/10.17632/r7mm9mtt3v.1#folder-26c6e562-7d8d-44c8-a65a-21fa0ae692eb |
| Oligonucleotides | ||
| Full list of plasmids and strains in Table S2 | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/download.html |
Highlights.
HtrA-LppZ is an essential protease-lipoprotein complex in Mycobacterium smegmatis
HtrA-LppZ is required to negatively regulate Ami3, a putative cell wall amidase
Without HtrA-LppZ, Ami3 can accumulate to toxicity when mannosylated by Pmt
ACKNOWLEDGMENTS
We thank the Taplin Mass Spectrometry facility at Harvard Medical School for help with mass spectrometry and the Microscopy Resources on the North Quad (MicRoN) core at Harvard Medical School for the use of microscopes and assistance with imaging. Additionally, we thank Sabine Ehrt and Helene Botella for the generous gift of the HtrA antibody. This research was supported by NIH grant F31 AI131502-01 and NSF graduate fellowship to K.J.W. and NIH grant U19 AI107774 to E.J.R. and T.R.I. These funding bodies had no part in the design of the study or collection, analysis, and interpretation of data or in writing the manuscript. Finally, we thank Thomas Bernhardt, Michael Chase, Simon Dove, Sarah Fortune, Darren Higgins, Marcia Goldberg, Christopher Sassetti, Lok To Sham, Matthew Waldor, and Junhao Zhu for helpful discussions.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2018.12.063.
REFERENCES
- Becker K, Haldimann K, Selchow P, Reinau LM, Dal Molin M, and Sander P (2017). Lipoprotein glycosylation by protein-O-mannosyltransferase (MAB_1122c) contributes to low cell envelope permeability and antibiotic resistance of Mycobacterium abscessus. Front. Microbiol 8, 2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botella H, Vaubourgeix J, Lee MH, Song N, Xu W, Makinoshima H, Glickman MS, and Ehrt S (2017). Mycobacterium tuberculosis protease MarP activates a peptidoglycan hydrolase during acid stress. EMBO J. 36, 536–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutte CC, Baer CE, Papavinasasundaram K, Liu W, Chase MR, Meniche X, Fortune SM, Sassetti CM, Ioerger TR, and Rubin EJ (2016). A cytoplasmic peptidoglycan amidase homologue controls mycobacterial cell wall synthesis. Elife 5, e14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MC, Kieser KJ, Minami S, Mavrici D, Aldridge BB, Fortune SM, Alber T, and Rubin EJ (2013). Protein complexes and proteolytic activation of the cell wall hydrolase RipA regulate septal resolution in mycobacteria. PLoS Pathog. 9, e1003197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen T, Southan C, and Ehrmann M (2002). The HtrA family of proteases: implications for protein composition and cell fate. Mol. Cell 10, 443–455. [DOI] [PubMed] [Google Scholar]
- Clausen T, Kaiser M, Huber R, and Ehrmann M (2011). HTRA proteases: regulated proteolysis in protein quality control. Nat. Rev. Mol. Cell Biol 12, 152–162. [DOI] [PubMed] [Google Scholar]
- DeJesus MA, and Ioerger TR (2013). A hidden Markov model for identifying essential and growth-defect regions in bacterial genomes from transposon insertion sequencing data. BMC Bioinformatics 14, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng G, Zhang F, Yang S, Kang J, Sha S, and Ma Y (2016). Mycobacterium tuberculosis Rv0431 expressed in Mycobacterium smegmatis, a potentially mannosylated protein, mediated the immune evasion of RAW 264.7 macrophages. Microb. Pathog 100, 285–292. [DOI] [PubMed] [Google Scholar]
- Egan AJ, Jean NL, Koumoutsi A, Bougault CM, Biboy J, Sassine J, Solovyova AS, Breukink E, Typas A, Vollmer W, and Simorre JP (2014). Outer-membrane lipoprotein LpoB spans the periplasm to stimulate the peptidoglycan synthase PBP1B. Proc. Natl. Acad. Sci. U S A 111, 8197–8202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Festa RA, McAllister F, Pearce MJ, Mintseris J, Burns KE, Gygi SP, and Darwin KH (2010). Prokaryotic ubiquitin-like protein (Pup) proteome of Mycobacterium tuberculosis [corrected]. PLoS ONE 5, e8589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, and Sassetti CM (2011). High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7, e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harriff MJ, Wolfe LM, Swarbrick G, Null M, Cansler ME, Canfield ET, Vogt T, Toren KG, Li W, Jackson M, et al. (2017). HLA-E Presents Glyco- peptides from the Mycobacterium tuberculosis Protein MPT32 to Human CD8+ T cells. Sci. Rep 7, 4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmans S, and De Bont JAM (1992). The genus Mycobacterium — Nonmedical In The Prokaryotes, Second Edition, Dowrkin M, Falkow S, Rosenberg E, Schleifer K-H, and Stackebrandt E, eds. (Springer-Verlag; ), pp.1214–1237. [Google Scholar]
