

Abstract
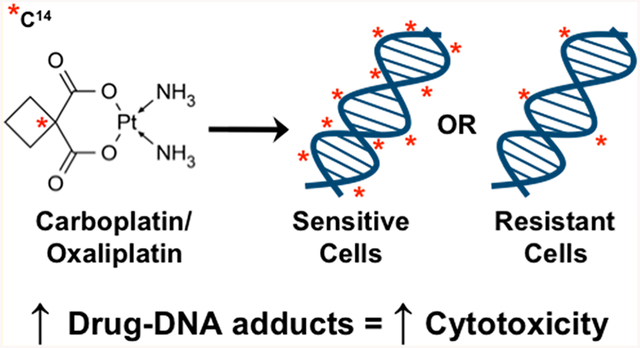
Platinum drugs, including carboplatin and oxaliplatin, are commonly used chemotherapy drugs that kill cancer cells by forming toxic drug-DNA adducts. These drugs have a proven, but modest, efficacy against several aggressive subtypes of breast cancer but also cause several side effects that can lead to the cessation of treatment. There is a clinical need to identify patients who will respond to platinum drugs in order to better inform clinical decision making. Diagnostic microdosing involves dosing patients or patient samples with subtherapeutic doses of radiolabeled platinum followed by measurement of platinum-DNA adducts in blood or tumor tissue and may be used to predict patient response. We exposed a panel of six breast cancer cell lines to 14C-labeled carboplatin or oxaliplatin at therapeutic and microdose (1% therapeutic dose) concentrations for a range of exposure lengths and isolated DNA from the cells. The DNA was converted to graphite, and measurement of radiocarbon due to platinum-DNA adduction was quantified via accelerator mass spectrometry (AMS). We observed a linear correlation in adduct levels between the microdose and therapeutic dose, and the level of platinum-DNA adducts corresponded to cell line drug sensitivity for both carboplatin and oxaliplatin. These results showed a clear separation in adduct levels between the sensitive and resistant groups of cell lines that could not be fully explained or predicted by changes in DNA repair rates or mutations in DNA repair genes. Further, we were able to quantitate oxaliplatin-DNA adducts in the blood and tumor tissue of a metastatic breast cancer patient. Together, these data support the use of diagnostic microdosing for predicting patient sensitivity to platinum. Future studies will be aimed at replicating this data in a clinical feasibility trial.
Graphical Abstract
INTRODUCTION
Over 255,000 new cases of breast cancer are diagnosed every year in the United States.1 Depending upon the stage and subtype of cancer, many of these patients will receive chemotherapy, which often includes a platinum drug, such as carboplatin. Carboplatin is an alkylating agent that kills cells by forming covalent carboplatin-DNA monoadducts and diadducts that induce DNA damage responses and halt cell proliferation (Figure 1a). The addition of carboplatin to combination therapies has been shown to increase response rates in several more aggressive types of breast cancers, including human epidermal growth factor receptor 2 (HER2)-positive breast cancers, triple negative breast cancers (TNBC), and metastatic breast cancers.2–7 Further, carboplatin is preferred over anthracycline-based therapies because of a more favorable risk to benefit ratio.2 Oxaliplatin is another platinum drug that kills cells in a manner similar to carboplatin, but may have efficacy in cisplatin- and carboplatin-resistant tumors.8 Though oxaliplatin is not currently approved for the treatment of breast cancer, it has been shown to be clinically efficacious as a single agent or in combination regimens in breast cancer patients. In metastatic breast cancer patients, oxaliplatin alone had an overall response rate of 21% and in combination therapies had a range of overall response rates from 6.7 to 59% with a median of 31.2%.9–27 In locally advanced breast cancer patients, the overall response rate was even higher at 69%.28
Figure 1.

Platinum-DNA adducts can be measured in breast cancer cell lines. (A) Structures of 14C-labeled carboplatin and oxaliplatin and formation of platinum mono- and diaducts. (B, C) Six breast cancer cell lines were treated with a carboplatin microdose (1 μM) or therapeutic dose (100 μM) supplemented with 0.3 μM 14C-labeled carboplatin for 4 h, followed by 20 h in drug-free medium. DNA was isolated from cells, and carboplatin-DNA monoadducts per 108 nt were determined by AMS at indicated times after the start of treatment. (E, F) Six breast cancer cell lines were treated with an oxaliplatin microdose (0.1 μM) or therapeutic dose (10 μM) supplemented with 0.03 μM 14C-labeled oxaliplatin for 24 h, followed by 24 h in drug-free medium. DNA was isolated from cells, and oxaliplatin-DNA adducts per 108 nt were determined by AMS at indicated times after the start of treatment. (D, G) Correlation analysis of adducts induced by a microdose compared to a therapeutic dose in the six breast cancer cell lines. Error bars indicated the standard deviation from three biological replicates.
Unfortunately, adding carboplatin or oxaliplatin to therapy regimens frequently increases toxic side effects, including nausea, low blood counts, and neuropathy, among others, and often leads to discontinuation of therapy, indicating a need to determine which patients will benefit from this therapy prior to treatment.5,6 Few biomarkers are known for predicting tumor response to platinum-based therapies, and none are currently used therapeutically for breast cancer. Because platinum agents form bulky drug-DNA adducts, much of the predictive biomarker research has focused on expression levels and mutations in nucleotide excision repair (NER) genes—the type of DNA repair responsible for the removal of bulky DNA damage.29 In particular, BRCA1/2 (breast cancer susceptibility gene), ERCC1/2 (excision repair cross-complementing group), and p53 have been investigated as potential biomarkers for platinum-response. BRCA1/2 is likely the most promising biomarker for platinum response in breast cancer patients, particularly TNBC patients, as BRCA1/2 germline mutations, low BRCA1/2 mRNA levels, and high BRCA1 promoter methylation levels all correspond to good responses to platinum-based therapies in cells, mice, and humans.7,30–35 The role of ERCC1/2 in predicting tumor response is not yet clear as there are conflicting results about the correlation of ERCC1 mRNA expression level and tumor response, but TNBC patients with an ERCC2 rs1799793 polymorphism had a better response to platinum-based chemotherapy.30,36,37 Finally, p53 frameshift or nonsense mutations, but not missense mutations, have been correlated with good responses to platinum-based therapies.30 However, mechanisms of chemoresistance are multifactorial, and analysis of one gene is unlikely to predict drug response in all patients.38,39 Rather than focus on single genes, some groups have created a biomarker score based on the hallmarks of homologous recombination deficiency (HRD), including loss of heterozygosity, telomeric allelic imbalance, and large-scale state transitions, which they used in combination to determine a score for each tumor and correlated high HRD scores with good responses to platinum-based therapies.40–44 By measuring HRD score, patients with nonmutated BRCA1/2 tumors that would similarly benefit from platinum-based therapies can be identified. One limitation to predicting drug response based on HRD score is that these values indicate HRD in the tumor at some point and do not account for the possibility that HR could be restored in the tumor through reversion mutations, pathways independent of DNA repair, or other mechanisms leading to more patients being predicted to respond than will actually benefit from therapy.45
To overcome the shortcomings of using mutational status and genomic instability to predict platinum response, we and other groups have developed phenotypic assays to predict response after treatment. For example, Saleh et al. treated cancer cell lines with cisplatin and quantitated the level of double strand breaks via constant-field gel electrophoresis and found a correlation between double strand breaks and cisplatin sensitivity.46 In the current study, we treated breast cancer cell lines with carboplatin or oxaliplatin and quantitated the formation of drug-DNA adducts via accelerator mass spectrometry (AMS) and found correlation with platinum sensitivity. The correlation between platinum-DNA adduct levels and drug sensitivity has been reported in patients with breast and other cancer types and supports the hypothesis that they can be used as a biomarker to predict patient platinum sensitivity or resistance.47–55 In this study, we combined the platinum-DNA adduct biomarker generation in cell lines with ultrasensitive AMS detection, with the goal of adapting the diagnostic microdosing to future clinical trials with breast cancer patients.
EXPERIMENTAL PROCEDURES
Carboplatin/Oxaliplatin.
