

Abstract
Extracellular mRNAs (ex-mRNAs) potentially supersede extracellular miRNAs (ex-miRNAs) and other RNA classes as biomarkers. We performed conventional small-RNA-sequencing (sRNA-seq) and sRNA-seq with T4 polynucleotide kinase (PNK) end treatment of total extracellular RNAs (exRNAs) isolated from serum and platelet-poor EDTA, acid citrate dextrose (ACD), and heparin plasma to study the effect on ex-mRNA capture. Compared with conventional sRNA-seq, PNK treatment increased the detection of informative ex-mRNAs reads up to 50-fold. The exRNA pool was dominated by RNA originating from hematopoietic cells and platelets, with additional contribution from the liver. About 60% of the 15- to 42-nt reads originated from the coding sequences, in a pattern reminiscent of ribosome profiling. Blood sample type had a considerable influence on the exRNA profile. On average approximately 350–1100 distinct ex-mRNA transcripts were detected depending on plasma type. In serum, additional transcripts from neutrophils and hematopoietic cells increased this number to near 2300. EDTA and ACD plasma showed a destabilizing effect on ex‑mRNA and noncoding RNA ribonucleoprotein complexes compared with other plasma types. In a proof-of-concept study, we investigated differences between the exRNA profiles of patients with acute coronary syndrome and healthy controls. The improved tissue resolution of ex‑mRNAs after PNK treatment enabled us to detect a neutrophil signature in ACS that escaped detection by ex‑miRNA analysis.
Keywords: Cardiology, Vascular Biology
Keywords: Bioinformatics, Molecular diagnosis, RNA processing
Small-RNA-sequencing coupled with T4 polynucleotide kinase end treatment of total extracellular RNA improves tissue and functional resolution.
Introduction
Extracellular RNAs (exRNAs) in biofluids were described as early as the first half of the 20th century (1) but underwent a more recent renaissance with the detection of circulating miRNAs (2). Despite the high nuclease activity in biofluids, extracellular miRNAs (ex-miRNAs) remain detectable due to protection by tightly bound RNA-binding proteins and/or inclusion into microvesicles (2–5). In recent years, especially with the advancement of RNA-seq, an extensive body of research accumulated regarding the role of ex‑miRNAs in a broad range of medical conditions and cardiovascular diseases, including advanced heart failure (6) and myocardial infarction (7).
Plasma or serum ex-miRNA signatures may be remarkably stable over time and informative as biomarkers, and we recently showed that a unique neuroendocrine ex‑miRNA signature could be followed for months in a healthy volunteer (8). However, a general limitation of ex-miRNAs is the relatively low number of miRNA genes, with only a few members with tissue-specific expression (9). In contrast, the number of mRNA-coding genes in the human genome is at least an order of magnitude higher (10), providing, in theory, a much better tissue and functional resolution for physiological conditions or disease states if captured extracellularly. While RNA-seq potentially offers the most comprehensive interrogation of extracellular mRNAs (ex-mRNAs) and ex-miRNAs and their changes in abundance, there is a lack of robust protocols and several challenges in the capture of fragmented exRNAs and the analysis of short sequencing reads.
Technical difficulties in exRNA profiling encompass the very low amounts of RNA in body fluids, and the influence of anticoagulants used for blood collection, increasing the likelihood for batch effects or spurious findings (11, 12). The type of blood sample used for RNA isolation can substantially influence the stability of certain ribonucleoprotein (RNP) complexes and associated RNAs. A striking example of differential stability of RNPs with different anticoagulants is the loss of 5′ transfer RNA (tRNA) fragments using magnesium ion–chelating EDTA or citrate salts for blood collection (6, 13). While it seems likely that these routinely used chelators for blood collection will effect the stability of other extracellular RNPs, the overall extent to which the sample types influence the exRNA profile remains unknown.
By design, small-RNA-sequencing (sRNA-seq) cDNA protocols enrich for miRNAs, which carry 5′ phosphate and 3′ hydroxyl groups. However, in body fluids, other classes of RNAs, including potentially mRNAs, most likely exist as degradation products due to the high nuclease activity (8). RNA degradation products commonly possess 5′ OH ends as well as 2′ or 3′ phosphate or 2′,3′ cyclic phosphate termini. These termini are incompatible with sRNA-seq, and fragments of those RNAs will largely escape detection. T4 polynucleotide kinase (PNK) is a 5′-kinase and 3′-phosphatase (14), and treatment of RNA by PNK rescues adapter ligation to RNA fragments devoid of the necessary termini. T4 PNK treatment has been used for different RNA-seq–based applications, including exRNA studies (15–17). However, an effect on ex-mRNA capture has not been shown thus far.
Here, we used a recently published RNA isolation protocol that quantitatively recovers exRNAs (8) and combined T4 PNK RNA treatment with sRNA-seq and stringent read annotation criteria to demonstrate effective and informative capture of ex-mRNAs. We investigated blood samples with different commonly used anticoagulants to identify confounding factors and finally tested the potential of ex-mRNAs in a proof-of-concept cohort of patients presenting with an acute coronary syndrome (ACS).
Results
PNK treatment of exRNA improves the capture of mRNA fragments by sRNA-seq.
To test if PNK treatment improves capture of ex-mRNA fragments, we performed sRNA-seq comparing untreated with PNK-treated total exRNA input from the same donors. Different anticoagulants were used to assess their influence on the exRNA profile (Figure 1).
Figure 1. Treatment of total extracellular RNA with T4 polynucleotide kinase followed by small-RNA-sequencing.

(A) Total RNA was isolated from 450 μl serum or platelet-depleted EDTA, acid citrate dextrose (ACD), and heparin plasma from 6 healthy individuals and purified using silica-based spin columns. Half of the RNA was treated with T4 polynucleotide kinase (T4 PNK) and repurified (PNK treated), and multiplexed small-RNA-sequencing (sRNA-seq) libraries were prepared separately for the untreated (libraries 1 and 3) and PNK-treated RNA (libraries 2 and 4). (B) Differences in read annotation in the 4 sample types for untreated RNA and PNK-treated RNA using initial annotation settings (reads 12–42 nt, up to 2 mismatches, multimapping). (C) Differences in ex‑mRNA capture between untreated and PNK-treated RNA using final annotation criteria (reads >15 nt, no mismatch and up to 2 mapping locations). Box plots show the median and first and third quartiles (bottom and top hinges). Whiskers extend at most ×1.5 interquartile range from the hinges; any data outside this are shown as individual outlier points. Shown are results from n = 6 individual samples per condition.
