

Abstract
Background:
Prodromal Alzheimer’s disease (AD) clinical trials enroll patients with Mild Cognitive Impairment (MCI) meeting biomarker criteria, but specific enrollment criteria vary among trials.
Methods:
We used data from AD Neuroimaging Initiative (ADNI) MCI participants to assess AD biomarker eligibility, variation in trial outcome measures, and statistical power.
Results:
Most (65%) participants meet eligibility criteria based on low cerebrospinal fluid (CSF) amyloid beta (Aβ). Relative to trials enrolling exclusively based on low CSF Aβ, trials including participants with a high ratio of phosphorylated tau (p-Tau) to Aβ would include an additional 15% of participants. Fewer (34–62%) participants met criteria for Aβ and tau. Differences in clinical and demographic characteristics of modeled trial samples were minimal. Those with low Aβ and high tau showed the greatest change over time on outcome measures.
Conclusions:
Eligibility rates for prodromal trials vary depending on the specific biomarker criteria, though differences in demographics and the variation associated with outcome measures are minimal. Broadening inclusion criteria beyond amyloid alone may facilitate recruitment but include patients showing slower progression over time. Biomarker criteria selection should be informed by the goal of enrolling individuals most likely to utilize and benefit from the intervention under investigation in a particular setting.
Keywords: Alzheimer’s disease, mild cognitive impairment, recruitment
INTRODUCTION
To intervene earlier in Alzheimer’s disease (AD), clinical trials enroll patients with Mild Cognitive Impairment (MCI).1 Successful disease slowing in MCI may prolong periods of highest function and lowest cost of care. Because heterogeneous etiologies may underlie MCI, these trials incorporate AD biomarker inclusion criteria, a concept termed prodromal AD.2,3 Enrolling only patients meeting AD biomarker criteria ensures on-target activity for candidate treatments with a mechanism of action related to AD pathophysiology, improves placebo group behavior,4 and optimizes statistical power.5,6
A query of clinicaltrials.gov reveals that markers of the 42-amino acid peptide beta amyloid (Aβ), including amyloid positron emission tomography (PET) imaging and cerebrospinal fluid (CSF) measures, are most frequently used as inclusion criteria in prodromal AD trials (Table 1). Fewer trials incorporate markers of neurofibrillary tangles, such as levels of CSF phosphorylated tau (p-tau) or total tau (t-tau). In a recently proposed framework, markers of Aβ are required for a diagnosis of AD.7
Table 1.
Frequency of biomarker criteria employed by recruiting or not yet recruiting prodromal AD (or MCI due to AD) trials identified through clinicaltrials.gov (01/11/18).*
| Biomarker inclusion criteria | Trials, n |
|---|---|
| Amyloid PET only | 11 |
| Amyloid PET or CSF Aβ | 5 |
| Amyloid PET or CSF Aβ or multiple downstream markers (e.g., MRI, FDG PET) | 2 |
| CSF Aβ or CSF t-Tau:Aβ ratio | 1 |
| Not reported | 4 |
Based on searches for prodromal AD, MCI due to AD, MCI, mild neurocognitive disorder, or mild cognitive disorder, refined based on reported inclusion criteria
A guiding principle of clinical trial inclusion criteria is to assess safety and efficacy in a controlled setting among individuals most likely to utilize and benefit from the intervention under investigation. While this implies that precise knowledge of specific drug mechanism of action may mandate more stringent criteria, overly restrictive biomarker criteria may exclude a proportion of otherwise eligible participants who may receive treatment once approved. The use of additional inclusive biomarker criteria may increase prodromal AD trial eligibility rates,8 whereas overly restrictive biomarker criteria may add to the already difficult challenge of completing prodromal trial accrual.9,10 Alternatively, too broad of enrollment criteria may risk inclusion of participants who are unlikely to progress to AD dementia or are less likely to benefit from the investigational intervention. More restrictive criteria may also increase the probability of cognitive decline during the trial, thereby potentially increasing statistical power.11,12
Few studies to date compare biomarker criteria in the setting of prodromal AD trials. We used AD Neuroimaging Initiative (ADNI) data to assess the impact of using different biomarker enrollment criteria. We first examined rates of prodromal AD trial eligibility and how differing criteria affected modeled trial participant demographics. We then assessed trial statistical power using commonly used clinical trial outcome measures. We did so in the real-world scenario in which the number of available eligible participants is limited.