Carboplatin (10 mg/mL) was generously supplied from the UC Davis Comprehensive Cancer Center pharmacy and oxaliplatin (5 mg/mL) was purchased from Sanofi-Aventis. 14C-labeled carboplatin (specific activity of 54 mCi/mmol) and 14C-labeled oxaliplatin (specific activity of 58 mCi/mmol) were purchased from Moravek Biochemicals. Radiolabeled drugs were combined with unlabeled drugs to reduce the amount of radiocarbon used. For injection, [14C]oxaliplatin was prepared under good manufacturing practices (GMP) at the GMP facility at UC Davis by dissolving the drug substance with sterile water for injection (WFI) followed by filter sterilization with 0.2 mm poly(ether sulfone) (PES) syringe filter into sterile glass vials and sealing with a rubber septum. Liquid scintillation counting (LSC) was used to confirm specific activity. 14C-labeled oxaliplatin was mixed with unlabeled oxaliplatin at indicated ratios to reduce the amount of radiocarbon used and to achieve the desired specific activities required for microdoses and therapeutic doses. Drug mixtures were prepared immediately before use.
Cell Lines.
Six human breast cancer cell lines were purchased from American Type Culture Collection (ATCC) and maintained in the recommended medium at 37 °C in a humidified incubator.
Cell Line Platinum Treatment and AMS Analysis.
Cells were dosed with radiocarbon-labeled carboplatin or oxaliplatin, and AMS analyses were performed as previously described.56 Briefly, one million cells were seeded in 60 mm dishes and allowed to attach overnight. For carboplatin dosing, cells were treated with a microdose (1 μM) or therapeutic dose (100 μM) of unlabeled carboplatin supplemented with 50,000 dpm/mL 14C-labeled carboplatin (0.3 μM) for 4 h to mimic the carboplatin half-life of 1.3−6 h observed in patients. After 4 h, the cells were washed with phosphate-buffered saline (PBS) and incubated with drug-free medium for 20 additional hours. Cells were pelleted at 0, 2, 4, 8, and 24 h post-treatment. For oxaliplatin dosing, the cells were treated with a microdose (0.1 μM) or therapeutic dose (10 μM) of unlabeled oxaliplatin supplemented with [14C]oxaliplatin at 5000 dpm/ml for 24 h at 37 °C to mimic the 16.8 h half-life in patients. Following treatment, the cells were washed in PBS and cultured in the absence of oxaliplatin for 24 additional hours. Cells were pelleted at 0, 2, 4, 8, 24, 26, 28, 32, and 48 h from the start of treatment. DNA was isolated from cell pellets using a Wizard Genomic DNA Purification system (Promega). Carboplatin DNA monoadducts are not stable at prolonged exposures to higher temperatures (e.g., 37 °C), therefore most steps of the DNA isolation were performed on ice. Collected cells and tissues were lysed by resuspending in 600 μL nuclei lysis buffer at 4 °C for 20 min. After RNase treatment of 20 min at 37 °C, proteins were precipitated by addition of 200 μL precipitation buffer, vortexing, and 5 min incubation on ice. Precipitate was cleared by centrifugation at 19,000g, and DNA was precipitated from the cleared supernatant by addition of 800 μL cold isopropanol. DNA was pelleted by centrifugation at 19,000g for 20 min and washed three times with 800 μL of cold 70% ethanol. DNA was then resuspended in nuclease-free water. DNA concentration and purity were determined via Nanodrop 1000 or 2000. The purity was ensured by obtaining a 260/ 280 nm OD ratio of approximately 1.9. Ten μg of DNA from each sample was converted to graphite, and the ratio of 14C to total carbon was determined by AMS as previously described.57 Briefly, DNA samples were dried under vacuum, combusted to CO2 and water, followed by cryogenic isolation of CO2 and conversion to graphite powder. The graphite samples were loaded into an array of aluminum anodes that was inserted into the AMS instrument. A cesium sputter source induced a fraction of the graphite sample to be converted into an ion beam that was subsequently analyzed for the ratio of 14C to total carbon, which is proportional to the drug concentration in the DNA sample. The instrument was calibrated with a NIST standard of 14C labeled oxalic acid. Since the AMS instrument provides absolute quantitation of the ration of 14C to total carbon, calibration curves were not needed.
For DNA repair calculations, the decrease in drug-DNA adducts was measured at several time points after the cells were placed in drug-free media and were used to calculate a rate of DNA repair in adducts/108 nt/h.
MTT Cell Sensitivity Assay.
The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was performed to determine the cell growth inhibition. Approximately, 5000−7000 cells per well for each cell line were seeded in 96-well plates. After overnight culture, cells were treated with increasing concentrations of carboplatin or oxaliplatin for 72 h. Cells were washed and incubated with MTT for 4 h. The absorption was measured at 570 and 690 nm using a SpectraMax M2 microplate reader (Molecular Devices).
Patient Oxaliplatin Treatment and analysis.
A pilot, Phase 0 clinical trial titled “Oxaliplatin in Treating Patients With Metastatic Breast Cancer” (Clinical-Trials.gov identifier NCT02077998) was initiated as a feasibility study of the diagnostic microdosing approach. The clinical trial was approved by the UC Davis Institutional Review Board (IRB) and conducted under an investigational new drug (IND) from the food and drug administration (FDA). Eligible patients had metastatic (Stage IV) breast cancer that had no standard therapy regimen and tumors that could be biopsied or resected 48 h after the microdose is administered. This study was completed after one patient due to low accrual and modified to a different indication to increase the number of eligible participants. The patient received an oxaliplatin microdose (1% of the published single agent therapeutic dose) of 1.3 mg/m2 (29.2 mg/kg) supplemented with 2 × 106 dpm/kg [14C]oxaliplatin by a 2 min intravenous (IV) infusion in the peripheral vein of one arm. Peripheral blood specimens were drawn into BD Vacutainer CPT tubes with sodium citrate (Becton Dickinson) from the other arm prior to and 5, 15, 30 min, 2, 4, 8, 24, and 48 h after the administration of the microdose. Tubes were immediately placed on ice, and peripheral blood mononuclear cells (PBMC) were isolated within 2 h of collection by centrifugation according to manufacturer’s instructions. A proportion of total plasma was used for oxaliplatin pharmacokinetic (PK) determination by LSC. A core biopsy from a breast cancer site of bone metastasis was collected 48 h after the microdose was administered. Approximately 15 mg of tissue was used for DNA isolation without further dissection. DNA was isolated from PBMCs and tumor tissue and analyzed by AMS to quantify oxaliplatin-DNA adducts as above. Toxicity of the [14C]oxaliplatin administered as a microdose was assessed using Common Terminology Criteria for Adverse Events (CTCAE). Patient response to chemotherapy was evaluated using RECIST criteria for correlation to oxaliplatin-DNA monoadduct frequency. Outcomes related to chemotherapy (including response and adverse events) were collected and correlated with oxaliplatin-DNA monoadduct data. This patient was a nonresponder.
For ex vivo DNA repair analysis, PBMC isolated from the 48 h post-microdose blood sample were washed with PBS and incubated with culture medium (RPMI-1640 with 10% fetal calf serum) for 4 and 24 h at 37 °C. DNA was isolated and analyzed by AMS as above. The repair velocity of DNA adducts was calculated based on the decrease of DNA adducts.
Statistical Analysis.
Statistical analyses were performed using GraphPad Prism software and SAS/STAT software. The data were analyzed by t test, correlation analysis, ANOVA, or Tukey’s studentized range test as indicated. P values <0.05 were considered significant.
RESULTS
Comparison of DNA Damage Induced by Microdoses and Therapeutic Doses of 14C-Labeled Platinum in Breast Cancer Cell Lines.
