

Abstract
In the unfolded protein response (UPR), Ire1 activates Hac1 to coordinate the transcription of hundreds of genes to mitigate ER stress. Recent work in C. elegans suggests that oxidative stress inhibits this canonical Ire1 signaling pathway, activating instead an anti-oxidant stress response. We sought to determine whether this novel mode of UPR function also existed in yeast, where Ire1 has been best characterized. We show that the yeast UPR is also subject to inhibition by oxidative stress. Inhibition is mediated by a single evolutionarily conserved cysteine, and affects both luminal and membrane pathways of Ire1 activation. In yeast, Ire1 appears dispensable for resistance to oxidative stress and, therefore, the physiologic significance of this pathway remains to be demonstrated.
Keywords: Ire1, unfolded protein response, cysteine, oxidative stress, arsenic
Introduction
Misfolded proteins are toxic and cells have developed complex stress responses to identify and eliminate them. In the unfolded protein response (UPR), activation of the endoplasmic reticulum transmembrane protein Ire1 promotes the unusual cytoplasmic splicing of HAC1 mRNA [1]. Hac1 (Xbp1 in mammals) then coordinates the transcription of hundreds of genes which adapt the cell to ER stress [2]. These include molecular chaperones within the ER, components of the ER-associated degradation (ERAD) pathway, genes involved in lipid metabolism, and others [2].
Two major pathways of Ire1 activation have been described. In the canonical luminal sensing pathway, Ire1 detects misfolded proteins within the ER. In yeast this appear to be mediated through direct interaction of Ire1 with their exposed hydrophobic domains [3–4], while in higher organisms the luminal chaperone BiP (Kar2) plays an important role in this process [5]. The second pathway of Ire1 activation, by contrast, senses defects in the ER membrane (so-called “bilayer stress”) and is mediated by Ire1’s transmembrane domain [6–7]. Various defects in lipid metabolism activate the UPR through this pathway, and the UPR in turn stimulates the expression of various genes involved in lipid metabolism [2, 8–10]. This emerging relationship between the UPR and lipid homeostasis is recapitulated at the level of human disease. Obesity, for still poorly understood reasons, is a major inducer of the UPR, and weight loss reverses UPR induction [11]. Conversely, defects in the UPR pathway lead to pre-diabetic insulin resistance [12–13].
Recent work suggests an entirely novel mode of regulation of the UPR by oxidative stress. In work done largely in C. elegans, oxidative stress was shown to result in a switch-like regulation of Ire1, inhibiting the canonical signaling pathway mediated by Hac1/Xbp1 while simultaneously stimulating an antioxidant response mediated by the p38 MAP kinase family and the transcription factor Nrf2 [14]. Ire1 and the UPR have been best characterized in the budding yeast S. cerevisiae. Therefore, it was important to determine whether this pathway also exists in yeast, its similarity to the pathway in higher organisms, and its physiologic significance.
Materials and Methods
Strains and Plasmids
Yeast strains are listed in Table 1. Standard techniques were used for strain construction and plasmid transformation. Cells were cultured at 30°C. YPD medium consisted of 1% yeast extract, 2% Bacto-peptone, and 2% dextrose. Synthetic medium consisted of 0.7% Difco yeast nitrogen base supplemented with amino acids, uridine, adenine, and 2% dextrose. Plasmid selection was by omission of uridine.
Table 1.
Yeast Strains
| Name | Genotype | Source |
|---|---|---|
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | RG collection |
| ire1Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ire1::KAN | RG collection |
| sGM223 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 [YCplac33] | This study |
| sGM224 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ire1::KAN [YCplac33] | This study |
| sGM225 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ire1::KAN [pJH233] | This study |
| sGM226 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ire1::KAN [pGM43] | This study |
| sGM252 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ire1::KAN [pGM57] | This study |
| hog1Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 hog1::KAN | RG collection |
Note: RG collection strains were obtained from ThermoFisher Scientific.
The following chemicals were used: sodium arsenite (1 mM; Sigma #S7400), tunicamycin (5 μg/ml; Sigma #T7765), dithiothreitol (1.5 mM; Gold Bio #DTT50), and hydrogen peroxide (5 mM; Sigma #H1009). All treatments were for one hour. Double drug treatments were applied concurrently.