Plasma and serum were collected from 6 healthy volunteers. Collection tubes for plasma samples contained the divalent metal ion chelators EDTA and acid citrate dextrose (ACD) or the polyanion heparin. All plasma samples were platelet depleted, and total exRNA was recovered using our recently published isolation protocol, which preserves RNA integrity and quantitatively recovers exRNA (8). The initially isolated total RNA for each donor was split, and half of it was treated with PNK. The average RNA yield from a 450-μl sample was 12 ng (minimum 7 ng, maximum 21 ng) in serum, 8 ng (minimum 4 ng, maximum 13 ng) in EDTA plasma, 9 ng (minimum 4 ng, maximum 14 ng) in ACD plasma, and 13 ng (minimum 9 ng, maximum 20 ng) in heparin plasma (P < 0.05 for differences between serum or heparin and EDTA/ACD plasma; for details see Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.127317DS1). Multiplexed sRNA-seq libraries were prepared from size-selected 19- to 45-nt RNA using untreated and PNK-treated RNA from each donor using barcoding (Figure 1). RNA from the 6 donors was split into 2 libraries, with all 4 sample types from the same donor in 1 library, thus resulting in 4 libraries total with matched libraries 1 (untreated) and 2 (PNK treated) for 3 donors and matched libraries 3 (untreated) and 4 (PNK treated) for the remaining 3 donors (Figure 1A and Supplemental Data 1).
The length distribution of adapter-trimmed reads was unexpectedly skewed toward shorter reads, with 13%–26% of reads in the untreated samples (libraries 1 and 3) and 26%–41% of reads in the PNK-treated samples less than 19 nt in size and less than 4% (7% for the PNK-treated serum samples) of reads longer than 35 nt in size. Nevertheless, 74%–87% of reads in the untreated samples and 59%–75% of reads in the PNK-treated samples were within the selected size range of 19–45 nt, and 95% of all trimmed reads were within 12–42 nt. A high proportion of reads shorter than 20 nt has been previously observed by Turchinovich and colleagues (15) and is likely indicative of substantial degradation of unprotected RNA in blood. Such short reads impose challenges for confident transcript assignment due to multimapping. For conventional sRNA-seq, i.e., miRNA studies, this is minimized by hierarchical mapping and requiring a minimum read length of 16 nt (18). Hierarchical mapping ensures that more abundant RNAs, such as rRNAs and tRNAs, take precedence over less abundant classes, such as mRNAs and miRNAs, if a sequence matches to more than one RNA class. To arrive at a comprehensive assessment, we initially retained reads <16 nt. With that, over 80% of reads mapped to established classes of human RNAs and the human genome, revealing the expected enrichment for miRNAs in the untreated samples (Figure 1B). The major difference after PNK treatment was increased rRNA content. A residual 3%–15% of reads mapped to the E. coli genome and approximately 1% to bacterial expression plasmids and diatoms (Supplemental Data 1). Bacterial RNA is a common contaminant in recombinantly produced ligase enzymes used for library preparation, and residual diatom RNA exists in commercial silica matrices (spin columns) used for nucleic acid isolation. In standard RNA-seq applications, which use higher amounts of input RNA, trace amounts of contaminating RNAs are unnoticeable. However, with limited input RNA samples and the use of amplification prior to sequencing, these traces contribute a sizeable fraction of sequence reads in body fluids (6, 8).
Reads annotated as mRNAs (ex‑mRNA) constituted 6.5%–20% of total reads, with some enrichment after PNK treatment in EDTA and ACD plasma but not in heparin plasma or serum (Figure 1B). Further review of read alignments, however, showed that untreated samples collected more mRNA reads with 1 or 2 mismatches, i.e., inflating the mRNA read count by inclusion of low-confidence reads (Supplemental Data 2). As expected, reads <15 nt had a high fraction of multimapping (Supplemental Figure 1B). Therefore, our final ex-mRNA analysis was restricted to perfectly mapping reads (0 mismatch) 15 nt or longer with at most 2 mapping locations. The latter was necessary to account for the identical coding sequences (CDS) of the hemoglobin paralogs HBA1 and HBA2 that otherwise would have been underrepresented.
Using these annotation criteria, PNK treatment unambiguously increased the percentage of ex‑mRNA reads and, even more, the number of unique transcripts captured. Compared with untreated samples, in PNK-treated samples, the mRNA read count increased approximately 4-fold in ACD samples and approximately 9-fold in all other sample types (Figure 1C). Requiring 5 unique reads per mRNA and donor sample, we captured an average of 2313 (minimum 452, maximum 4634), 583 (minimum 162, maximum 1192), 350 (minimum 75, maximum 625), and 1108 (minimum 591, maximum 1760) distinct mRNA transcripts in serum, EDTA plasma, ACD plasma, and heparin plasma samples, respectively. This compared with only 46 (minimum 2, maximum 182), 33 (minimum 1, maximum 86), 27 (minimum 5, maximum 70), and 43 (minimum 0, maximum 140) distinct mRNAs in the corresponding untreated samples (P < 8 × 10–9, Wilcoxon rank-sum test), representing a 13- to 50-fold increase.
Each of the 4 libraries contained 1 water control sample that was carried through the RNA isolation process step without (libraries 1 and 3) or followed by PNK treatment (libraries 2 and 4). As expected, these samples had a high fraction of size marker, adapter, and bacterial RNA species. While they did not have a notable fraction of reads annotated as miRNAs (<1%), up to 18% of reads in the water samples were annotated as mRNAs. These sequences were almost exclusively short, multimapping repetitive sequences inflating transcript counts for biologically implausible transcripts.
Ex-mRNAs in circulation originate mostly from the CDSs and not UTRs.
It has been previously reported that ex-mRNAs in cell culture media mostly originate from the 3′ UTR of mRNA transcripts (17). Review of read alignments in our study, however, indicated that most of the ex‑mRNA reads originated from the transcript CDS, a pattern that was only observable in PNK-treated samples due to better transcript coverage. Read distribution and read length were reminiscent of ribosome-profiling data, which indicated that ex-mRNA fragments are ribosome protected and circulate as polysome or monosome complexes. This observation was confirmed by a metagene analysis that was, depending on sample type, based on an average of 12,789–16,486 ex-mRNA transcripts in the PNK-treated samples. This showed that approximately 60% of the reads originated from the CDS and approximately 30% from the 3′ UTR (Figure 2).
Figure 2. Read distribution of ex‑mRNA reads across the full-length mRNA transcripts.

(A and B) Read coverage for the hemoglobin A2 transcript (A) and the albumin transcript (B) by sample type for untreated and T4 PNK end-treated samples. Exon boundaries (HBA2: 3 exons, ALB: 15 exons) are indicated by alternating intensities of gray, and UTRs are distinguished from CDS by thinner bars. (C) Metagene analysis with relative read coverage (percentage) across 5′ UTRs, CDSs, and 3′ UTRs for untreated and PNK-treated samples as well as corresponding data obtained after 100 random simulations (across an average of 2342–3500 captured transcripts for untreated samples and an average of 12,789–16,487 captured transcripts for PNK-treated samples, depending on sample type). Shown are results from n = 6 individual samples per condition.