We began by determining if (1) microdoses of 14C-labeled carboplatin or oxaliplatin can induce measurable drug-DNA adducts and if (2) levels of drug-DNA adducts induced by microdoses are linearly proportional to the those caused by therapeutic doses of platinum in six breast cancer cell lines. We chose cell lines that were isolated from patients which would be expected to receive platinum-based therapy, including cell lines derived from metastatic breast cancer (MCF7, MDA-MB-468, MDA-MB-231, and T-47D) and cell lines derived from triple negative breast cancer (Hs 578T, MDA-MB-468, MDA-MB-231, and BT-549). To determine carboplatin-DNA adduct formation, cells from each cell line were treated with 1 μM (microdose) or 100 μM (therapeutic dose) carboplatin supplemented with 0.3 μM of 14C-labeled carboplatin (Figure 1A) at a final concentration of 50,000 dpm/mL for 4 h, washed in PBS, and incubated in culture medium free of carboplatin for an additional 20 h. This procedure mimicked the in vivo IV dosing of carboplatin in which a bolus injection is followed by a rapid decrease in carboplatin concentration after a few hours and a half-life of 1.3−6 h. The number of carboplatin-DNA monoadducts was calculated based on the 14C content in genomic DNA as measured by AMS. In all cell lines, the number of carboplatin-DNA monoadducts increased up to the 4 h time point and then decreased, although at different rates (Figure 1B,C). The dose−response of carboplatin-DNA monoadduct formation was significantly linear at all time points for all cell lines at both microdose and therapeutic doses (R2 = 0.90, P < 0.001). The DNA damage concentrations ranged from ~1 to 15 monoadducts per 108 nt for the microdose and ~100 to 1500 monoadducts per 108 nt for the therapeutic dose, demonstrating an approximate 100-fold difference in the DNA damage with a 100-fold difference in drug concentration. A linear regression analysis was performed to determine if the levels of carboplatin-DNA monoadducts induced by a microdose of carboplatin can be used to predict the levels of monoadducts induced by a therapeutic dose of carboplatin (Figure 1D). The carboplatin-DNA monoadducts induced from the microdose were significantly correlated to those induced by the therapeutic dose (r = 0.9863, p < 0.0001). Similarly, to determine oxaliplatin-DNA adducts, cells from each cell line were treated with 0.1 μM (microdose) or 10 μM (therapeutic dose) oxaliplatin supplemented with 0.03 μM of 14C-labeled oxaliplatin (Figure 1a) at a final concentration 5000 dpm/mL for 24 h, washed in PBS, and incubated in culture medium free of oxaliplatin for an additional 24 h followed by AMS analysis. The longer dosing time mimicked the in vivo IV dosing and longer half-life of oxaliplatin (16.8 h) in patients. Oxaliplatin-DNA mono- and diadducts were both measured in this assay in all cell lines and time points tested (Figure 1E,F). Adduct levels increased in all cell lines up to the 24 h time point and then decreased, but at different rates. Again, the therapeutic dose caused an approximate 100-fold increase in adducts (~100−6000 adducts per 108 nt) compared to the microdose (~1−60 adducts per 108 nt). Further, the microdose-induced oxaliplatin-DNA adducts significantly correlated to those induced by the therapeutic dose (Figure 1G, r = 0.9745, p < 0.0001).
Comparison of Adducts to Platinum Sensitivity.
We next determined if the amount of carboplatin- or oxaliplatin-DNA adducts correlated with cellular sensitivity to carboplatin or oxaliplatin. Carboplatin and oxaliplatin IC50 values were determined for each of the six breast cancer cell lines by MTT assay (Table 1). The cell lines with carboplatin IC50 < 100 μM and oxaliplatin IC50 < 10 μM were classified as sensitive, based on the maximum platinum serum concentration attainable in patients.58 The six breast cancer cell lines used in this study had an ~6-fold range in carboplatin sensitivity and an ~200-fold range in oxaliplatin sensitivity, and carboplatin sensitivity did not correlate with oxaliplatin sensitivity in two-thirds of the cell lines.
Table 1.
Carboplatin Cell Line Sensitivities and Monoadduct AUC Levels
| carboplatin | oxaliplatin | ||||
|---|---|---|---|---|---|
| status | cell line | IC50 (μM) |
sensitivity | cell line | IC50 (μM) |
| sensitive IC50 < 100 μM | Hs 578T | 43.0 | sensitive IC50 < 10 μM | MCF7 | 1.03 |
| MDA- MB- 468 |
44.0 | T-47D | 2.35 | ||
| BT-549 | 68.3 | BT-549 | 6.71 | ||
| MCF7 | 136 | MDA- MB- 468 |
150 | ||
| resistant IC50 > 100 μM | MDA- MB- 231 T-47D |
248 | resistant IC50 > 10 μM | Hs 578T MDA- MB- 231 |
177 |
| 250 | 199 | ||||
We then determined the area under the curve (AUC) measured in h*adducts/108 nt induced by a carboplatin (Figure 2A,B) or oxaliplatin (Figure 2E,F) microdose and therapeutic dose in each cell line. Though there was variation in AUC values between the different cell lines, the general trend showed sensitive cell lines having higher AUC values and the resistant cell lines having lower AUC values. Furthermore, AUC values inversely correlated with cellular IC50 values, although these were only significant in the oxaliplatin-treated samples (Figure 2C,D,G,H).
Figure 2.

Levels of carboplatin- and oxaliplatin-DNA adduct formation correspond to cellular platinum sensitivity. (A, B, E, F) The six breast cancer cell lines were treated as in Figure 1B, C, E, F and AUC levels determined in h*adducts/108 nt for each cell line. Each cell line was classified as sensitive (blue) or resistant (red) to carboplatin and oxaliplatin (Table 1). (C, D, G, H) Correlation analysis of microdose- or therapeutic dose-induced AUC compared to cellular IC50 in the six breast cancer cell lines. Error bars indicated the standard deviation from three biological replicates.
We next compared the platinum-DNA adduct levels over time between the cells when grouped as sensitive or resistant, as outlined in Table 1. In the carboplatin-treated cells, the sensitive group of cell lines had significantly higher adduct levels at the time points between 2−24 h for both the microdose and the therapeutic dose as determined by t test (Figure 3A,B). Further, the microdose and therapeutic dose AUC values were significantly higher for the sensitive group of cell lines (Figure 3C,D). Similarly, in the oxaliplatin-treated cells, the sensitive group of cell lines had significantly higher adduct levels at the time points between 8 and 48 h for both the microdose and therapeutic dose (Figure 3E,F). The AUC levels for the oxaliplatin microdose and therapeutic doses were also significantly higher in the sensitive group of cell lines (Figure 3G,H).
Figure 3.

Grouped sensitive breast cancer cell lines have significantly higher adduct levels than resistant cell lines. The six breast cancer cell lines were grouped as sensitive (blue) or resistant (red) to carboplatin and oxaliplatin as described in Table 1. (A, B, E, F) Sensitive and resistant groups of breast cancer cell lines were treated as in Figure 1B, C, E, F and adduct levels determined by AMS at indicated times after the start of treatment. t tests were used to determine statistical differences in adduct levels between the groups at each time point. (C, D, G, H) AUCs in h*adducts/108 nt were determined for the sensitive and resistant groups of breast cancer cell lines and displayed as box and whiskers plots with the black bar representing the median, the box representing the middle quartiles, and the whiskers representing the remaining data at indicated time points for each group. t tests were used to determine statistical differences in AUC between the groups. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
DNA Repair Rates and Genetic Analysis of Six Breast Cancer Cell Lines.
To determine if increased DNA repair was responsible for carboplatin or oxaliplatin resistance in our breast cancer cell line panel, we determined the rates of DNA repair for each cell line. After a period of time in which cells were exposed to drug, they were washed with PBS and maintained in drug-free medium after 4 h (carboplatin) or 24 h (oxaliplatin). By measuring the decrease in adduct levels after the removal of the drug, we were able to calculate DNA repair rates. The DNA repair rates in the carboplatin-treated cells ranged from 0.0143 to 0.800 (microdose) and from 0.581 to 114 (therapeutic dose) adducts/108 nt/h with the most sensitive cell line, Hs 578T, having the highest DNA repair rate and the least sensitive cell line having the lowest DNA repair rate (Figure 4A,B). When the cell lines were grouped as sensitive or resistant, the sensitive group of cell lines had higher rates of DNA repair for both the carboplatin microdose (0.405 vs 0.0897 adducts/108 nt/h) and therapeutic dose (48.8 vs 7.04 adducts/108 nt/h) (Figure 4C,D). The DNA repair rates in the oxaliplatin-treated cells were slightly higher than the carboplatin-treated cells with a range from 0.103 to 2.16 (microdose) and from 5.52 to 425 (therapeutic dose) adducts/108 nt/h (Figure 4E,F) and did not appear to correlate with cell sensitivity. When the cell lines were grouped as sensitive or resistant, the sensitive group of cell lines had higher rates of DNA repair for both the oxaliplatin microdose (1.45 vs 0.758 adducts/108 nt/h) and therapeutic dose (182 vs 73.0 adducts/108 nt/h) (Figure 4G,H).