All plasmids used were based on ycPlac33, a low copy centromeric vector (CEN) bearing the URA3 auxotrophic marker [15]. IRE1 was cloned with its endogenous promoter (500 bp upstream and 200 bp downstream) into ycPlac33 (pJH233). Site-directed mutagenesis of pJH233 was performed using the QuikChange method (Agilent). Ire1-C832S (pGM43) was generated using the following primers: 5’-CAGACTTTGGTCTTTcCAAAAAACTAGACTCTGG-3’, 5’-CCAGAGTCTAGTTTTTTGgAAAGACCAAAGTCTG-3’. Ire1-C748S (pGM54) was generated using the following primers: 5’-TTTTGTATATTGCTTTAGAGCTCaGCAATTTGAACCTTCAAGATTTG-3’, 5’-CAAATCTTGAAGGTTCAAATTGCtGAGCTCTAAAGCAATATACAAAA-3’. All plasmids were verified by sequencing
Proteomic Analysis
The tandem mass tag-based mass spectrometry proteomic analysis of the cellular response to sodium arsenite was previously described in detail [16], and quantitated 4,563 proteins (of ~6,000 in yeast) at 0, 1, and 4 hours after arsenic treatment (1 mM) and with biologic triplicates. The data presented here represent further original analysis of that data set. We previously showed that cells treated under these conditions resumed growth with a normal doubling time within just 1-2 hours of arsenic wash-out, indicating that the observed proteomic changes likely reflect a physiologic stress response rather than non-specific changes in dying or dead cells [16].
RT-PCR
Reverse-transcription polymerase chain reaction (RT-PCR) was performed as previously described [17]. HAC1 was amplified using the primers 5’-CACTCGTCGTCTGATACG-3’ and 5’-CATTCAATTCAAATGAATTCAAACCTG-3’. These primers amplify the region encompassing base pairs 373-949 of the open reading frame, allowing for detection of both the unspliced and spliced forms of HAC1. KAR2 (BiP in mammals) was amplified using the primers 5’-GTGTCTTATCCGGTGAAGAAG- 3’ and 5’-CTAGATTCAACCTTGGCCTTG-3’ which amplify the region encompassing base pairs 1268-1745 of the open reading frame. ACT1 was amplified with primers 5’-CTGGTATGTTCTAGCGCTTG-3’ and 5’-GATACCTTGGTGTCTTGGTC-3’ which amplify the region encompassing base pairs 8-439 of the open reading frame.
Immunoblot Analysis
Whole cell lysates were prepared as previously described from logarithmic phase cultures that had been untreated or chemically treated as indicated [18]. In brief, cells were normalized by optical density (OD600) and collected by centrifugation. Pellets were then resuspended in cold 2 M lithium acetate and incubated on ice for 5 min, followed by a 5 min incubation in cold 0.4 M sodium hydroxide on ice. After centrifugation, pellets were resuspended in 1X Laemmli buffer and boiled at 100°C for 5 min. Standard SDS-PAGE and immunoblotting were performed.
The following antibodies were used in this study: anti-Kar2 (BiP) (Santa Cruz; #sc33630), anti-Hog1 (Santa Cruz; #sc-165978), anti-phospho-38 MAPK T180/Y182 (Cell Signaling, #9211S), and anti-Pgk1 (Novex; #459250).
Sequence and Structural Analysis
Sequence alignments were performed using Clustal Omega. The Ire1 crystal structure (PDB: 3FBV) was visualized using Cn3D.
Results
Inhibition of the UPR by Oxidative Stress
We recently carried out a proteomic analysis of the cellular response to trivalent arsenic (arsenite) in yeast. This analysis quantitated 4,563 proteins (of approximately 6,000) at 0, 1, and 4 hours after arsenic treatment [16]. Widespread proteomic changes were observed, with approximately 1,000 proteins showing a significant change in abundance. A large number of these proteins related to pathways of protein quality control, with significant upregulation of molecular chaperones, proteasome subunits, and autophagy components. This profile suggested that these cells were under significant proteotoxic stress. We were therefore surprised that components of the UPR were not induced under these conditions. For example, the UPR transcriptional targets Kar2, Lhs1, and Pdi1 showed little or no change in protein abundance (Fig. 1A). Stress-induced proteasome biogenesis is induced by a separate proteotoxic stress response mediated by the transcription factor Rpn4 [19]. In contrast to the UPR, Rpn4 protein levels increased by approximately twenty-fold [16], resulting in the increased abundance of proteasome subunits such as Pre2 (Fig. 1A).
Figure 1. Inhibition of the UPR by oxidative stress.