METHODS
Study population.
Data used in the preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). For up-to-date information, see www.adni-info.org. Data were downloaded on January 14, 2017 from http://adni.loni.usc.edu/data-samples/access-data/.
This study included MCI participants in ADNI-1, ADNI-GO (including early and late MCI), and ADNI-2, who had participated in the lumbar puncture aspects of the studies and had available measures of Aβ, t-tau, and p-tau at baseline. All participants had a modified Hachinski scale score of ≤4, a Geriatric Depression Scale (abbreviated 15-item version) score ≤6, were fluent in English or Spanish, had a suitable study partner who could accompany them to study visits, and lived at home. They had no significant neurologic or psychiatric disease; no history of alcohol or substance abuse; no clinically significant laboratory abnormalities; and no contraindication to neuroimaging.13 MCI was defined using Petersen criteria.14 Patients were required to demonstrate objective impairment on psychometric tests without impaired activities of daily living or fulfilling criteria for dementia. Additionally, patients were required to score 24–30 on the Mini-Mental State Examination (MMSE) and 0.5 on the global Clinical Dementia Rating scale (CDR), with a memory box score ≥0.5. Cognitive impairment was assessed through the Logical Memory II subscale (Delayed Paragraph Recall) from the Wechsler Memory Scale–Revised. Patients were required to score 0.5–1.5 standard deviations below education-adjusted norms for early MCI and >1.5 standard deviations below education-adjusted norms for late MCI.
Biomarker measures.
Our analyses focused on CSF and amyloid PET measures as trial inclusion criteria. We applied established cutoffs for identifying AD CSF signatures: Aβ<192 pg/mL; t-Tau>93 pg/mL; tau phosphorylated at threonine 181 (p-Tau)>23pg/mL; ratio of t-Tau:Aβ (t-Tau:Aβ ratio)>0.39; ratio of p-Tau:Aβ (p-Tau:Aβ ratio)>0.1 and combinations of these criteria.15 Similarly, we applied a quantitative eligibility criterion for florbetapir amyloid PET, mean standardized uptake value ratios (SUVR) across four regions of interest (frontal, temporal, parietal, and cingulate cortex) of 1.11 or greater, using whole cerebellum as a reference region.16,17
Analyses.
To examine the impact of varying CSF protein level criteria, we applied specific inclusion criteria to all available MCI participants from the ADNI studies for whom CSF data were available at baseline. We hypothesized that incorporating high ratios of tau to Aβ, in addition to low CSF Aβ, as inclusion criteria would increase the proportion of trial eligible participants compared to using Aβ criteria alone. We explored the relationship between CSF and amyloid PET inclusion criteria by assessing the proportion of eligible participants based on CSF who also met amyloid PET criteria, among those participants with data available for both biomarkers. We investigated the impact of various inclusion criteria on trial population demographics and trial power for two clinical outcome measures commonly used in clinical trials: the AD Assessment Scale-cognitive subscale (ADAS-cog)18 and the CDR.19 Specifically, because of increased sensitivity demonstrated in MCI,5,20 we used the ADAS-cog including additional elements for delayed recall and executive function (ADAS13) and, in line with FDA guidance,21 the sum-of-the-boxes score for the CDR (CDRSB). Two-year follow-up data were available for 502 participants for the ADAS13 and 510 participants for the CDRSB.