Anticoagulants have a widespread effect on the exRNA profile.
The anticoagulants we studied are the predominant ones used to collect blood samples in clinical practice and for research purposes. All of them influence blood cells ex vivo (19–21), and heparin may not be removed sufficiently by common extraction protocols and as a result interfere with downstream applications (22). This is especially relevant if patient populations are studied that often receive high doses of heparin as part of their medical treatment.
We therefore looked at how sample type influenced the exRNA composition of untreated and PNK-treated samples. We noted the previously reported destabilization of 5′ tRNA fragments in EDTA and ACD samples (Supplemental Figure 2) (6, 13) and described alterations in miRNA composition between serum and platelet-depleted EDTA plasma (12). In a comparison across all groups (analogous to ANOVA), we observed abundant differences for 86 miRNAs in the untreated samples and for 1458 mRNA transcripts in the PNK-treated samples among the 3 plasma types and serum (Supplemental Data 3 and 4). Serum generally had a higher abundance of ex‑miRNAs (e.g., miR-223 and -142) and ex‑mRNAs (e.g., S100A8) enriched in myeloid cells and platelets. In a gene set analysis, ex-mRNAs abundant in serum were associated with inflammation and leukocyte activation, whereas plasma ex-mRNAs were more related to general cellular processes, such as translation (Supplemental Data 5). Although there was a high degree of similarity between the exRNA profiles of EDTA and ACD plasma (Supplemental Figure 3B), there were distinctive differences as well. For instance, EDTA plasma had increased levels of erythropoietic transcripts, i.e., miR-451 and hemoglobin mRNAs, compared with all other samples. ACD had 3- to 4-fold higher levels of miR-150, a lymphocyte-restricted miRNA, than the other sample types (Supplemental Figure 3A and Supplemental Data 3 and 4).
The destabilizing effect of ACD and EDTA on RNP complexes was not restricted to tRNAs, and also affected read coverage signatures of other RNAs. Human small nuclear RNAs U1 and U2 are approximately 164 nt and approximately 190 nt, respectively, and assemble with proteins into small nuclear RNPs. Biochemical studies demonstrated that U1 and U2 possess core structures that are relatively resistant to nuclease digestion (23). At high magnesium ion concentrations several U1 domains are protected from nuclease digestion, whereas at lower magnesium ion concentrations, i.e., after the addition of EDTA or similar chelating reagents to plasma, only the core region remained relatively resistant to digestion. Our sRNA-seq data agreed well with these earlier observations (Supplemental Figure 3C). In addition, the coverage of the more protected core region was 4- to 8-fold lower in EDTA and ACD plasma, respectively, than in the other two sample types. There was no difference in read coverage patterns for small nuclear RNAs U2, U4, U5, and U6 or the large ribosomal subunits, 18S and 28S, between the different sample types.
Hematopoietic cells, platelets, and liver are the major sources of exRNAs in healthy individuals.
We next sought to identify sources contributing to the tissue exRNA pool in the physiological state. We generated a polyA mRNA-seq tissue atlas comprising major human cell and tissue types and calculated a tissue specificity score (TSS) (10) for all of the 19,828 mRNAs as defined in Ensembl release 88 (Supplemental Data 6). Genes restricted to a few tissues or cell types had a TSS greater than 3, e.g., aldolase B (ALDOB) expressed in liver and kidney, while classic marker genes like albumin (ALB) or cardiac troponin T (TNNT2) had a TSS greater than 4.
To compare the ex‑mRNA profile to the tissue atlas, we considered ex‑mRNAs in the PNK-treated samples with at least 5 unique reads in at least 3 of the 6 donors per sample type and took the union of ex‑mRNAs in all 4 samples types. A total of 3427 ex-mRNAs entered comparative analysis, and, of those, 169 had a TSS >3, therefore being most informative regarding tissue of origin (116 ex‑mRNAs >3 but <4, 53 ex‑mRNAs >4; Supplemental Data 7). About 30% of the 169 mRNAs were most abundant in neutrophils, 10% in liver, and 5% each in RBCs, platelets, and skeletal muscle. Conversely, when we compared the 1000 highest expressed mRNAs for each tissue in the atlas to the 3427 ex-mRNAs, we found a much higher fraction of the top 1000 transcripts from RBCs, platelets, neutrophils, PBMCs, and monocytes captured in circulation than from any of the other tissue (Figures 3 and 4, Supplemental Figure 4, and Supplemental Data 8).
Figure 3. Tissue sources of ex‑mRNAs.

Heatmap with the top the 821 most abundant ex-mRNAs in circulation for untreated and PNK-treated samples (left), together with the corresponding expression in selected cells or tissues (right). Selected tissue-specific/enriched mRNAs are labeled together with the tissue specificity score. Shown are results from n = 6 individual samples per condition. Tissue and cell RNA-seq data used for the tissue heatmap are listed in Supplemental Data 6.
Figure 4. Top expressed transcripts from hematopoietic tissues captured in circulation.

The 1000 most abundant cellular mRNA transcripts (excluding mRNAs encoded on the mitochondrial genome) from the selected cell types that collected 5 unique reads in at least 3 of the 6 donors per sample type were considered captured. The captured transcripts (x axis) were ordered in descending order by the tissue specificity score (TSS; y axis). Transcripts with a TSS greater than 3 are highlighted in red and listed, space permitting. Shown are results from n = 6 individual samples per condition. Tissue and cell RNA-seq data used for TSS calculation are listed in Supplemental Data 6.
Our annotation criteria led to the detection of certain highly tissue-specific genes from other tissues, e.g., MYBPC3 (myocardium), SFTPB (lung), or MIOX (kidney; Supplemental Figure 4) in some serum or plasma sample types. However, the underlying reads were repetitive and short and therefore highly suggestive of annotation artefacts.
In EDTA and ACD plasma, we detected 12%–21% of the 1000 most highly expressed cellular mRNAs among the hematopoietic cells investigated. This percentage increased to 27%–49% in heparin plasma and 38%–81% in serum. Particularly striking was the difference for neutrophils, for which we detected 12%, 17%, 49%, and 81% of the 1000 most highly expressed transcripts in ACD, EDTA, heparin, and serum, respectively, as ex‑mRNAs (Figure 4, Supplemental Figure 4, and Supplemental Data 8). The increase of ex‑mRNAs in serum compared with the other samples is likely related to in vitro neutrophil degranulation and cell death during coagulation. On the mRNA level, this was much more pronounced for neutrophil than platelets transcripts, of which we detected 35% and 42% of the top 1000 cellular transcripts in heparin plasma and serum, respectively. Although miRNAs have been reported as markers for platelet activation (12), our data suggest that neutrophils contribute similarly to coagulation-dependent exRNA changes.