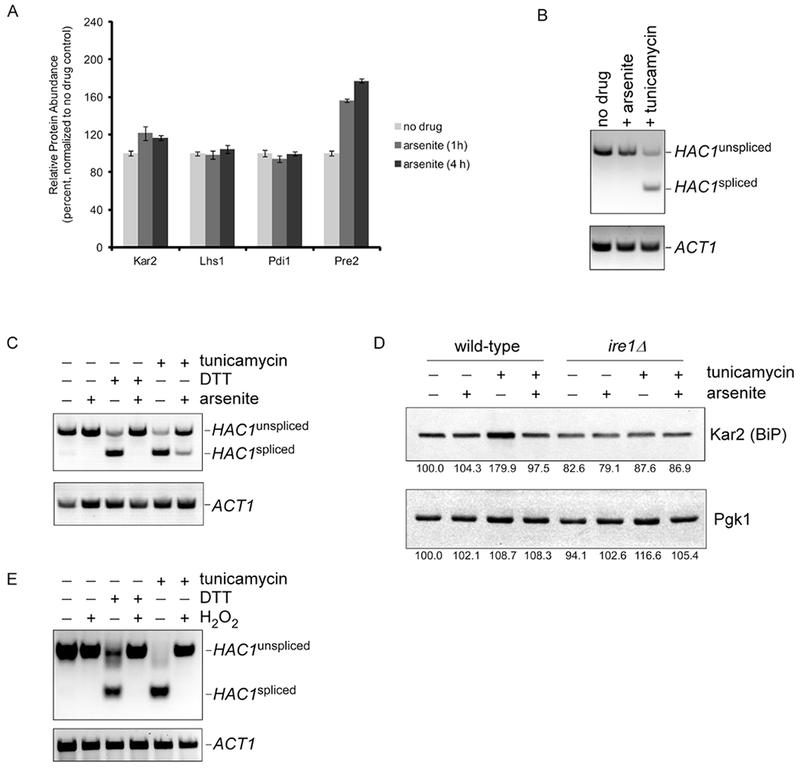
A) Relative protein abundance of UPR targets Kar2, Lhs1, and Pdi1 at 0, 1, and 4 h after treatment with sodium arsenite (1 mM). The data were generated using a quantitative mass spectrometry-based proteomic approach [16]. Pre2 is a proteasome subunit and a transcriptional target of the Rpn4 proteotoxic stress response which is best known for controlling proteasome abundance [16]. Error bars represent standard deviations from triplicate cultures.
B) Splicing of the Ire1 target HAC1 in response to treatment with sodium arsenite (1 mM) or tunicamycin (5 μg/ml) for 1 hour, as determined by RT-PCR. Spliced and unspliced forms of HAC1 are indicated. ACT1 serves as a loading control (lower panel).
C) Inhibition of tunicamycin-induced (5 μg/ml) or DTT-induced (1.5 mM) HAC1 splicing by concurrent treatment with sodium arsenite (1 mM), as determined by RT-PCR. Chemical treatment was for one hour. Lower panel, ACT1 loading control.
D) Inhibition of tunicamycin-induced (5 μg/ml) expression of Kar2 (BiP) protein by concurrent treatment with sodium arsenite (1 mM) for one hour. Whole cell extracts were prepared and analyzed by SDS–PAGE followed by immunoblot with anti-Kar2 antibody (upper panel) or anti-Pgk1 antibody (lower panel; loading control). There was no tunicamycin-induced expression of Kar2 in the ire1Δ mutant. Images were quantitated using NIH Image J, and the relative expression of Kar2 and Pgk1 is indicated as a percentage of the untreated wild-type control (i.e. lane).
E) Inhibition of tunicamycin-induced (5 μg/ml) or DTT-induced (1.5 mM) HAC1 splicing by concurrent treatment with hydrogen peroxide (5 mM), as determined by RT-PCR. Chemical treatment was for one hour. Lower panel, ACT1 loading control.
To confirm the lack of UPR induction under these conditions, we looked directly at HAC1 splicing by RT-PCR. Tunicamycin, an inhibitor of ER glycosylation, resulted in splicing of HAC1 as expected (Fig. 1B). By contrast, there was no detectable HAC1 splicing upon arsenic treatment (Fig. 1B). In principle, this lack of UPR activation could reflect simply a failure to induce the UPR or active inhibition of the UPR by arsenic. To distinguish between these possibilities, we simultaneously treated cells with both tunicamycin and arsenic. Surprisingly, the tunicamycin-mediated induction of the UPR was strongly inhibited by the presence of arsenic (Fig. 1C). To determine whether this inhibition was specific to tunicamycin, we employed dithiothreitol (DTT), which also induces the UPR but is structurally and mechanistically distinct from tunicamycin. DTT strongly induced the UPR, however arsenic again was able to largely inhibit this induction (Fig. 1C). We confirmed this UPR inhibition at the protein level by visualizing the Hac1 target Kar2. Tunicamycin induced Kar2 expression at the protein level, but this effect was largely abrogated if cells had been simultaneously treated with arsenic (Fig. 1D). Thus, arsenic is capable of inhibiting the UPR.