To assess power relative to the differential inclusion criteria, we considered a hypothetical two-year, two-arm randomized clinical trial assessing the within-subject change in CDRSB or ADAS13. We assumed that study resources would be constrained so that only a finite pool of potentially eligible MCI patients would be available for recruitment into the trial (in this case the sample of MCI participants in ADNI). For the varying inclusion criteria, we assessed the variance of CDRSB or ADAS13 after two years of follow-up. We estimated the correlation coefficient (ρ) for repeated measures within subjects using a continuous auto-regressive covariance model. Using these empirically based estimates of variance and correlation, we computed the minimally detectable difference in the within-subject change over two years between treatment and control groups that a trial would be powered to achieve for a sample size of n per group, where n was taken to be 50% of the number of ADNI MCI participants eligible based on given CSF criteria. Power calculations assumed that the analysis would employ an analysis of covariance (ANCOVA) model22 of the form:
where Txi denotes the indicator of whether patient i received the experimental treatment or control, i=1,…,2n, and and denote pre- and post-treatment measurements, respectively. In this case Δ denotes the treatment effect, interpreted as the expected difference in the two-year within-subject change in response comparing treatment to control. It was further assumed that the pre- and post-treatment measurements within a subject followed a bivariate normal distribution with variance and correlation From the above specification, power for detecting a true difference of Δ can be computed as
To illustrate the impact of the varying inclusion criteria on trial power, we plotted absolute power curves, demarcating the necessary true effect size to achieve 90% power. We also computed relative differences between power curves obtained from each of the inclusion criteria, using trials incorporating low CSF Aβ inclusion criteria as a reference group.
To compare the change over time for subjects satisfying high p-Tau:Aβ ratio and low Aβ criteria to those only satisfying one criterion, we fit linear mixed effects models with random slopes and random intercepts to each of the groups. We then compared the fixed effects for the three groups for ADAS13 and CDRSB. We also fit a continuation ratio model to investigate progression to dementia in subjects who satisfy Aβ and p-Tau:Aβ ratio, those satisfying only low Aβ, and subjects satisfying only p-Tau:Aβ ratio, compared to those meeting no biomarker criteria. We adjusted for age, education, race, gender, and apolipoprotein E (APOE) status (at least one ε4 allele compared to none). Progression was defined as two consistent visits with a diagnosis of dementia. Time to progression was taken to be the time from baseline until the first diagnosis of dementia among those who progressed.
RESULTS
Description of the sample.
In total, 623 MCI participants were enrolled in ADNI-1, ADNI-GO, or ADNI-2 and had baseline CSF data available (Table 2). ADNI-1 participants were older and demonstrated slightly poorer performance on the MMSE and ADAS13 at baseline.
Table 2.
Baseline description of the ADNI MCI participants with CSF.
| Characteristic | ADNI-1 | ADNI-GO | ADNI-2 | Combined Sample |
|---|---|---|---|---|
| N | 199 | 117 | 307 | 623 |
| Age, mean (SD) | 74.37 (7.5) | 71.11 (7.8) | 71.64 (7.3) | 72.41 (7.5) |
| Female, mean (SD) | 66 (33.2) | 52 (44.4) | 138 (45.0) | 256 (41.1) |
| White, n (%) | 190 (95.5) | 104 (88.9) | 292 (95.1) | 586 (94.1) |
| Education, mean (SD) | 15.78 (3.0) | 15.67 (2.7) | 16.38 (2.6) | 16.05 (2.8) |
| APOE ε4 alleles 0, n (%) 1, n (%) 2, n (%) |
92 (46.2) 86 (43.2) 21 (10.6) |
71 (60.7) 38 (32.5) 8 (6.8) |
153 (49.8) 119 (38.8) 35 (11.4) |
316 (50.7) 243 (39.0) 64 (10.3) |
| MMSE, mean (SD) | 26.93 (1.8) | 28.3 (1.5) | 27.99 (1.8) | 27.71 (1.8) |
| CDRSB, mean (SD) | 1.56 (0.9) | 1.23 (0.7) | 1.53 (0.9) | 1.48 (0.9) |
| ADAS13, mean (SD) | 18.89 (6.3) | 12.33 (5.1) | 15.84 (7.1) | 16.15 (6.9) |
| Aβ CSF Eligible, n (%)* | 147 (73.9) | 54 (46.2) | 202 (65.8) | 403 (64.7) |
| t-Tau CSF Eligible, n (%)* | 90 (45.2) | 31 (26.5) | 101 (32.9) | 222 (35.6) |
| p-Tau CSF Eligible, n (%) | 141 (70.9) | 77 (65.8) | 234 (76.2) | 452 (72.6) |
| Aβ PET Eligible, n (%) | NA | 50 (42.7) | 178 (58.0) | 228 (36.6) |
| Proportion with visits at 24 Months | 0.804 | 0.855 | 0.847 | 0.835 |
CSF, cerebrospinal fluid; ADNI, Alzheimer’s Disease Neuroimaging Initiative; APOE, apolipoprotein; MMSE, Mini-Mental State Exam; CDRSB, Clinical Dementia Rating Scale Sum of Boxes; ADAS13, Alzheimer’s Disease Assessment Scale-cognitive subscale.