In summary, these results indicated that hematopoietic cells, platelets, and the liver are main contributors to the ex-mRNA profile, and, based on our data, there was little support that other solid tissues contribute substantially.
RNA end treatment increases the diagnostic potential of exRNA in disease.
To evaluate the clinical potential of ex-mRNAs in patients we studied exRNA changes in a pilot cohort of patients with an ACS (n = 6) and age- and gender-matched healthy controls (n = 10; Supplemental Data 1 and 9). All patients had evidence of myocardial tissue–damage based on elevated cardiac troponin I levels, a highly sensitive and routinely used marker for myocardial damage. Patients with myocardial injury represent a proof-of-concept cohort, as the myocardium is one of the few tissues expressing tissue-specific miRNAs (myomirs miR-208a, -208b, and -499), which have been shown to be elevated in the circulation of these patients. Routine laboratory parameters measured on the same day were available for all patients and controls (Supplemental Data 9 and 10). In comparison with the controls, the ACS group had higher white blood cell counts.
Because the ACS group received high doses of heparin before sample collection, all patient and control samples were collected in heparin plasma to avoid any biases associated with different anticoagulants, as discussed previously herein (6, 12, 13). sRNA-seq libraries were generated from untreated (library 5) and PNK-treated (library 6) total RNA (Supplemental Figure 5), each library barcoding the ACS and control samples of the corresponding RNA treatment group. As before, one water sample was included per library; however, here the water was added in lieu of the RNA input at the beginning of the cDNA preparation and was not carried through a mock RNA isolation process.
Unsupervised hierarchical clustering of the calibrator spike-in small RNAs (see Methods) did not separate the 2 groups, arguing against any potential bias due to residual heparin in the samples (Supplemental Figure 6). However, 18 ex‑miRNAs were differentially altered in the untreated samples; 11 were higher and 7 were lower in ACS than controls (Figure 5A and Supplemental Data 11). The myocardium-specific miR-208b in the ACS group was 17-fold higher than in the controls; the 2 other myocardium-specific miRNAs, miR-208a (FDR 0.07%) and miR-499 (FDR 0.15%), were elevated 8-fold in ACS. These changes were in line with release due to myocardial injury and in magnitude they were similar to what we reported for patients in advanced heart failure (6) and, again, suggested no heparin-associated bias. Individual myeloid-enriched miRNAs were elevated in ACS as well, e.g., miR-223, while platelet miRNAs in general were not changed (Figure 5A).
Figure 5. Changes in ex‑mRNAs and ex‑miRNAs in patients with ACS compared with controls.

(A) MA plot of ex-miRNA changes with color coding of miRNAs highly expressed in platelets, defined as the top 85% miRNAs. (B and C) MA plots of ex-mRNA changes with color coding highly of expressed neutrophil genes (B) or platelet genes (C). Navy blue: highly expressed and FDR >5%; light blue: highly expressed and FDR <5%; red: not highly expressed and FDR <5%; gray: all other. Highlighted miRNAs (A) include the myocardium-specific miR-208b; miR-223, which is highly but not specifically expressed in neutrophils; and miR-24, which is highly but not specifically expressed in megakaryocytes (platelet precursor). Highlighted mRNAs are selected highly enriched neutrophils (B) or platelets (C) transcripts. (D) Heatmap showing altered ex‑mRNAs in the ACS group compared with healthy controls. Selected neutrophil-enriched mRNAs are indicated on the right. (E) RNA-seq read coverage of the 523 nt S100A8 transcript in ACS group and healthy controls (downsampled to 600,000 reads). Transcript structure indicated at the bottom with the 3 exons in alternating intensities of gray, and the 5′/3′ UTRs as thin bars.
PNK treatment improved the detection of distinct ex‑mRNAs 30-fold, with an average of 1124 (minimum 47, maximum 4825) unique transcripts in the PNK-treated samples compared with an average of 38 (minimum 6, maximum 313) in the untreated samples. Differential analysis identified 209 changes for ex‑mRNAs; 167 were higher and 42 were lower in ACS than controls. Neutrophil ex‑mRNAs broadly increased in ACS (Figure 5B, Supplemental Figure 7, and Supplemental Data 12), while platelet transcripts, including the highly specific PF4 and PPBP, were unchanged between the 2 groups (Figure 5C). The top 6 elevated ex‑mRNAs in the ACS group by FDR (Figure 5D) were IFITM2 (4.2-fold, TSS 2.25), MGAM (10-fold, TSS 4.3), CXCR2 (4.5-fold, TSS 4.1), H3F3A (3.6-fold, TSS 0.74), GCA (3.8-fold, TSS 3.2), and S100A8 (3.7-fold, TSS 3.2), all of which are highly expressed in neutrophils (Supplemental Data 6). The reads of these neutrophil transcripts mainly mapped to the CDSs (Figure 5E).
In contrast to the capture of myocardium-specific ex‑miRNAs, we did not detect myocardium-specific ex‑mRNAs in circulation.
The exRNA profile of the water samples did not differ substantially from water samples in libraries 1–4.
Taken together, the improved capture of ex‑mRNAs by PNK treatment allowed us to detect a neutrophil signature in ACS patients that is not detectable based on ex‑miRNAs, demonstrating improved tissue-resolution of ex‑mRNAs in the diseased state.
Discussion
Here, we showed that mRNA fragments in circulation (ex-mRNAs) can be efficiently captured by T4 PNK end treatment of total exRNA followed by sRNA-seq. Ex-mRNAs provide superior tissue and functional resolution for most conditions compared with other RNA classes because of the higher number of comparatively well annotated, highly expressed tissue-restricted transcripts. Tissue-specific ex-miRNAs, in selected cases, offer complementary information.
Ex-miRNAs have been widely studied as biomarkers in many types of diseases and conditions (6, 7, 24, 25). They perform well in the detection of tissue damage of organs with tissue-specific miRNAs, such as the liver (miR-122) (26) or the heart (myomirs) (6, 7). Individual miRNAs alone or in combination are also used for risk prediction for chronic conditions (25), and characteristic ex‑miRNA changes have been shown to be stable over months, even in the absence of detectable illness (8). But the precise tissue source or etiology of such differences based on the ex-miRNA profile alone remain unclear. Many tissues do not possess specifically expressed miRNAs, and measurements of ubiquitously or weakly expressed miRNAs in biofluids are prone to misinterpretation.