An important aspect of arsenic toxicity is its ability to covalently interact with free thiol groups in the amino acid sidechains of proteins [20–21]. This may contribute to some of arsenic’s proteotoxic effects [16, 22–23]. However, arsenic is also a potent oxidizing agent capable of generating oxygen free radicals and other oxidizing species [24]. If the UPR inhibitory effect of arsenic derived from its ability to directly bind Ire1, this effect would likely not be shared with other oxidizing agents. Alternately, Ire1 inhibition by arsenic could reflect a more general feature of oxidizing agents. To distinguish between these possibilities, we employed the oxidizing agent hydrogen peroxide which is not known to covalently modify proteins. Hydrogen peroxide potently inhibited both tunicamycin- and DDT-mediated UPR induction (Fig. 1E). Thus, the effects observed are not specific to arsenic, but appear to reflect a general capacity for oxidative stress to inhibit the UPR.
Thiol-Based Regulation of Ire1 in Response to Oxidative Stress
In C. elegans, a critical cysteine residue (C663 in C. elegans) located within the kinase activation loop was essential for oxidative stress-induced UPR inhibition [14]. That residue is conserved in yeast (Fig. 2A; ref. 14) and therefore we sought to test whether it functioned similarly. We cloned IRE1 into a low-copy centromeric vector expressed from the endogenous Ire1 promoter. This plasmid fully complemented the ire1Δ mutant (see Fig. 4A below). We prepared an ire1-C832S mutant and tested its function in the HAC1 splicing assay. Arsenic-induced inhibition of the UPR was markedly attenuated in the ire1-C832S mutant for both tunicamycin and DTT (Fig. 2B-C). As a measure of downstream Hac1 function, we looked at transcription of Kar2 by RT-PCR. In wild-type cells, tunicamycin-mediated Kar2 transcription was strongly inhibited by simultaneous arsenic treatment (Fig. 2D). However, this inhibitory effect was again largely lost in the ire1-C832S mutant (Fig. 2D). Finally, the capacity for hydrogen peroxide to inhibit the UPR was also lost in the ire1-C832S mutant (Fig. 2E). Thus, a critical cysteine residue mediates control of the UPR by oxidative stress in an evolutionarily conserved manner.
Figure 2. Cysteine-832 mediates oxidative stress-induced inhibition of Ire1.
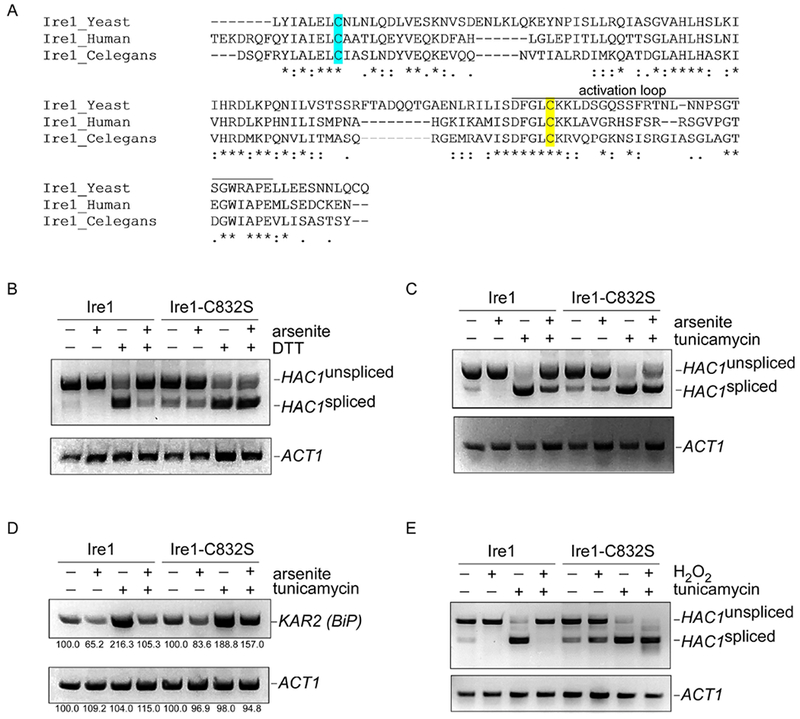
A) Sequence alignment of the critical region of Ire1 from S. cerevisiae, C. elegans, and humans. Cysteine-832 is highlighted in yellow. Cysteine-748 is highlighted in cyan. Asterisks, identical residues; double dots, highly similar residues; single dots, similar residues.