- He H, Hovey R, Kane J, Singh V, and Zahrt TC (2006). MprAB is a stress- responsive two-component system that directly regulates expression of sigma factors SigB and SigE in Mycobacterium tuberculosis. J. Bacteriol 188, 2134–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidrich C, Templin MF, Ursinus A, Merdanovic M, Berger J, Schwarz H, de Pedro MA, and Holtje JV (2001). Involvement of N-acetylmuramyl-L-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol. Microbiol 41, 167–178. [DOI] [PubMed] [Google Scholar]
- Ingmer H, and Brondsted L (2009). Proteases in bacterial pathogenesis. Res. Microbiol 160, 704–710. [DOI] [PubMed] [Google Scholar]
- Kapopoulou A, Lew JM, and Cole ST (2011). The MycoBrowser portal: a comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis (Edinb). 91, 8–13. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJ (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieser KJ, Boutte CC, Kester JC, Baer CE, Barczak AK, Meniche X, Chao MC, Rego EH, Sassetti CM, Fortune SM, and Rubin EJ (2015). Phosphorylation of the Peptidoglycan Synthase PonA1 Governs the Rate of Polar Elongation in Mycobacteria. PLoS Pathog. 11, e1005010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew JM, Kapopoulou A, Jones LM, and Cole ST (2011). Tubercu-List—10 years after. Tuberculosis (Edinb). 91, 1–7. [DOI] [PubMed] [Google Scholar]
- Lewis JA, and Hatfull GF (2003). Control of directionality in L5 integrase- mediated site-specific recombination. J. Mol. Biol 326, 805–821. [DOI] [PubMed] [Google Scholar]
- Li X, He J, Fu W, Cao P, Zhang S, and Jiang T (2018). Effect of Mycobacterium tuberculosis Rv3717 on cell division and cell adhesion. Microb. Pathog 117, 184–190. [DOI] [PubMed] [Google Scholar]
- Liu CF, Tonini L, Malaga W, Beau M, Stella A, Bouyssie D, Jackson MC, Nigou J, Puzo G, Guilhot C, et al. (2013). Bacterial protein-O-mannosylating enzyme is crucial for virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U S A 110, 6560–6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupoli TJ, Lebar MD, Markovski M, Bernhardt T, Kahne D, and Walker S (2014). Lipoprotein activators stimulate Escherichia coli penicillin-binding proteins by different mechanisms. J. Am. Chem. Soc 136, 52–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machowski EE, Senzani S, Ealand C, and Kana BD (2014). Comparative genomics for mycobacterial peptidoglycan remodelling enzymes reveals extensive genetic multiplicity. BMC Microbiol. 14, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganelli R, Fattorini L, Tan D, Iona E, Orefici G, Altavilla G, Cusatelli P, and Smith I (2004). The extra cytoplasmic function sigma factor sigma(E) is essential for Mycobacterium tuberculosis virulence in mice. Infect. Immun 72, 3038–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee P, Sureka K, Datta P, Hossain T, Barik S, Das KP, Kundu M, and Basu J (2009). Novel role of Wag31 in protection of mycobacteria under oxidative stress. Mol. Microbiol 73, 103–119. [DOI] [PubMed] [Google Scholar]
- Paradis-Bleau C, Markovski M, Uehara T, Lupoli TJ, Walker S, Kahne DE, and Bernhardt TG (2010). Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell 143, 1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Kim YT, Choi C, and Kim S (2017). Tripodal lipoprotein variants with C-terminal hydrophobic residues allosterically modulate activity of the DegP protease. J. Mol. Biol 429, 3090–3101. [DOI] [PubMed] [Google Scholar]
- Pashley CA, and Parish T (2003). Efficient switching of mycobacteriophage L5-based integrating plasmids in Mycobacterium tuberculosis. FEMS Microbiol. Lett 229, 211–215. [DOI] [PubMed] [Google Scholar]
- Peng J, and Gygi SP (2001). Proteomics: the move to mixtures. J. Mass Spectrom 36, 1083–1091. [DOI] [PubMed] [Google Scholar]