Figure 4.

Increased DNA repair is not a major mechanism of resistance in these cell lines. (A, B, E, F) The six breast cancer cell lines were treated as in Figure 1B, C, E, F. The decrease in adduct levels at several time points after removal of drug at 4 h (carboplatin) or 24 h (oxaliplatin) was used to determine a rate of DNA repair in adducts/108 nt/h for each cell line. (C, D, G, H) The six cell lines were classified as sensitive (blue) or resistant (red) to carboplatin and oxaliplatin (Table 1) and combined to determine average DNA repair rates.
We next analyzed the six breast cancer cell lines for any mutations in genes involved in DNA repair using the publically available Catalogue of Somatic Mutations in Cancer (COSMIC) database.59 As shown in Figure 5 and explained in detail in Table S1, each cell line had at least three mutations in different genes related to DNA repair with missense mutations being the most common type. There appeared to be no correlation between the number of mutated genes and cellular sensitivity to carboplatin or oxaliplatin or between the mutation of one gene and platinum sensitivity. The MDA-MB-468 cell line had the highest number of mutated genes in this data set (9) and was considered sensitive to carboplatin but resistant to oxaliplatin. A missense mutation in p53 was the most frequent mutation in this panel of cell lines and was found in five out of the six cell lines. Mutations in other DNA repair genes with the potential to predict platinum-sensitivity, including BRCA2, ERCC6, ERCC8, and PIK3CA were observed in both sensitive and resistant cell lines.
Figure 5.

Genetic analysis of DNA repair genes. DNA repair gene mutations in the six breast cancer cell lines were obtained from the publically available COSMIC database. Mutated genes labeled in bold were mentioned in this study as potential biomarkers for platinum sensitivity.
Pilot Patient Study.
We performed a small, oxaliplatin diagnostic microdosing feasibility study in a metastatic breast cancer patient following the schematic model in Figure 6A. Prior to receiving a platinum-containing therapeutic regimen, the patient was given a radiolabeled microdose, and the number of adducts in the plasma and tumor were determined at several time points. The adduct levels were compared to the patient response to determine a threshold level of adducts which would predict patient responders and nonresponders to oxaliplatin-containing therapy regimens. As part of a Phase 0 clinical trial and under IRB- and IND-approval, a Stage IV metastatic breast cancer patient was given a [14C]oxaliplatin microdose (1% of the published single agent therapeutic dose) of 1.3 mg/m2 (29.2 mg/kg) supplemented with 2 × 106 dpm/kg [14C]oxaliplatin by a 2 min intravenous (IV) infusion in the peripheral vein of one arm. Prior to and 5, 15, 30 min, 2, 4, 8, 24, and 48 h after the administration of the microdose, peripheral blood specimens were drawn from the other arm to determine plasma [14C]oxaliplatin concentration and PBMC oxaliplatin-DNA adduct levels. In this patient, the [14C]-oxaliplatin was rapidly cleared from the blood with a half-life of 0.2211 h (Figure 6B). The published half-life of oxaliplatin in patients is 14.1 min.60 In contrast, oxaliplatin-DNA adducts in the PBMC rapidly increased from 0 to 4 h with a peak of 8.029 adducts/108 nt and then slowly declined over the next 44 h. Forty-8 h after administration of the microdose, a tumor biopsy was collected and cut into three sections for AMS analysis. As shown in Figure 6D, the three tumor pieces had a fairly large range between 1.71 and 19.5 adducts/108 nt, and the mean was higher than that determined in the PBMC at the same collection time. PBMC collected in the 4 h peripheral blood sample were cultured ex vivo in drug-free media for an additional 4 or 20 h to determine the rate of DNA repair in this patient (Figure 6e). During follow-up care, this patient was determined as a nonresponder to treatment with oxaliplatin.
Figure 6.

Oxaliplatin-DNA adducts can be measured in the PBMC and tumor of a breast cancer patient. (A) Schematic of diagnostic microdosing strategy. (B) A metastatic breast cancer patient was given a diagnostic microdose of oxaliplatin [1.3 mg/m2 (29.2 mg/kg) supplemented with 2 × 106 dpm/kg [14C]oxaliplatin] by a 2 min intravenous (IV) infusion in the peripheral vein of one arm. Peripheral blood was drawn from the other arm prior to and 5, 15, 30 min, 2, 4, 8, 24, and 48 h after the administration of the microdose and oxaliplatin concentration determined by LSC. (C) PBMC was isolated from the peripheral blood samples above, and the number of adducts determined by AMS. (D) DNA was isolated from a tumor biopsy collected 48 h after administration of the microdose and adducts determined by AMS. (E) DNA repair rates were determined in PBMC isolated from the blood samples collected at 4 h after microdose administration that were cultured in drug-free media for an additional 4 or 20 h.
DISCUSSION
In this study we report a linearly proportional relationship between drug-DNA adducts formed from drug microdoses and therapeutic doses in six breast cancer cell lines exposed to either [14C]carboplatin or [14C]oxaliplatin. This result is similar to our previous observations in ovarian, lung, and bladder cancer cell lines as well as patient-derived xenograft models of bladder cancer and in bladder cancer patients, suggesting that the use of diagnostic microdosing to determine platinum sensitivity is applicable in multiple types of cancers in spite of them having differing etiologies and biologies.61–64 Our AMS data provided information on the total drug-DNA adduct load without molecular information on the actual distribution of specific adduct types. Both carboplatin and oxaliplatin predominantly bind to N7 of guanine and adenine, but other adduction products are possible. Unlike other N7 adducts that are formed from alkylating agents, such and nitrogen mustards, platinum-DNA adducts are stable at 37 °C with minimal depurination.65–67 Adduct distribution has previously been studied using AMS by exposing cancer cell cultures to [14C]oxaliplatin, followed by enzymatic DNA digestion and HPLC-AMS analysis.68 However, this approach is labor intensive and would not provide much additional useful information for proving the feasibility of diagnostic microdosing for translational research applications.
The [14C]carboplatin used in this study has the 14C label located on the cyclobutane dicarboxylic acid moiety, which is displaced upon carboplatin diadduct formation (Figure 1A). This chemistry has the potential to confound the measurement of carboplatin adduct formation and repair. However, carboplatin-DNA monoadducts persist over a few days, with a subset of monoadducts remaining stable even longer, likely as a consequence of sequence context effects.69 Therefore, we measured carboplatin monoadduct formation and repair as a surrogate for total drug exposure of cellular DNA here and in our previous studies.56,61,70 In the case of oxaliplatin, the 14C label is located on the cyclohexane ring, and the label is retained in both the monoadduct and cross-linked species (Figure 1A). For this reason, we are able to use a lower specific activity and still observe sufficient signal-to-noise in the AMS data. The high sensitivity and precision of AMS for detecting the microdose-induced DNA damage and the linear relationship to the damage induced by the therapeutic dose suggest that subtoxic microdoses of platinum can be used to predict the levels of DNA damage induced by therapeutic chemo-therapy in vivo.