B-C) The ire1-C832S mutant is refractory to arsenic-induced inhibition of HAC1 splicing, as determined by RT-PCR. HAC1 splicing was induced by either DTT (1.5 mM; panel B) or tunicamycin (5 μg/ml; panel C). Arsenite was used at 1 mM. Treatment was for one hour. Lower panels, ACT1 controls.
D) The ire1-C832S mutant is refractory to arsenic-induced inhibition of transcription of the HAC1 target KAR2, as determined by RT-PCR. Treatment was with tunicamycin (5 μg/ml) and/or arsenite (1 mM) for one hour. Lower panel, ACT1 control. Images were quantitated using NIH Image J, and the relative expression of KAR2 and PGK1 is indicated as a percentage of the respective untreated control.
E) The ire1-C832S mutant is refractory to hydrogen peroxide-induced inhibition of HAC1 splicing, as determined by RT-PCR. HAC1 splicing was induced by tunicamycin (5 μg/ml). Hydrogen peroxide was used at 5 mM. Chemical treatment was for one hour. Lower panel, ACT1 control.
Figure 4. The ire1-C832S Mutant Does not Show Obvious Phenotypes or Alteration of MAP Kinase Activation.
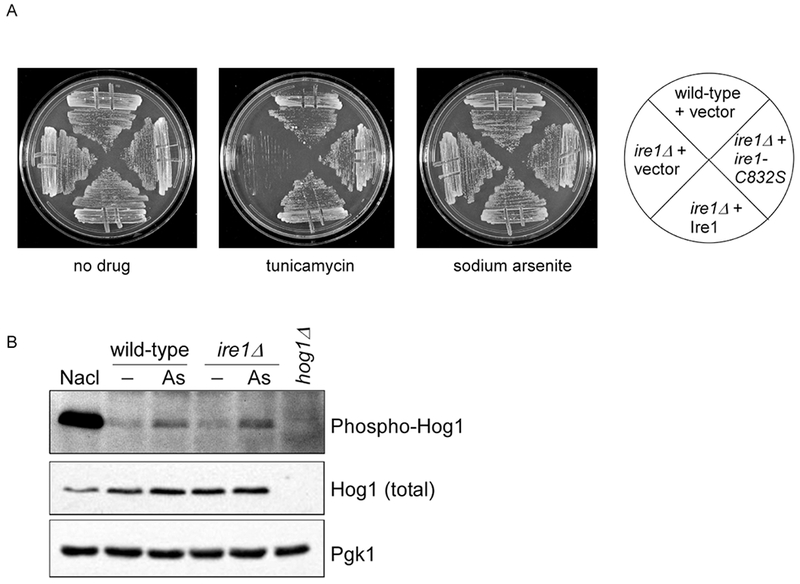
A) Growth of wild-type and ire1Δ strains expressing an empty vector, IRE1 wild-type, and ire1-C832S, as indicated. Cells were spotted in three-fold serial dilutions onto plates containing no drug, tunicamycin (1 μg/ml), or sodium arsenite (1 mM) and cultured for 2-4 days at 30°C.
B) Arsenite-induced activation of the Hog1 is unaffected in the ire1Δ mutant. Cells were treated with sodium arsenite (1 mM) for hour. Whole cell extracts were prepared and analyzed by SDS-PAGE followed by immunoblotting for phosphorylated and total Hog1. Lower panel, Pgk1 (loading control). A parallel treatment with osmotic stress (0.4 M NaCl for 5 min) confirms the assignment of the phosphorylated Hog1 species. Parallel analysis of the hog1Δ strain confirms the accuracy of the Hog1 antibodies.
The experiments of Fig. 2B-C were conducted in synthetic (i.e. minimal) media, which is relatively deficient compared to rich media in a number of lipid precursors including choline, ethanolamine, and inositol. Under these conditions, a mild constitutive induction of the UPR is present even in the absence of an exogenous proteotoxic stressor. Interestingly, arsenic was also capable of inhibiting this constitutive UPR induction (Fig. 2B-C, compare lanes 1 and 2), and Cysteine-832 was required for UPR inhibition (Fig. 2B-C, compares lanes 1and 2 with lanes 5 and 6). These data suggest that oxidative stress inhibits both the luminal (tunicamycin, DTT) and membrane (minimal media) pathways of Ire1 activation, and inhibition of both pathways is mediated by Cysteine-832.