We looked at patients with ACS as benchmark population to evaluate our analysis of ex‑mRNAs, given the consistently reported elevations of myomirs in circulation of patients with otherwise confirmed myocardial damage (6, 7). As expected, myomirs were elevated in ACS, but, aside from these changes, few alterations were detectable between ACS and healthy controls on the ex‑miRNA level. However, the ACS group had a characteristic neutrophil ex‑mRNA signature, i.e., elevated exRNA levels of neutrophil-enriched genes. Although this finding needs validation in larger cohorts and might potentially be confounded by the higher leukocyte count in the ACS group, the results are in line with the increasing recognition of inflammation and neutrophil activation in atherosclerotic disease. Endothelial damage and neutrophil activation have been linked to thrombus formation in animal studies (27), and neutrophils in atherosclerotic plaques are detectable in vessels from animal models as well as human samples (28). Irrespective of the reason for the neutrophil signature in the ACS cohort, i.e., an inflammatory response to ACS or due to higher neutrophil counts, the results clearly emphasize the superior tissue resolution of ex-mRNAs compared with ex‑miRNAs. The lack of detectable myocardial ex‑mRNAs in any of the samples used in this study is most likely due to the low-sequencing depth of ex‑mRNAs caused mainly by large rRNA fractions, but the differential stability of ex‑miRNAs and ex‑mRNA fragments likely contributes (5).
Previous exRNA-seq studies also commented on ex-mRNAs that were detectable by their approaches. Yuan et al. (29) studied the exRNA composition of extracellular vesicles in the plasma of 50 healthy individuals and 142 cancer patients without RNA end treatment. While in their study the by far most abundant RNAs were miRNAs and Piwi-interacting RNAs, they also detected 1,338 unique mRNAs representing 2.1% of all included RNA categories. The 3 most abundant ex‑mRNAs were CCDC9, ST8SIA1, and MTRNR2L5. They described 7 ex‑mRNAs associated with aging as well as CDHR1 and PAQR5, associated with prostate and pancreatic cancer, respectively. Despite some overlap between the ex‑mRNAs detected in the study by Yuan et al. and the ones detected in ours, highly abundant ex‑mRNAs in total plasma, such as the hemoglobin transcripts or platelet transcripts, were not detected in their cohort. We assume that these and other differences are not only due to the fact that they studied a subset of exRNAs in vesicles but also to the lack of RNA end treatment, which Turchinovich and colleagues noted to be important to improve cDNA library preparation from plasma RNA (15). Moreover, differences in results also arise from differences in the analyses. While most investigators used hierarchical mapping strategies similar to ours, RNA reference sequences differed and the priorities that the included RNA classes were given in the annotation process were often dissimilar. Both may have a substantial influence on the results. For instance, up to 30% of reads using untreated input RNA in our study mapped to the expression plasmid of Rnl2 ligase, the recombinant protein of which is used for sRNA-seq cDNA library preparation. Omitting this plasmid reference from the mapping hierarchy during sequence read alignments resulted in a substantial amount of plasmid sequences aligning perfectly to other RNA classes, including mRNA transcripts, even using our most stringent mapping criteria. Wei et al. (17) used T4 PNK treatment in combination with tobacco acid pyrophosphatase treatment for exRNA analysis of cell culture supernatant of glioblastoma cell cultures. Interestingly, they observed that most of their RNA-seq reads originated from the 3′ UTRs of mRNAs and not the CDSs. We speculate that the differences in transcript coverage between their and our observations are due to different sRNA-seq protocols and analyses or reflect the different environments studied.
While our work did not address different mechanisms of exRNA release or the different compartments of exRNAs currently discussed, a few findings suggest that ex-mRNAs and probably a large part of all exRNA circulate within polysome complexes. First, for ex‑mRNAs sequenced with good coverage, i.e., high abundance transcripts, read length (~28 nt) and read distribution across the transcripts with better coverage across coding regions were reminiscent of sequencing data from ribosome-profiling studies (30). Second, the loss of 5′ tRNA halves in EDTA and ACD samples (6, 13) is consistent with loss of protection by tRNA-binding proteins (31) due to magnesium ion chelation. Chelation of magnesium ion by EDTA, traditionally used experimentally for that purpose (23, 32), and ACD in blood collection tubes will lead to disassembly of polysomes, rendering the associated tRNAs vulnerable to nuclease digestion. The more widespread effect of RNP destabilization after magnesium ion chelation is furthermore evident by loss of RNA fragments from certain regions of U1 RNA and, overall, fewer captured transcripts in EDTA and ACD samples. It ultimately remains unclear at this point how much of an effect the different anticoagulants had on the hematopoietic cells ex vivo (19–21) and whether they potentially confounded the exRNA composition due to ongoing or changed RNA release. But aside from their value to the study in vivo changes of exRNAs and in the development of diagnostic applications, the discriminatory value of ex-mRNAs compared with other RNA classes can also be utilized to assess such changes and biases related to blood collection and processing to a greater detail.
The adoption of ex‑mRNAs and other exRNAs as clinical biomarkers will require quantitative and reasonably fast assays, such as qPCR. However, primer design for short fragments is challenging, and qPCR, similar to other assays not based on sequencing, does not easily allow for the verification of the amplified signal (i.e., read sequence). The diminutive amounts of RNA in body fluids carry a great risk of introducing biases, as described here, and such assays will have to be carefully developed. Similar considerations will have to be taken into account with different methods or further refinements, e.g., using heparinase treatment to reduce possible interference of heparin with enzymatic reactions or enzymatic rRNA removal.
In conclusion, total exRNA PNK treatment followed by sRNA-seq allows for robust investigation of ex‑mRNA changes for biomarker discovery and other studies, but sample collection and analysis strategies have to be carefully planned. Future method refinements, such as depletion of rRNA and tRNA fragments, will further increase the potential of this approach.
Methods
Overall experimental design
sRNAs-seq data from serum and plasma samples were prepared in 2 batches, representing 2 different experiments; batch 1 comprised samples for libraries 1–4, and batch 2 comprised samples for libraries 5 and 6. For each batch, total extracellular RNA was isolated in parallel using identical reagent lots, and sRNA-seq cDNA libraries were prepared in parallel. Each batch was sequenced on 1 flow cell on a HiSeq 2500: high-output mode was used for libraries 1–4 and rapid-run mode was used for libraries 5 and 6.
Tissue and cell samples were prepared independently from each other.
Sample procurement
Peripheral vein blood from arm veins (hands or antecubital) was collected from healthy volunteers and from patients with ACS at The Rockefeller University and Mannheim University Medical Center, respectively. Human tissue samples for bulk mRNA-seq of myocardium and kidney were obtained from the National Disease Research Interchange, and other samples were obtained from biopsies or discarded surgical waste.
Serum and plasma sample processing and platelet depletion
Serum samples were allowed to coagulate before further processing at room temperature; plasma samples were processed within 30 minutes of collection by centrifugation at 2500 g for 15 minutes in the blood collection tube, followed by another centrifugation of the supernatant at the same conditions. The resulting supernatant was aliquoted into 500-μl aliquots avoiding the residual pellet, flash-frozen in liquid nitrogen, and stored at –80°C until use. Samples were thawed only once.