Moreover, our breast cancer cell line data support our hypothesis that the levels of platinum-DNA adduct formation (both at multiple time points and calculated as an AUC) correlate with cellular drug sensitivity and that determination of platinum-DNA adduct levels in patients prior to the start of treatment may allow us to predict response to platinum-based therapy. We previously observed a similar correlation between carboplatin-DNA monoadducts and cellular sensitivity in ovarian and lung cancer cells lines, where the sensitive cell lines had significantly higher monoadduct levels than the resistant cell lines.56,61,64 The correlation between carboplatin-DNA adducts and tumor response was also observed in patient-derived xenograft models of bladder cancer and in bladder cancer and lung cancer patients, although a larger confirmatory trial is needed.63,64 Interestingly, carboplatin-DNA monoadduct levels in bladder cancer cell lines did not correlate with sensitivity, but oxaliplatin-DNA adduct levels and carboplatin-DNA adducts in the tumors of bladder cancer patients did, highlighting the need for preclinical and clinical testing for each cancer type and drug combination.56,62,63 Furthermore, Poirier et al. observed a significantly lower number of adducts in patients with progressive disease compared to nonprogressive disease using a less sensitive, ELISA-based method to quantitate carboplatin-DNA adducts in peripheral blood cells isolated from breast cancer patients (51 vs 248 amol/μg DNA, P = 0.047).55 These findings suggest the possibility that breast cancer sensitivity to carboplatin could be predicted by measuring carboplatin monoadducts in peripheral blood cells—a much less invasive biomarker test than measuring adduct levels in tumor biopsy samples—but this will need to be verified in a clinical trial setting. Though more patient data are needed for confirmation, we have demonstrated the feasibility of measuring oxaliplatin PK and oxaliplatin-DNA adduct levels in both the PBMC and tumor tissue of a metastatic breast cancer patient. This patient had a low peak level of oxaliplatin-DNA adducts which may correspond with her lack of response to oxaliplatin therapy, although measurement of oxaliplatin-DNA adduct levels from multiple responder and nonresponder breast cancer patients is necessary to confirm these results. However, comparatively, the peak oxaliplatin-DNA adduct level of the nonresponding patient (8.03 adducts/108 nt) falls within the range of peak adduct levels we observed in the oxaliplatin resistant cell lines (range: 1.76−20.2, mean: 8.57 adducts/108 nt) after receiving an oxaliplatin microdose than the oxaliplatin sensitive cell lines (range: 23.0−53.7, mean: 39.3 adducts/108 nt). These data, in combination with our previous clinical studies, show the potential promise of diagnostic microdosing for multiple types cancer types.
Additionally, we determined rates of DNA repair and analyzed mutations found in DNA repair genes in our breast cancer cell line panel. In the six breast cancer cell lines, the carboplatin-sensitive cells had a higher rate of carboplatin-DNA adduct repair than the resistant cell lines. The oxaliplatin-sensitive cell lines showed a similar trend, but the difference between sensitive and resistant is smaller. These findings are in contrast to our previous results where the carboplatin and oxaliplatin-resistant bladder cancer cell line had higher levels of platinum-DNA adduct repair than the sensitive cell line.56 These DNA repair data indicate that the rate of repair does not strongly influence oxaliplatin or carboplatin cytotoxicity, which is consistent with previous reports on cisplatin for other cancer cell culture studies.71 However, our approach to measuring DNA repair rates via loss of drug-DNA adducts will also include loss of adducts due to hydrolysis and cell division in addition to DNA repair, so further studies are necessary to confirm these results. Missense mutations of p53 were the most frequent mutation observed in this cell line panel but did not correlate with cellular sensitivity, as was previously observed by Silver et al.30 Two carboplatin-resistant, oxaliplatin-sensitive cell lines, MCF7 and T-47D, had missense mutations in the phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) gene. The relationship between mutations in this gene and response to platinum has not been described for breast cancer, but Arjumand et al. found that cervical cancer cells harboring the E545 K mutation, the same mutation observed in the MCF7 cells was more resistant to cisplatin than wild-type, which corresponds to the carboplatin-resistance of the MCF7 cell line.72 The MDA-MB-468 cell line (carboplatin-sensitive, oxaliplatin-resistant) was the only cell line in our panel to have a mutation in the BRCA1/2 genes. Germline BRCA1/2 mutations, which impair DNA repair in the tumor cells, have previously been observed to correlate with good responses to platinum-based therapies.7,30–34 We observed this cell line to be sensitive to carboplatin but resistant to oxaliplatin, indicating that simply having mutated BRCA1/2 genes is not sufficient for causing or predicting platinum sensitivity. Further, the two other carboplatin-sensitive cell lines and all three oxaliplatin-sensitive cell lines do not contain BRCA1/2 mutations, suggesting that the lack of mutated BRCA1/2 genes alone does not predict resistance to platinum-based therapies. Finally, one of the carboplatin-resistant cell lines, MCF7, contains a mutation in both ERCC6 and ERCC8, but the role of the ERCC family in predicting response to platinum is not yet clear.30,36 The mutational analysis of this panel of six breast cancer cell lines that have a nearly 6-fold range in carboplatin IC50 values and 200-fold range in oxaliplatin IC50 values highlights that cellular drug resistance mechanisms are multifactorial and the use of DNA repair rates and genetic mutations in DNA repair genes to predict cellular sensitivity is complicated and not well understood.
Here we have demonstrated that we can dose a panel of six breast cancer cell lines with a platinum microdose to calculate levels of platinum-DNA adduct formation via AMS, that adducts formed with a microdose are proportional to those formed with a therapeutic dose, and that adduct levels correlate with cellular sensitivities. These data collectively support the use of platinum microdosing and quantification of adduct formation as a means to determine patient therapeutic response.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank the LLNL staff for analyzing the AMS samples and the UC Davis patient for participating in our clinical trial.
Funding
AMS samples were analyzed at Lawrence Livermore National Laboratory under the auspices of the DOE contract DE-AC52–07NA27344 and supported by NIH/NCRR Resource for Biomedical Accelerator Mass Spectrometry P41 RR013461 and DOE LDRD grant 08-LW-100 (P.T.H. and K.T.) and by the American Cancer Society Institutional Research Grant (C.X.P.). This study was also supported by the VA Career Development Award-2 (C.X.P.), an NCI Cancer Center Support Grant (R.D.W.) a Cancer Clinical Investigator Team Leadership Award (C.X.P.), and NIH awards HHSN26120 1200048C, HHSN2 61201200 084C, R01CA155642 (P.T.H.). The authors gratefully acknowledge the Susan and Gerry Knapp Family Fund. This work was performed, in part, under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under contract DE-AC52–07NA27344. The work reported here does not represent the views or opinions of the Department of Veterans Affairs or the United States Government.
The authors declare the following competing financial interest(s): Paul T. Henderson and Chong-Xian Pan have ownership stakes in Accelerated Medical Diagnostics Incorpo rated.
ABBREVIATIONS
- amol
attomole
- AMS
accelerator mass spectroscopy
- ATCC
American Type Culture Collection
- AUC
area under the curve
- BRCA1/2
breast cancer susceptibility gene
- COSMIC
Catalogue of Somatic Mutations in Cancer
- ERCC1/2
excision repair cross-complementing group
- FDA
Food and Drug Administration
- GMP
good manufacturing practice
- HER2
human epidermal growth factor receptor 2
- HRD
homologous recombination deficiency
- IND
investigational new drug
- IRB
Institutional Review Board
- LSC
liquid scintillation counter
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NER
nucleotide excision repair
- nt
nucleotide
- PBMC
peripheral blood mononuclear cells
- PES
poly(ether sulfone)
- PIK3CA
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
- PK
pharmaco-kinetic
- TNBC
triple negative breast cancer
- WFI
water for injection
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemrestox.8b00170.