Specificity of Cysteine-832 in Regulating Ire1
Cysteine oxidation may result in a number of distinct modifications, including sulfenylation, sulfinylation, sulfonylation, and disulfide bonding [25] . The transcription factor Yap1, for example, is a major mediator of the oxidative stress response in yeast and is regulated by disulfide bonding which controls the protein’s localization [26]. We wanted to determine whether other cysteines might contribute to oxidative regulation of Ire1 in this manner. We noticed that a second cysteine (C748) is located near C832 and also shows strong evolutionary conservation from yeast to humans (Fig. 2A, 3A). We mutated this residue to serine and determined the ability of this mutant to mediate arsenic-induced inhibition of the UPR. The ire1-C748S mutant showed normal induction of the UPR in response to DTT, and unlike the C832S mutant, showed no defect in arsenic-mediated UPR inhibition (Fig. 3B). This result emphasizes the specificity of the effects seen with C832, and makes a disulfide bonding event unlikely, as no other potential disulfide binding partner was present in the close vicinity of C832.
Figure 3. An Adjacent Cysteine (748) Does not Contribute to Arsenic-Induced Inhibition of Ire1.
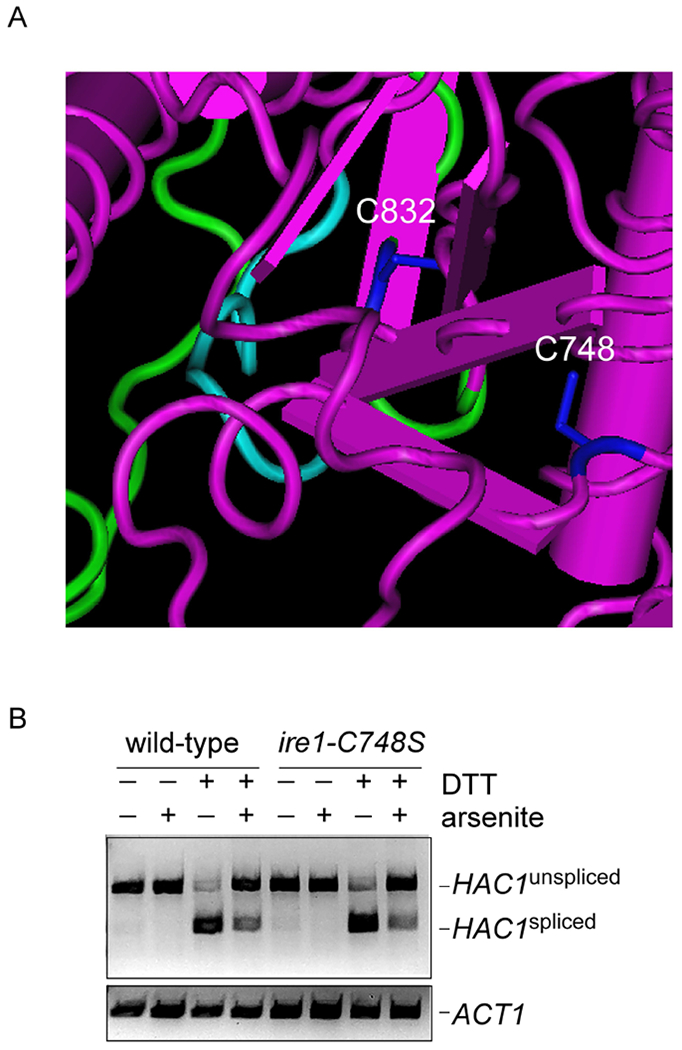
A) Structural features of S. cerevisiae Ire1 (PDB: 3FBV). Cysteines 748 and 832 are indicated in blue. Cyan, catalytic loop. Green, activation loop.
B) Inhibition of DTT-induced (1.5 mM) HAC1 splicing by concurrent treatment with sodium arsenite (1 mM) persists in the ire1-C748S mutant, as determined by RT-PCR. Chemical treatment was for one hour. Lower panel, ACT1 loading control.
Phenotypic Analysis of the Ire1-C832S Mutant
In C. elegans, the corresponding cysteine residue (C663) is required for survival upon arsenic exposure [14]. Furthermore, inhibition of the canonical UPR signaling pathway is accompanied by stimulation of an alternate anti-oxidant response mediated by the p38 MAP kinase pathway [14]. We sought to determine whether similar functions of Ire1 could be detected in yeast. We expressed wild-type and the C832S mutant of Ire1 in the ire1Δ strain and measured its survival against ER stress. Both the wild-type and mutant Ire1 plasmids fully complemented the growth defect of the ire1Δ mutant upon tunicamycin exposure, confirming that Cysteine-832 is not required for normal UPR function (Fig. 4A). By contrast, the ire1Δ mutant showed no growth defect upon exposure to arsenic, even at high doses (Fig. 4B; ref. 27), and the C832S mutant did not confer a dominant negative phenotype. Simultaneous treatment with both tunicamycin and arsenic also did not reveal a phenotype for the C832S mutant that was different from the wild-type (data not shown). Finally, we considered that redundancy in oxidative stress responses might have masked a phenotype for the C832S mutant. We therefore knocked out Yap1, a major mediator of the oxidative stress response in yeast [26], in the ire1Δ mutant. However, this double mutant did not show a synthetic negative phenotype (data not shown).