RNA isolation
Isolation of total cell-free exRNA.
For a detailed protocol, with recipes and catalog numbers, see the supplemental information of Max et al. (8). All steps were carried out in Eppendorf LoBind 2 ml tubes (catalog 022431048), and samples were collected in G-Tube Snap Cap siliconized microcentrifuge tubes (catalog 22179-004). Samples for libraries 1–4 were eluted using Zymo-Spin I columns (Zymo-Research, catalog C1003-50); samples for libraries 5 and 6 were eluted using Qiagen MinElute columns (catalog 74204). Briefly, for exRNA isolation a 450 μl sample was added to 105 μl buffer P (30% v/v sodium dodecyl sulfate in RNase-free water, 66 mM Tris-HCl, 19.8 mM EDTA, freshly added β-mercaptoethanol to 2% v/v) preheated to 60°C in a dry block incubator (ThermoMixer, Eppendorf). Buffer P was supplemented with 2 attomole each of the 10 calibrator ribonucleotides of cocktail 1 (see Supplemental Data 13). The tube was immediately mixed for 3 seconds at 1200 rpm, followed by the addition of 28 μl proteinase K solution (2.1 mg/ml proteinase K, 5.4% v/v glycerol, 3.2 mM CaCl2, 5.4 mM Tris-HCl, 4.5 M NaCl), and incubation for 10 minutes at 550 rpm. After that, 513 μl buffer ED2 (80% v/v acidic phenol saturated with 0.1 M citrate buffer at pH 4.3, 1.6 M guanidinium-isothiocyanate, 37.4 mM sodium citrate, 0.4% w/v sarcosyl, freshly added β-mercaptoethanol to 0.35% v/v) was added and the tubes mixed for 30 seconds at 1400 rpm. The samples were centrifuged for 30 seconds at 50 g and room temperature to collect the fluid and transferred to a ThermoMixer set to 10°C. For RNA extraction, 103 μl chloroform was added to each tube, and the samples mixed for 30 seconds at 10°C and 1400 rpm, followed by centrifugation at 16,000 g and 4°C in a microcentrifuge. The upper aqueous phase (600 μl) was transferred to 2-ml siliconized tubes containing 1200 μl buffer VB2G (98.2% v/v isopropanol, 7.2 mM MgCl2, 2.4 mM CaCl2, 1 M guanidinium-isothiocyanate, 5 mM TCEP) and inverted 5 times. The mixed samples were then applied to silica spin columns in a vacuum manifold and washed twice with 900 μl buffer EWL (70% v/v isopropanol in RNase-free water, 0.4 M GITC, 3 mM CaCl2, 3 mM MgCl2, 15 mM NaCl, 0.3% Triton-X 100, 5 mM TCEP), once with 900 μl 100% ethanol, and once with 500 μl 80% ethanol (in RNase-free water). The spin columns were then spun at 16,000 g and room temperature for 5 minutes to dry the matrix and transferred to a new 1.5-ml siliconized tube, followed by the addition of 20 μl RNase-free water and incubation for 1 minute. The samples were eluted by centrifugation at 16,000 g and room temperature for 1 minute, resulting in about 18 μl untreated total exRNA. The isolated RNA was stored at –80°C until usage.
exRNA yield was measured using the Qubit RNA HS assay with modifications (for details see Supplemental Information).
Isolation of cell and tissue RNAs.
Cellular or tissue total RNA was extracted using TRIzol with an additional phenol/chloroform extraction step and concentrated by alcohol precipitation or purified using the miRNeasy kit (Qiagen) according to the manufacturer’s instructions. Total RNA from cells and tissues was quantified on a NanoDrop UV spectrophotometer, and the RNA integrity was determined on an Agilent Bioanalyzer 2100 with Agilent RNA 6000 Pico Chips.
PNK treatment of total exRNA
To half of the eluted exRNA, i.e., 14 μl, we added 6 μl of a master mix corresponding to the equivalent of 2 μl ×10 T4 PNK buffer, 2 μl 10 mM ATP, 1 μl RNase-free water, and 1 μl T4 PNK (NEB, catalog M0201S) for a final reaction volume of 20 μl in a 1.5 ml siliconized microcentrifuge tube. The reaction was incubated for 30 minutes at 37°C followed by the addition of 40 μl buffer VB2G (for composition see above) and reapplied to the same silica column used for the initial purification. The column was washed twice with 900 μl buffer EWL (for composition see above), once with 900 μl 100% ethanol, and once with 500 μl 80% ethanol. The column was then placed in a collection tube and spun at 13,000 rpm and room temperature for 5 minutes to dry the silica matrix. The silica column was then transferred to a new siliconized 1.5-ml tube, 15 μl RNase-free water was applied to the column, and the samples were incubated for 1 minute and finally centrifuged at 13,000 rpm and room temperature for 1 minute to elute the PNK-treated RNA. This yielded approximately 13 μl, and 8.5 μl was used for sRNA-seq cDNA library preparation.
RNA-seq
sRNA-seq cDNA library preparation from the serum and plasma samples to profile exRNAs was done as described previously (33) using 8.5 μl untreated or PNK-treated RNA with size selection from 19 to 45 nt. Tissue and cell RNA-seq for comparison with the exRNA profile was done using the Illumina Stranded mRNA-seq TruSeq protocol following the manufacturer’s instructions. All sequencing was done in the Genomics Core Facility at The Rockefeller University. For details, see Supplemental Information. The raw data for all newly generated sRNA-seq and cellular mRNA-seq libraries have been deposited in NCBI’s Sequence Read Archive (bioproject PRJNA474043).
Bioinformatics analysis
Read mapping and annotation.
Read processing and annotation for sRNA-seq of serum and plasma samples was done as described previously (18), with modifications for PNK-treated samples. Long RNA-seq reads from tissues or cells were aligned to the human genome build 38 using the STAR aligner version 2.0.4j (34) and quantified using the featureCounts version 1.5.1 (35) program based on Ensembl release 88. For details, see Supplemental Information.
Clinical laboratory parameters
Standard clinical laboratory assays were performed by the Central Laboratories of the University Medical Centre Mannheim, Mannheim, Germany, and Memorial Sloan Kettering Cancer Center, New York, New York, USA. For details, please see Supplemental Data 9 and 10.
Metagene analysis
To determine if the mRNA read distribution was biased toward certain transcript regions, i.e., 5′ UTR, CDS, and 3′ UTR, we compared the experimental data with 100 random simulations. From the reference transcriptome, we only considered mRNA transcripts with UTRs 15 nt or longer (subset transcriptome), and we only included ex‑mRNA reads 15 nt or longer without mismatch and up to 2 mapping locations to the subset transcriptome. Reads from each subsample (i.e., 1–6) per sample type were mapped using the Bowtie aligner (version 1.2.1, for details see Supplemental Information).