Table S1. Details of mutations in DNA repair genes. Table of nucleotide and amino acid mutations present in the DNA repair genes of all six breast cancer cell lines used in this study (PDF)
REFERENCES
- (1).Society AC (2017) Cancer Facts & Figures 2017, American Cancer Society, Atlanta, GA. [Google Scholar]
- (2).Slamon D, Eiermann W, Robert N, Pienkowski T, Martin M, Press M, Mackey J, Glaspy J, Chan A, Pawlicki M, Pinter T, Valero V, Liu MC, Sauter G, von Minckwitz G, Visco F, Bee V, Buyse M, Bendahmane B, Tabah-Fisch I, Lindsay MA, Riva A, Crown J, and Breast Cancer International Research, G. (2011) Adjuvant trastuzumab in HER2-positive breast cancer. N. Engl. J. Med 365, 1273–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Schneeweiss A, Chia S, Hickish T, Harvey V, Eniu A, Hegg R, Tausch C, Seo JH, Tsai YF, Ratnayake J, McNally V, Ross G, and Cortes J (2013) Pertuzumab plus trastuzumab in combination with standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer: a randomized phase II cardiac safety study (TRYPHAENA). Ann. Oncol 24, 2278–2284. [DOI] [PubMed] [Google Scholar]
- (4).Robert N, Leyland-Jones B, Asmar L, Belt R, Ilegbodu D, Loesch D, Raju R, Valentine E, Sayre R, Cobleigh M, Albain K, McCullough C, Fuchs L, and Slamon D (2006) Randomized phase III study of trastuzumab, paclitaxel, and carboplatin compared with trastuzumab and paclitaxel in women with HER-2-overexpressing metastatic breast cancer. J. Clin. Oncol 24, 2786–2792. [DOI] [PubMed] [Google Scholar]
- (5).von Minckwitz G, Schneeweiss A, Loibl S, Salat C, Denkert C, Rezai M, Blohmer JU, Jackisch C, Paepke S, Gerber B, Zahm DM, Kummel S, Eidtmann H, Klare P, Huober J, Costa S, Tesch H, Hanusch C, Hilfrich J, Khandan F, Fasching PA, Sinn BV, Engels K, Mehta K, Nekljudova V, and Untch M (2014) Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): a randomised phase 2 trial. Lancet Oncol. 15, 747–756. [DOI] [PubMed] [Google Scholar]
- (6).Sikov WM, Berry DA, Perou CM, Singh B, Cirrincione CT, Tolaney SM, Kuzma CS, Pluard TJ, Somlo G, Port ER, Golshan M, Bellon JR, Collyar D, Hahn OM, Carey LA, Hudis CA, and Winer EP (2015) Impact of the addition of carboplatin and/or bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin and cyclophosphamide on pathologic complete response rates in stage II to III triple-negative breast cancer: CALGB 40603 (Alliance). J. Clin. Oncol 33, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Isakoff SJ, Mayer EL, He L, Traina TA, Carey LA, Krag KJ, Rugo HS, Liu MC, Stearns V, Come SE, Timms KM, Hartman AR, Borger DR, Finkelstein DM, Garber JE, Ryan PD, Winer EP, Goss PE, and Ellisen LW (2015) TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy With Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol 33, 1902–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Raymond E, Faivre S, Woynarowski JM, and Chaney SG (1998) Oxaliplatin: mechanism of action and antineoplastic activity. Semin Oncol 25, 4–12. [PubMed] [Google Scholar]
- (9).Garufi C, Nistico C, Brienza S, Vaccaro A, D’Ottavio A, Zappala AR, Aschelter AM, and Terzoli E (2001) Single-agent oxaliplatin in pretreated advanced breast cancer patients: a phase II study. Ann. Oncol 12, 179–182. [DOI] [PubMed] [Google Scholar]
- (10).Zelek L, Cottu P, Tubiana-Hulin M, Vannetzel JM, Chollet P, Misset JL, Chouaki N, Marty M, Gamelin E, Culine S, Dieras V, Mackenzie S, and Spielmann M (2002) Phase II study of oxaliplatin and fluorouracil in taxane- and anthracycline-pretreated breast cancer patients. J. Clin. Oncol 20, 2551–2558. [DOI] [PubMed] [Google Scholar]
- (11).Pectasides D, Pectasides M, Farmakis D, Bountouroglou N, Nikolaou M, Koumpou M, Mylonakis N, and Kosmas C (2003) Oxaliplatin plus high-dose leucovorin and 5-fluorouracil in pretreated advanced breast cancer: a phase II study. Ann. Oncol 14, 537–542. [DOI] [PubMed] [Google Scholar]
- (12).Thuss-Patience PC, von Minckwitz G, Kretzschmar A, Loibl S, Schaller G, Dorken B, and Reichardt P (2003) Oxaliplatin and 5-fluorouracil for heavily pretreated metastatic breast cancer: a preliminary phase II study. Anti-Cancer Drugs 14, 549–553. [DOI] [PubMed] [Google Scholar]
- (13).Kakolyris S, Kalbakis K, Potamianou A, Malamos N, Vamvakas L, Christophillakis C, Tselepatiotis E, Giassas S, Mavroudis D, Amarantidis K, and Georgoulias V (2006) Salvage chemotherapy with gemcitabine and oxaliplatin in heavily pretreated patients with metastatic breast cancer: a multicenter phase II study. Oncology 70, 273–279. [DOI] [PubMed] [Google Scholar]
- (14).Airoldi M, Cattel L, Passera R, Pedani F, Delprino L, and Micari C (2006) Gemcitabine and oxaliplatin in patients with metastatic breast cancer resistant to or pretreated with both anthracyclines and taxanes: clinical and pharmacokinetic data. Am. J. Clin. Oncol 29, 490–494. [DOI] [PubMed] [Google Scholar]
- (15).Delozier T, Guastalla JP, Yovine A, Levy C, Chollet P, Mousseau M, Delva R, Coeffic D, Vannetzel JM, Zazzi ES, Brienza S, and Cvitkovic E (2006) A phase II study of an oxaliplatin/vinorelbine/5-fluorouracil combination in patients with anthracycline-pretreated and taxane-pretreated metastatic breast cancer. Anti-Cancer Drugs 17, 1067–1073. [DOI] [PubMed] [Google Scholar]
- (16).Caruba T, Cottu PH, Madelaine-Chambrin I, Espie M, Misset JL, and Gross-Goupil M (2007) Gemcitabine-oxaliplatin combination in heavily pretreated metastatic breast cancer: a pilot study on 43 patients. Breast J. 13, 165–171. [DOI] [PubMed] [Google Scholar]
- (17).Polyzos A, Gogas H, Markopoulos C, Tsavaris N, Papadopoulos O, Polyzos K, and Giannopoulos A (2009) Salvage chemotherapy with oxaliplatin and capecitabine for breast cancer patients pretreated with anthracyclines and taxanes. Anticancer Res. 29, 2851–2856. [PubMed] [Google Scholar]
- (18).Yardley DA, Daniel D, Stipanov M, Drosick DR, Mainwaring M, Peyton J, Shastry M, and Hainsworth JD (2010) A phase II trial of oxaliplatin and trastuzumab in the treatment of HER2-positive metastatic breast cancer. Cancer Invest. 28, 865–871. [DOI] [PubMed] [Google Scholar]
- (19).Delpeuch A, Leveque D, Rob L, and Bergerat JP (2011) Off-label use of oxaliplatin in patients with metastatic breast cancer. Anticancer Res. 31, 1765–1767. [PubMed] [Google Scholar]
- (20).Guerrero A, Servitja S, Rodriguez-Lescure A, Calvo L, del Barco S, Quintanar MT, Juarez JI, Gayo J, Llombart A, and Tusquets I (2011) Phase I/II study of biweekly vinorelbine and oxaliplatin as first-line treatment in patients with metastatic breast cancer. Anti-Cancer Drugs 22, 283–289. [DOI] [PubMed] [Google Scholar]
- (21).Sun S, Wang LP, Zhang J, Yang XY, Zhang QL, Jia Z, Hu XC, and Wang BY (2012) Phase II study of oxaliplatin plus leucovorin and 5-fluorouracil in heavily pretreated metastatic breast cancer patients. Med. Oncol 29, 418–424. [DOI] [PubMed] [Google Scholar]
- (22).Nielsen DL, Norgaard H, Vestermark LW, Pfeiffer P, Jensen BK, Nelausen KM, Bergenfeldt M, Hermann KL, and Jensen BV (2012) Intrahepatic and systemic therapy with oxaliplatin combined with capecitabine in patients with hepatic metastases from breast cancer. Breast 21, 556–561. [DOI] [PubMed] [Google Scholar]