Hog1 is generally considered to be the yeast ortholog of the p38 MAP kinase family, and functions in a variety of stress responses including the cellular response to arsenic [28]. Activation of Hog1 is typically accompanied by phosphorylation of Hog1 at two key residues within its kinase activation segment [29]. Hog1 is best known for its role in the osmotic stress response, and is rapidly and strongly phosphorylated in response to sodium chloride (Fig. 4B). Arsenic-induced phosphorylation of Hog1 is much weaker but is readily detectable (ref. 30). Hog1 phosphorylation in response to arsenic was completely unaffected in the ire1Δ mutant (Fig. 4B).
Discussion
The UPR is a widely studied and evolutionarily conserved stress response that detects misfolded proteins within the endoplasmic reticulum and orchestrates a complex program of cellular remodeling to mitigate this threat. Recent work suggested an exciting and entirely novel mode of UPR regulation by oxidative stress. In this pathway, oxidation of a key cysteine residue within the kinase activation loop of Ire1 inhibits the canonical signaling pathway through Xbp1 (the ortholog of Hac1) and instead activates an anti-oxidant stress response mediated by p38 kinase signaling and the transcription factor Skn1/Nrf2 [14]. Ire1 has been best characterized in yeast, and therefore it was important to determine whether this pathway also existed in yeast. Our results support and extend those of Blackwell and colleagues [14]. We find that the UPR is not induced by arsenic in yeast, consistent with a prior report [27]. This was surprising since proteotoxicity appears to represent an important aspect of arsenic’s toxicity [16, 22]. In fact, arsenic was capable of potently inhibiting Ire1 in a manner that was dominant to known UPR inducers such as tunicamycin and DTT. Inhibition was also seen under conditions of minimal media, indicating that arsenic can inhibit both the luminal and membrane pathways of threat sensing by Ire1. UPR inhibition was not specific to arsenic, but was also observed with hydrogen peroxide, suggesting that it is a consequence of oxidative stress generally rather than reflecting specific aspects of arsenic toxicity such as its ability to covalently modify thiol groups. The mechanism of inhibition also appears to be highly conserved as it is mediated by the same cysteine residue in yeast (C832) and C. elegans (C663). This novel mode of UPR regulation thus extends to yeast and may represent a universal aspect of Ire1 function.
In contrast to the results from higher organisms, we do not yet understand the physiologic significance of this regulatory program. We have not yet been able to demonstrate a physiologic defect of the ire1-C832S mutant (or the ire1Δ mutant) upon exposure to arsenic or other oxidizing agents, even under conditions where Ire1 is strongly inhibited (see also ref. 27). There are two possible explanations for this. First, it may be that Ire1 performs an essential role in response to oxidative stress, but that additional functional redundancies in yeast have prevented us from visualizing this aspect of Ire1 function. Alternately, it may be that inhibitory cysteine oxidation of Ire1 also occurs in yeast, but that the coupling of this function to a physiologically efficacious Ire1-mediated antioxidant stress response represents an important development in higher organisms.
Acknowledgments
The authors thank members of the Hanna laboratory for comments on the manuscript.
Funding
This work was supported by NIH grant DP5-OD019800 (to J.H.).