The mapped reads for each region per distinct transcript were tallied and normalized by region length and averaged across all transcripts. Finally, the transcript regions were scaled to 100%. For the simulation, the transcript region mapping positions of the previously mapped reads were randomly shuffled and counted as above. This simulation was repeated 100 times and the results were averaged.
TSS
A TSS was calculated as the difference between the maximal possible information content and the Shannon entropy (10).
(Equation 1)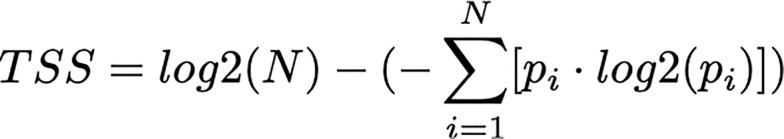
where N is the number of samples and pi (relative gene expression) is defined as follows:
(Equation 2),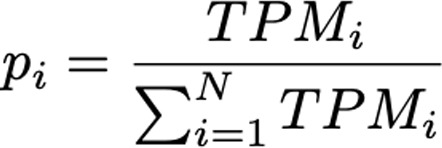
where TPM represents transcripts per million.
In this study, we used 25 tissue and cell samples for comparison with the exRNA profile, therefore the maximum possible TSS was log2(25) = 4.64. For details on the included samples, see Supplemental Data 6. The TSS was calculated in the R language.
Statistics
Analysis of RNA-seq data.
Differential analysis of miRNA and mRNA changes was done with the Bioconductor package edgeR (version 3.17.5) in the R statistical language version 3.5.1 (36). We used edgeR’s quasilikelihood (QL) framework (37, 38) to fit a generalized linear model comparing the conditions of interest. The QL dispersion distribution was estimated robustly, other parameters were kept at their default setting. miRNAs or mRNAs with fewer than 1 and 0.5 counts per million, respectively, in the number of samples comprising the smallest group for each comparison were excluded from the differential analysis and all downstream analyses.
Gene set analysis based on curated gene sets (C2), as defined in the Broad Institute’s Molecular Signatures Database (MSigDB, version 5.1), comparing the 4 different sample types (EDTA, ACD, and heparin plasma, and serum) used in libraries 1–4 was done using the mroast function of the Bioconductor package edgeR (39). Gene set analysis was based on the same edgeR object used for the differential analysis. The MSigDB C2 gene set was downloaded from the Walter and Eliza Hall Institute (http://bioinf.wehi.edu.au/software/MSigDB/), where gene sets are provided as “rdata” files. The original Entrez Gene IDs in the rdata files were mapped to Ensembl Gene IDs to match our annotation. Finally, gene sets containing the terms “ribosome,” “translation,” or “inflammation” were used as input for the analysis. An FDR of <5% was used as a cutoff.
Other statistical analysis.
Differences in the number of captured transcripts between untreated and PNK-treated samples were tested using the Wilcoxon test. Differences between clinical variables, as indicated in the text, were tested using the 2-tailed t test for normally distributed values and Kruskal-Wallis for data with nonnormal distribution. Differences in the RNA yield of the 4 different sample types were tested by fitting a linear fixed-effect model with Tukey’s post-hoc test. All analysis was done in R (for details see Supplemental Information). P < 0.05 was considered significant.
Study approval
The study was approved by the institutional review boards of The Rockefeller University; the Medical Faculty Mannheim, University of Heidelberg; the Friedrich-Alexander-Universität Erlangen-Nuremberg, Erlangen, Germany; and the Columbia University Medical Center. Written informed consent was received from all participating human subjects.
Author contributions
KMA and TT conceived the study and wrote the manuscript. KMA, PM, and MB analyzed the data. KMA, KB, AS, and KEAM performed experiments. KMA, YAL, AH, KH, MFN, TGD, and MB recruited human subjects and collected samples.
Supplementary Material
Acknowledgments
This work was supported by the NIH (UH3 TR000933, UH2 AR06768902, R01HD086327) and in part by a grant (UL1 TR000043) from the National Centre for Advancing Translational Sciences, NIH Clinical and Translational Science Award program. We thank the Rockefeller Genomics Resource Center for performing the sequencing and Tasos Gogakos from the Tuschl laboratory for scientific input and helpful discussions.
Version 1. 04/11/2019
In-Press Preview
Version 2. 05/02/2019
Electronic publication
Footnotes
TGD’s present address is: Department of Pediatrics and Pharmacology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania.
Conflict of interest: TT is a cofounder of and adviser to Alnylam Pharmaceuticals.
Copyright: © 2019 American Society for Clinical Investigation
Reference information: JCI Insight. 2019;4(9):e127317. https://doi.org/10.1172/jci.insight.127317.
Contributor Information
Kemal M. Akat, Email: kakat@rockefeller.edu.
Youngmin A. Lee, Email: ylee04@rockefeller.edu.
Arlene Hurley, Email: hurleya@rockefeller.edu.
Pavel Morozov, Email: pmorozov@mail.rockefeller.edu.
Klaas E.A. Max, Email: kmax@mail.rockefeller.edu.
Miguel Brown, Email: brownm28@email.chop.edu.
Kimberly Bogardus, Email: kbogardus14@gmail.com.
Anuoluwapo Sopeyin, Email: ansopeyin@gmail.com.
Kai Hildner, Email: kai.hildner@uk-erlangen.de.
Thomas G. Diacovo, Email: td2142@columbia.edu.
Markus F. Neurath, Email: markus.neurath@uk-erlangen.de.
Martin Borggrefe, Email: martin.borggrefe@umm.de.
Thomas Tuschl, Email: ttuschl@rockefeller.edu.