- (23).Njiaju UO, Tevaarwerk AJ, Kim K, Chang JE, Hansen RM, Champeny TL, Traynor AM, Meadows S, Van Ummersen L, Powers K, and Stewart JA (2013) Capecitabine and oxaliplatin in combination as first- or second-line therapy for metastatic breast cancer: a Wisconsin Oncology Network trial. Cancer Chemother. Pharmacol 71, 613–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhang J, Wang L, Wang Z, Hu X, Wang B, Cao J, Lv F, Zhen C, Zhang S, and Shao Z (2015) A phase II trial of biweekly vinorelbine and oxaliplatin in second- or third-line metastatic triple-negative breast cancer. Cancer Biol. Ther 16, 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Liu J, Xiao Y, Wei W, Guo JX, Liu YC, Huang XH, Zhang RX, Wu YJ, and Zhou J (2015) Clinical efficacy of administering oxaliplatin combined with S-1 in the treatment of advanced triple-negative breast cancer. Exp. Ther. Med 10, 379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Li T, Wang B, Wang Z, Ragaz J, Zhang J, Sun S, Cao J, Lv F, Wang L, Zhang S, Ni C, Wu Z, Xie J, and Hu X (2015) Bevacizumab in Combination with Modified FOLFOX6 in Heavily Pretreated Patients with HER2/Neu-Negative Metastatic Breast Cancer: A Phase II Clinical Trial. PLoS One 10, e0133133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rizzi A, Aroldi F, Bertocchi P, Prochilo T, Mutti S, Savelli G, Fraccon AP, and Zaniboni A (2016) GEMOX: An Active Regimen for the Treatment of Luminal and Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer. Chemotherapy 62, 30–33. [DOI] [PubMed] [Google Scholar]
- (28).Fei F, Chen C, Xue J, Di GH, Lu JS, Liu GY, Shao ZM, and Wu J (2013) Efficacy and safety of docetaxel combined with oxaliplatin as a neoadjuvant chemotherapy regimen for Chinese triple-negative local advanced breast cancer patients. A prospective, open, and unicentric Phase II clinical trial. Am. J. Clin. Oncol 36, 545–551. [DOI] [PubMed] [Google Scholar]
- (29).Hoeijmakers JH (2001) Genome maintenance mechanisms for preventing cancer. Nature 411, 366–374. [DOI] [PubMed] [Google Scholar]
- (30).Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, Juul N, Leong CO, Calogrias D, Buraimoh A, Fatima A, Gelman RS, Ryan PD, Tung NM, De Nicolo A, Ganesan S, Miron A, Colin C, Sgroi DC, Ellisen LW, Winer EP, and Garber JE (2010) Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J. Clin. Oncol 28, 1145–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Evers B, Schut E, van der Burg E, Braumuller TM, Egan DA, Holstege H, Edser P, Adams DJ, Wade-Martins R, Bouwman P, and Jonkers J (2010) A high-throughput pharmaceutical screen identifies compounds with specific toxicity against BRCA2-deficient tumors. Clin. Cancer Res 16, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bhattacharyya A, Ear US, Koller BH, Weichselbaum RR, and Bishop DK (2000) The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J. Biol. Chem 275, 23899–23903. [DOI] [PubMed] [Google Scholar]
- (33).Byrski T, Gronwald J, Huzarski T, Grzybowska E, Budryk M, Stawicka M, Mierzwa T, Szwiec M, Wisniowski R, Siolek M, Dent R, Lubinski J, and Narod S (2010) Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J. Clin. Oncol 28, 375–379. [DOI] [PubMed] [Google Scholar]
- (34).Vollebergh MA, Lips EH, Nederlof PM, Wessels LF, Schmidt MK, van Beers EH, Cornelissen S, Holtkamp M, Froklage FE, de Vries EG, Schrama JG, Wesseling J, van de Vijver MJ, van Tinteren H, de Bruin M, Hauptmann M, Rodenhuis S, and Linn SC (2011) An aCGH classifier derived from BRCA1-mutated breast cancer and benefit of high-dose platinum-based chemotherapy in HER2-negative breast cancer patients. Ann. Oncol 22, 1561–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Vollebergh MA, Lips EH, Nederlof PM, Wessels LF, Wesseling J, Vd Vijver MJ, de Vries EG, van Tinteren H, Jonkers J, Hauptmann M, Rodenhuis S, and Linn SC (2014) Genomic patterns resembling BRCA1- and BRCA2-mutated breast cancers predict benefit of intensified carboplatin-based chemotherapy. Breast Cancer Res. 16, R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lu J, Zhao H, Li S, Tian Z, Zhu X, Wang H, and Fu H (2015) Correlation of rs1799793 polymorphism in ERCC2 and the clinical response to platinum-based chemotherapy in patients with triple negative breast cancer. Int. J. Clin Exp Med. 8, 2934–2938. [PMC free article] [PubMed] [Google Scholar]
- (37).MA ELB, and El Kashef WF (2017) ERCC1 Expression in Metastatic Triple Negative Breast Cancer Patients Treated with Platinum-Based Chemotherapy. Asian Pac J. Cancer Prev 18, 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Gyorffy B, Hatzis C, Sanft T, Hofstatter E, Aktas B, and Pusztai L (2015) Multigene prognostic tests in breast cancer: past, present, future. Breast Cancer Res. 17, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Cimino GD, Pan CX, and Henderson PT (2013) Personalized medicine for targeted and platinum-based chemotherapy of lung and bladder cancer. Bioanalysis 5, 369–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, Szallasi Z, Barry WT, Winer EP, Tung NM, Isakoff SJ, Ryan PD, Greene-Colozzi A, Gutin A, Sangale Z, Iliev D, Neff C, Abkevich V, Jones JT, Lanchbury JS, Hartman AR, Garber JE, Ford JM, Silver DP, and Richardson AL (2016) Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 22, 3764–3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Marquard AM, Eklund AC, Joshi T, Krzystanek M, Favero F, Wang ZC, Richardson AL, Silver DP, Szallasi Z, and Birkbak NJ (2015) Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark Res. 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Timms KM, Abkevich V, Hughes E, Neff C, Reid J, Morris B, Kalva S, Potter J, Tran TV, Chen J, Iliev D, Sangale Z, Tikishvili E, Perry M, Zharkikh A, Gutin A, and Lanchbury JS (2014) Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 16, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Telli ML, Jensen KC, Vinayak S, Kurian AW, Lipson JA, Flaherty PJ, Timms K, Abkevich V, Schackmann EA, Wapnir IL, Carlson RW, Chang PJ, Sparano JA, Head B, Goldstein LJ, Haley B, Dakhil SR, Reid JE, Hartman AR, Manola J, and Ford JM (2015) Phase II Study of Gemcitabine, Carboplatin, and Iniparib As Neoadjuvant Therapy for Triple-Negative and BRCA1/2 Mutation-Associated Breast Cancer With Assessment of a Tumor-Based Measure of Genomic Instability: PrECOG 0105. J. Clin. Oncol 33, 1895–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kaklamani VG, Jeruss JS, Hughes E, Siziopikou K, Timms KM, Gutin A, Abkevich V, Sangale Z, Solimeno C, Brown KL, Jones J, Hartman AR, Meservey C, Jovanovic B, Helenowski I, Khan SA, Bethke K, Hansen N, Uthe R, Giordano S, Rosen S, Hoskins K, Von Roenn J, Jain S, Parini V, and Gradishar W (2015) Phase II neoadjuvant clinical trial of carboplatin and eribulin in women with triple negative early-stage breast cancer (NCT01372579). Breast Cancer Res. Treat 151, 629–638. [DOI] [PubMed] [Google Scholar]
- (45).Watkins JA, Irshad S, Grigoriadis A, and Tutt AN (2014) Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 16, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Saleh EM, El-Awady RA, Anis N, and El-Sharkawy N (2012) Induction and repair of DNA double-strand breaks using constant-field gel electrophoresis and apoptosis as predictive markers for sensitivity of cancer cells to cisplatin. Biomed. Pharmacother 66, 554–562. [DOI] [PubMed] [Google Scholar]