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.Cox JS, Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 1996. November 1;87(3):391–404. [DOI] [PubMed] [Google Scholar]
- 2.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000. April 28;101(3):249–58. [DOI] [PubMed] [Google Scholar]
- 3.Kimata Y, Ishiwata-Kimata Y, Ito T, Hirata A, Suzuki T, Oikawa D, Takeuchi M, Kohno K. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J Cell Biol. 2007. October 8;179(1):75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gardner BM, Walter P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science. 2011. September 30;333(6051):1891–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oikawa D, Kimata Y, Kohno K, Iwawaki T. Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp Cell Res. 2009. September 10;315(15):2496–504. [DOI] [PubMed] [Google Scholar]
- 6.Promlek T, Ishiwata-Kimata Y, Shido M, Sakuramoto M, Kohno K, Kimata Y. Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol Biol Cell. 2011. September;22(18):3520–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Halbleib K, Pesek K, Covino R, Hofbauer HF, Wunnicke D, Hänelt I, Hummer G, Ernst R. Activation of the Unfolded Protein Response by Lipid Bilayer Stress. Mol Cell. 2017. August 17;67(4):673–684.e8. [DOI] [PubMed] [Google Scholar]
- 8.Cox JS, Chapman RE, Walter P. The unfolded protein response coordinates the production of endoplasmic reticulum protein and endoplasmic reticulum membrane. Mol Biol Cell. 1997. September;8(9):1805–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jonikas MC, Collins SR, Denic V, Oh E, Quan EM, Schmid V, Weibezahn J, Schwappach B, Walter P, Weissman JS, Schuldiner M. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science. 2009. March 27;323(5922):1693–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thibault G, Shui G, Kim W, McAlister GC, Ismail N, Gygi SP, Wenk MR, Ng DT. The membrane stress response buffers lethal effects of lipid disequilibrium by reprogramming the protein homeostasis network. Mol Cell. 2012. October 12;48(1):16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS, Klein S. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009. March;58(3):693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011. May 26;473(7348):528–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004. October 15;306(5695):457–61. [DOI] [PubMed] [Google Scholar]
- 14.Hourihan JM, Moronetti Mazzeo LE, Fernández-Cárdenas LP, Blackwell TK. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Mol Cell. 2016. August 18;63(4):553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gietz RD, Sugino A. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 74, 527–34. [DOI] [PubMed] [Google Scholar]
- 16.Guerra-Moreno A, Isasa M, Bhanu MK, Waterman DP, Eapen VV, Gygi SP, Hanna J. 2015. Proteomic Analysis Identifies Ribosome Reduction as an Effective Proteotoxic Stress Response. J Biol Chem. 290, 29695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guerra-Moreno A, Hanna J. 2016. Tmc1 Is a Dynamically Regulated Effector of the Rpn4 Proteotoxic Stress Response. J Biol Chem. 291, 14788–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weisshaar N, Welsch H, Guerra-Moreno A, Hanna J. Phospholipase Lpl1 links lipid droplet function with quality control protein degradation. Mol Biol Cell. 2017. March 15;28(6):716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie Y, Varshavsky A. RPN4 is a ligand, substrate, and transcriptional regulator of the 26S proteasome: a negative feedback circuit. Proc Natl Acad Sci U S A. 2001. March 13;98(6):3056–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosen BP, Bhattacharjee H, Shi W. Mechanisms of metalloregulation of an anion-translocating ATPase. J Bioenerg Biomembr. 1995. February;27(1):85–91. [DOI] [PubMed] [Google Scholar]
- 21.Shen S, Li XF, Cullen WR, Weinfeld M, Le XC. Arsenic binding to proteins. Chem Rev. 2013. October 9;113(10):7769–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobson T, Navarrete C, Sharma SK, Sideri TC, Ibstedt S, Priya S, Grant CM, Christen P, Goloubinoff P, Tamás MJ. Arsenite interferes with protein folding and triggers formation of protein aggregates in yeast. J Cell Sci. 2012. November 1;125(Pt 21):5073–83. [DOI] [PubMed] [Google Scholar]
- 23.Hanna J, Guerra-Moreno A, Ang J, Micoogullari Y. Protein Degradation and the Pathologic Basis of Disease. Am J Pathol. 2019. January;189(1):94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hughes MF. Arsenic toxicity and potential mechanisms of action. Toxicol Lett. 2002. July 7;133(1):1–16. [DOI] [PubMed] [Google Scholar]
- 25.Lo Conte M, Carroll KS. The redox biochemistry of protein sulfenylation and sulfinylation. J Biol Chem. 2013. September 13;288(37):26480–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wood MJ, Storz G, Tjandra N. Structural basis for redox regulation of Yap1 transcription factor localization. Nature. 2004. August 19;430(7002):917–21. [DOI] [PubMed] [Google Scholar]
- 27.Gardarin A, Chédin S, Lagniel G, Aude JC, Godat E, Catty P, Labarre J. Endoplasmic reticulum is a major target of cadmium toxicity in yeast. Mol Microbiol. 2010. May;76(4):1034–48. [DOI] [PubMed] [Google Scholar]
- 28.Saito H, Posas F. Response to hyperosmotic stress. Genetics. 2012. October;192(2):289–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brewster JL, de Valoir T, Dwyer ND, Winter E, Gustin MC. An osmosensing signal transduction pathway in yeast. Science. 1993. March 19;259(5102):1760–3. [DOI] [PubMed] [Google Scholar]
- 30.Sotelo J, Rodríguez-Gabriel MA. Mitogen-activated protein kinase Hog1 is essential for the response to arsenite in Saccharomyces cerevisiae. Eukaryot Cell. 2006. October;5(10):1826–30. [DOI] [PMC free article] [PubMed] [Google Scholar]