- Pham TT, Jacobs-Sera D, Pedulla ML, Hendrix RW, and Hatfull GF (2007). Comparative genomic analysis of mycobacteriophage Tweety: evolutionary insights and construction of compatible site-specific integration vectors for mycobacteria. Microbiology 153, 2711–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju RM, Goldberg AL, and Rubin EJ (2012a). Bacterial proteolytic complexes as therapeutic targets. Nat. Rev. Drug Discov 11, 777–789. [DOI] [PubMed] [Google Scholar]
- Raju RM, Unnikrishnan M, Rubin DH, Krishnamoorthy V, Kandror O, Akopian TN, Goldberg AL, and Rubin EJ (2012b). Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathog. 8, e1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju RM, Jedrychowski MP, Wei JR, Pinkham JT, Park AS, O’Brien K, Rehren G, Schnappinger D, Gygi SP, and Rubin EJ (2014). Posttranslational regulation via Clp protease is critical for survival of Mycobacterium tuberculosis. PLoS Pathog. 10, e1003994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego EH, Audette RE, and Rubin EJ (2017). Deletion of a mycobacterial divisome factor collapses single-cell phenotypic heterogeneity. Nature 546, 153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwechheimer C, Rodriguez DL, and Kuehn MJ (2015). NlpI-mediated modulation of outer membrane vesicle production through peptidoglycan dynamics in Escherichia coli. MicrobiologyOpen 4, 375–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senzani S, Li D, Bhaskar A, Ealand C, Chang J, Rimal B, Liu C, Joon Kim S, Dhar N, and Kana B (2017). An amidase_3 domain-containing N-acetylmuramyl-L-alanine amidase is required for mycobacterial cell division. Sci. Rep 7, 1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A,Wilm M,Vorm O, and Mann M (1996). Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem 68, 850–858. [DOI] [PubMed] [Google Scholar]
- Singh SK, Parveen S, SaiSree L, and Reddy M (2015). Regulated proteolysis of across-link-specific peptidoglycan hydrolase contributes to bacterial morphogenesis. Proc. Natl. Acad. Sci. U S A 112, 10956–10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadokoro A, Hayashi H, Kishimoto T, Makino Y, Fujisaki S, and Nishimura Y (2004). Interaction of the Escherichia coli lipoprotein NlpI with periplasmic Prc (Tsp) protease. J. Biochem 135, 185–191. [DOI] [PubMed] [Google Scholar]
- Typas A, Banzhaf M, van den Berg van Saparoea B, Verheul J, Biboy J, Nichols RJ, Zietek M, Beilharz K, Kannenberg K, von Rechenberg M, et al. (2010). Regulation of peptidoglycan synthesis by outer-membrane proteins. Cell 143, 1097–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heijenoort J (2011). Peptidoglycan hydrolases of Escherichia coli. Microbiol. Mol. Biol. Rev 75, 636–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kessel JC, and Hatfull GF (2008). Mycobacterial recombineering. Methods Mol. Biol 435, 203–215. [DOI] [PubMed] [Google Scholar]
- VanderVen BC, Harder JD, Crick DC, and Belisle JT (2005). Exportmediated assembly of mycobacterial glycoproteins parallels eukaryotic pathways. Science 309, 941–943. [DOI] [PubMed] [Google Scholar]
- White MJ, He H, Penoske RM, Twining SS, and Zahrt TC (2010). PepD participates in the mycobacterial stress response mediated through MprAB and SigE. J. Bacteriol 192, 1498–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MJ, Savaryn JP, Bretl DJ, He H, Penoske RM, Terhune SS, and Zahrt TC (2011). The HtrA-like serine protease PepD interacts with and modulates the Mycobacterium tuberculosis 35-kDa antigen outer envelope protein. PLoS ONE 6, e18175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2017). Global Tuberculosis Report (Geneva: World Health Organization; ). [Google Scholar]
- Zhang YJ, Ioerger TR, Huttenhower C, Long JE, Sassetti CM, Sacchettini JC, and Rubin EJ (2012). Global assessment of genomic regions required for growth in Mycobacterium tuberculosis. PLoS Pathog. 8, e1002946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N, Darby CM, Small J, Bachovchin DA, Jiang X, Burns-Huang KE, Botella H, Ehrt S, Boger DL, Anderson ED, et al. (2015). Target- based screen against a periplasmic serine protease that regulates intrabacterial pH homeostasis in Mycobacterium tuberculosis. ACS Chem. Biol 10, 364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Full, uncropped Western blots for relevant figures have been deposited to the Mendeley database under the following DOI: https://doi.org/10.17632/r7mm9mtt3v.1#folder-26c6e562-7d8d-44c8-a65a-21fa0ae692eb.