- (47).Fichtinger-Schepman AM, van Oosterom AT, Lohman PH, and Berends F (1987) cis-Diamminedichloroplatinum(II)-induced DNA adducts in peripheral leukocytes from seven cancer patients: quantitative immunochemical detection of the adduct induction and removal after a single dose of cisdiamminedichloroplatinum(II). Cancer Res. 47, 3000–3004. [PubMed] [Google Scholar]
- (48).Reed E, Ostchega Y, Steinberg SM, Yuspa SH, Young RC, Ozols RF, and Poirier MC (1990) Evaluation of platinum-DNA adduct levels relative to known prognostic variables in a cohort of ovarian cancer patients. Cancer Res. 50, 2256–2260. [PubMed] [Google Scholar]
- (49).Reed E, Ozols RF, Tarone R, Yuspa SH, and Poirier MC (1988) The measurement of cisplatin-DNA adduct levels in testicular cancer patients. Carcinogenesis 9, 1909–1911. [DOI] [PubMed] [Google Scholar]
- (50).Oshita F, Yamamoto N, Fukuda M, Ohe Y, Tamura T, Eguchi K, Shinkai T, and Saijo N (1995) Correlation of therapeutic outcome in non-small cell lung cancer and DNA damage assayed by polymerase chain reaction in leukocytes damaged in vitro. Cancer Res. 55, 2334–2337. [PubMed] [Google Scholar]
- (51).Schellens JH, Ma J, Planting AS, van der Burg ME, van Meerten E, de Boer-Dennert M, Schmitz PI, Stoter G, and Verweij J (1996) Relationship between the exposure to cisplatin, DNA-adduct formation in leucocytes and tumour response in patients with solid tumours. Br. J. Cancer 73, 1569–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).van de Vaart PJ, Belderbos J, de Jong D, Sneeuw KC, Majoor D, Bartelink H, and Begg AC (2000) DNA-adduct levels as a predictor of outcome for NSCLC patients receiving daily cisplatin and radiotherapy. Int. J. Cancer 89, 160–166. [PubMed] [Google Scholar]
- (53).Sar DG, Montes-Bayon M, Blanco-Gonzalez E, and Sanz-Medel A (2010) Quantitative methods for studying DNA interactions with chemotherapeutic cisplatin. TrAC, Trends Anal. Chem 29, 1390–1398. [Google Scholar]
- (54).Darcy KM, Tian C, and Reed E (2007) A Gynecologic Oncology Group study of platinum-DNA adducts and excision repair cross-complementation group 1 expression in optimal, stage III epithelial ovarian cancer treated with platinum-taxane chemotherapy. Cancer Res. 67, 4474–4481. [DOI] [PubMed] [Google Scholar]
- (55).Poirier MC, Reed E, Shamkhani H, Tarone RE, and Gupta-Burt S (1993) Platinum drug-DNA interactions in human tissues measured by cisplatin-DNA enzyme-linked immunosorbent assay and atomic absorbance spectroscopy. Environ. Health Perspect 99, 149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Wang S, Zhang H, Scharadin TM, Zimmermann M, Hu B, Pan AW, Vinall R, Lin TY, Cimino G, Chain P, Vuyisich M, Gleasner C, McMurry K, Malfatti M, Turteltaub K, de Vere White R, Pan CX, and Henderson PT (2016) Molecular Dissection of Induced Platinum Resistance through Functional and Gene Expression Analysis in a Cell Culture Model of Bladder Cancer. PLoS One 11, e0146256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Ognibene TJ, Bench G, Vogel JS, Peaslee GF, and Murov S (2003) A high-throughput method for the conversion of CO2 obtained from biochemical samples to graphite in septa-sealed vials for quantification of 14C via accelerator mass spectrometry. Anal. Chem 75, 2192–2196. [DOI] [PubMed] [Google Scholar]
- (58).Jacobs SS, Fox E, Dennie C, Morgan LB, McCully CL, and Balis FM (2005) Plasma and cerebrospinal fluid pharmacokinetics of intravenous oxaliplatin, cisplatin, and carboplatin in nonhuman primates. Clin. Cancer Res 11, 1669–1674. [DOI] [PubMed] [Google Scholar]
- (59).Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, Teague JW, Stratton MR, McDermott U, and Campbell PJ (2015) COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 43, D805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Ehrsson H, Wallin I, and Yachnin J (2002) Pharmacokinetics of oxaliplatin in humans. Med. Oncol 19, 261–265. [DOI] [PubMed] [Google Scholar]
- (61).Henderson PT, Li T, He M, Zhang H, Malfatti M, Gandara D, Grimminger PP, Danenberg KD, Beckett L, de Vere White RW, Turteltaub KW, and Pan CX (2011) A microdosing approach for characterizing formation and repair of carboplatin-DNA monoadducts and chemoresistance. Int. J. Cancer 129, 1425–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Wang S, Zhang H, Malfatti M, de Vere White R, Lara PN Jr., Turteltaub K, Henderson P, and Pan CX (2010) Gemcitabine causes minimal modulation of carboplatin-DNA monoadduct formation and repair in bladder cancer cells. Chem. Res. Toxicol 23, 1653–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Zimmermann M, Wang SS, Zhang H, Lin TY, Malfatti M, Haack K, Ognibene T, Yang H, Airhart S, Turteltaub KW, Cimino GD, Tepper CG, Drakaki A, Chamie K, de Vere White R, Pan CX, and Henderson PT (2017) Microdose-induced Drug-DNA Adducts as Biomarkers of Chemotherapy Resistance in Humans and Mice. Mol. Cancer Ther 16, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Wang SS, Zimmermann M, Zhang H, Lin TY, Malfatti M, Haack K, Turteltaub KW, Cimino GD, de Vere White R, Pan CX, and Henderson PT (2017) A diagnostic microdosing approach to investigate platinum sensitivity in non-small cell lung cancer. Int. J. Cancer 141, 604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Wing RM, Pjura P, Drew HR, and Dickerson RE (1984) The primary mode of binding of cisplatin to a B-DNA dodecamer: C-G-C-G-A-A-T-T-C-G-C-G. EMBO J. 3, 1201–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Fichtinger-Schepman AM, van der Veer JL, den Hartog JH, Lohman PH, and Reedijk J (1985) Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: formation, identification, and quantitation. Biochemistry 24, 707–713. [DOI] [PubMed] [Google Scholar]
- (67).Baik MH, Friesner RA, and Lippard SJ (2002) Theoretical study on the stability of N-glycosyl bonds: why does N7-platination not promote depurination? J. Am. Chem. Soc 124, 4495–4503. [DOI] [PubMed] [Google Scholar]
- (68).Hah SS, Sumbad RA, de Vere White RW, Turteltaub KW, and Henderson PT (2007) Characterization of oxaliplatin-DNA adduct formation in DNA and differentiation of cancer cell drug sensitivity at microdose concentrations. Chem. Res. Toxicol 20, 1745–1751. [DOI] [PubMed] [Google Scholar]
- (69).Hah SS, Stivers KM, de Vere White RW, and Henderson PT (2006) Kinetics of carboplatin-DNA binding in genomic DNA and bladder cancer cells as determined by accelerator mass spectrometry. Chem. Res. Toxicol 19, 622–626. [DOI] [PubMed] [Google Scholar]
- (70).Zimmermann M, Wang SS, Zhang H, Lin TY, Malfatti M, Haack K, Ognibene T, Yang H, Airhart S, Turteltaub KW, Cimino GD, Tepper CG, Drakaki A, Chamie K, de Vere White R, Pan CX, and Henderson PT (2017) Microdose-Induced Drug-DNA Adducts as Biomarkers of Chemotherapy Resistance in Humans and Mice. Mol. Cancer Ther 16, 376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Wang D, and Lippard SJ (2005) Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discovery 4, 307–320. [DOI] [PubMed] [Google Scholar]
- (72).Arjumand W, Merry CD, Wang C, Saba E, McIntyre JB, Fang S, Kornaga E, Ghatage P, Doll CM, and Lees-Miller SP (2016) Phosphatidyl inositol-3 kinase (PIK3CA) E545K mutation confers cisplatin resistance and a migratory phenotype in cervical cancer cells. Oncotarget 7, 82424–82439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.