References
- 1.Mandel P, Metais P. [Not Available] C R Seances Soc Biol Fil. 1948;142(3-4):241–243. [PubMed] [Google Scholar]
- 2.Mitchell PS, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA. 2008;105(30):10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arroyo JD, et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci USA. 2011;108(12):5003–5008. doi: 10.1073/pnas.1019055108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turchinovich A, Weiz L, Langheinz A, Burwinkel B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011;39(16):7223–7233. doi: 10.1093/nar/gkr254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elkayam E, et al. The structure of human argonaute-2 in complex with miR-20a. Cell. 2012;150(1):100–110. doi: 10.1016/j.cell.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akat KM, et al. Comparative RNA-sequencing analysis of myocardial and circulating small RNAs in human heart failure and their utility as biomarkers. Proc Natl Acad Sci USA. 2014;111(30):11151–11156. doi: 10.1073/pnas.1401724111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corsten MF, et al. Circulating MicroRNA-208b and MicroRNA-499 reflect myocardial damage in cardiovascular disease. Circ Cardiovasc Genet. 2010;3(6):499–506. doi: 10.1161/CIRCGENETICS.110.957415. [DOI] [PubMed] [Google Scholar]
- 8.Max KEA, et al. Human plasma and serum extracellular small RNA reference profiles and their clinical utility. Proc Natl Acad Sci USA. 2018;115(23):E5334–E5343. doi: 10.1073/pnas.1714397115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landgraf P, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129(7):1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet. 2014;15(12):829–845. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giraldez MD, et al. Comprehensive multi-center assessment of small RNA-seq methods for quantitative miRNA profiling. Nat Biotechnol. 2018;36(8):746–757. doi: 10.1038/nbt.4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willeit P, et al. Circulating microRNAs as novel biomarkers for platelet activation. Circ Res. 2013;112(4):595–600. doi: 10.1161/CIRCRESAHA.111.300539. [DOI] [PubMed] [Google Scholar]
- 13.Dhahbi JM, et al. 5’ tRNA halves are present as abundant complexes in serum, concentrated in blood cells, and modulated by aging and calorie restriction. BMC Genomics. 2013;14:298. doi: 10.1186/1471-2164-14-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das U, Shuman S. Mechanism of RNA 2’,3’-cyclic phosphate end healing by T4 polynucleotide kinase-phosphatase. Nucleic Acids Res. 2013;41(1):355–365. doi: 10.1093/nar/gks977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turchinovich A, Surowy H, Serva A, Zapatka M, Lichter P, Burwinkel B. Capture and Amplification by Tailing and Switching (CATS). An ultrasensitive ligation-independent method for generation of DNA libraries for deep sequencing from picogram amounts of DNA and RNA. RNA Biol. 2014;11(7):817–828. doi: 10.4161/rna.29304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Danielson KM, Rubio R, Abderazzaq F, Das S, Wang YE. High Throughput Sequencing of Extracellular RNA from Human Plasma. PLoS One. 2017;12(1):e0164644. doi: 10.1371/journal.pone.0164644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei Z, et al. Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nat Commun. 2017;8(1):1145. doi: 10.1038/s41467-017-01196-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farazi TA, et al. Bioinformatic analysis of barcoded cDNA libraries for small RNA profiling by next-generation sequencing. Methods. 2012;58(2):171–187. doi: 10.1016/j.ymeth.2012.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brunialti MK, Kallás EG, Freudenberg M, Galanos C, Salomao R. Influence of EDTA and heparin on lipopolysaccharide binding and cell activation, evaluated at single-cell level in whole blood. Cytometry. 2002;50(1):14–18. doi: 10.1002/cyto.10049. [DOI] [PubMed] [Google Scholar]
- 20.Duvigneau JC, et al. Heparin and EDTA as anticoagulant differentially affect cytokine mRNA level of cultured porcine blood cells. J Immunol Methods. 2007;324(1-2):38–47. doi: 10.1016/j.jim.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 21.Shalekoff S, Page-Shipp L, Tiemessen CT. Effects of anticoagulants and temperature on expression of activation markers CD11b and HLA-DR on human leukocytes. Clin Diagn Lab Immunol. 1998;5(5):695–702. doi: 10.1128/cdli.5.5.695-702.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ukita T, Terao T, Irie M. Inhibition of pancreatic ribonuclease-I activity by heparin. J Biochem. 1962;52:455–457. doi: 10.1093/oxfordjournals.jbchem.a127644. [DOI] [PubMed] [Google Scholar]
- 23.Reveillaud I, Lelay-Taha MN, Sri-Widada J, Brunel C, Jeanteur P. Mg2+ induces a sharp and reversible transition in U1 and U2 small nuclear ribonucleoprotein configurations. Mol Cell Biol. 1984;4(9):1890–1899. doi: 10.1128/MCB.4.9.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeller T, et al. Assessment of microRNAs in patients with unstable angina pectoris. Eur Heart J. 2014;35(31):2106–2114. doi: 10.1093/eurheartj/ehu151. [DOI] [PubMed] [Google Scholar]
- 25.Karakas M, et al. Circulating microRNAs strongly predict cardiovascular death in patients with coronary artery disease-results from the large AtheroGene study. Eur Heart J. 2017;38(7):516–523. doi: 10.1093/eurheartj/ehw250. [DOI] [PubMed] [Google Scholar]
- 26.Ward J, et al. Circulating microRNA profiles in human patients with acetaminophen hepatotoxicity or ischemic hepatitis. Proc Natl Acad Sci USA. 2014;111(33):12169–12174. doi: 10.1073/pnas.1412608111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Franck G, et al. Flow Perturbation Mediates Neutrophil Recruitment and Potentiates Endothelial Injury via TLR2 in Mice: Implications for Superficial Erosion. Circ Res. 2017;121(1):31–42. doi: 10.1161/CIRCRESAHA.117.310694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee TD, Gonzalez ML, Kumar P, Chary-Reddy S, Grammas P, Pereira HA. CAP37, a novel inflammatory mediator: its expression in endothelial cells and localization to atherosclerotic lesions. Am J Pathol. 2002;160(3):841–848. doi: 10.1016/S0002-9440(10)64907-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan T, et al. Plasma extracellular RNA profiles in healthy and cancer patients. Sci Rep. 2016;6:19413. doi: 10.1038/srep19413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324(5924):218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garzia A, et al. The E3 ubiquitin ligase and RNA-binding protein ZNF598 orchestrates ribosome quality control of premature polyadenylated mRNAs. Nat Commun. 2017;8:16056. doi: 10.1038/ncomms16056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Penman S, Vesco C, Penman M. Localization and kinetics of formation of nuclear heterodisperse RNA, cytoplasmic heterodisperse RNA and polyribosome-associated messenger RNA in HeLa cells. J Mol Biol. 1968;34(1):49–60. doi: 10.1016/0022-2836(68)90234-9. [DOI] [PubMed] [Google Scholar]
- 33.Hafner M, Renwick N, Farazi TA, Mihailović A, Pena JT, Tuschl T. Barcoded cDNA library preparation for small RNA profiling by next-generation sequencing. Methods. 2012;58(2):164–170. doi: 10.1016/j.ymeth.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 36. R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ Accessed April 3, 2019.
- 37.Lun AT, Chen Y, Smyth GK. It’s DE-licious: A Recipe for Differential Expression Analyses of RNA-seq Experiments Using Quasi-Likelihood Methods in edgeR. Methods Mol Biol. 2016;1418:391–416. doi: 10.1007/978-1-4939-3578-9_19. [DOI] [PubMed] [Google Scholar]
- 38.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu D, Smyth GK. Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 2012;40(17):e133. doi: 10.1093/nar/gks461. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.