Effect of varying CSF criteria on enrollment eligibility.
Table 3 displays the proportion of MCI participants meeting CSF enrollment criteria and combinations of those criteria. Compared to the proportion of participants meeting the low Aβ criterion (65%), an additional 15% of the sample would qualify for a trial also enrolling participants with adequately high p-Tau:Aβ ratios. In contrast, only an additional 2% would qualify for a trial also accepting participants with high t-Tau:Aβ ratios. Approximately 75% of subjects who met the p-Tau:Aβ ratio criterion but not the Aβ criterion were within one standard deviation of Aβ eligibility (73/95). Similarly, 65% of subjects who met the t-Tau:Aβ ratio criterion but not the Aβ criterion were within one standard deviation of Aβ eligibility (9/14). Restricting to those who met the low Aβ criterion as well as one of the tau criteria reduced the overall eligible sample by 4% - 48%.
Table 3.
Prodromal trial eligibility.
| Aβ | Aβ | |||||||
|---|---|---|---|---|---|---|---|---|
| Eligible (≤192; n=403) | Not eligible (>192; n=220) | Eligible (≤192; n=403) | Not eligible (>192; n=220) | |||||
| p-Tau:Aβ ratio | Eligible (≥0.10; n=483) | N= 388 (62%) | N= 95 (15%) | p-Tau:Aβ ratio | Eligible (≥0.39;n=360) | N= 346 (56%) | N= 14 (2%) | |
| Not eligible (<0.10; n=140) | N= 15 (3%) | N= 125 (20%) | Not eligible (<0.39;n=263) | N= 57 (9%) | N= 206 (33%) | |||
| p-Tau | Eligible (≥23; n=452) | N= 358 (57%) | N= 94 (15%) | t-Tau | Eligible (≥93; n=222) | N= 210 (34%) | N= 12 (2%) | |
| Not eligible (<23; n=171) | N= 45 (7%) | N= 126 (20%) | Not eligible (<93; n=401) | N= 193 (31%) | N= 208 (33%) | |||
Among participants who had amyloid PET data available, 94% of those eligible based on amyloid PET were also eligible based on low CSF Aβ (Table 4). Among those with discrepant Aβ eligibility, 75% (39/52) were eligible based on CSF but ineligible by PET; 25% (13/52) were eligible by PET but ineligible by CSF. Ninety-one percent of those ineligible based on CSF Aβ who met the p-Tau:Aβ ratio criterion were ineligible based on amyloid PET. Compared to a trial enrolling based only on amyloid PET, incorporating the additional p-Tau:Aβ ratio criterion would increase the pool of eligible participants by 46%.
Table 4.
Relationship between CSF and amyloid PET. The proportion of amyloid PET negative and positive participants among those with PET data who were eligible and not eligible based CSF criteria, all cells n (%)
| Amyloid PET SUVR <1.1 (n=192) | Amyloid PET SUVR >1.1 (n=228) | |||||||
|---|---|---|---|---|---|---|---|---|
| low CSF Aβ | low CSF Aβ | |||||||
| Eligible (n = 39) | Not eligible (n = 153) | Eligible (n = 215) | Not eligible (n = 13) | |||||
| high p-Tau:Aβ ratio | Eligible (n = 105) | 30 (15.6%) | 75 (39.1%) | high p-tau ratio | Eligible (n = 220) | 213 (93.4%) | 7 (3.1%) | |
| Not eligible (n = 87) | 9 (4.7%) | 78 (40.6%) | Not eligible (n = 8) | 2 (0.9%) | 6 (2.6%) | |||
| high p-tau | Eligible (n = 97) | 20 (10.4%) | 77 (40.1%) | high p-tau | Eligible (n = 211) | 204 (89.5%) | 7 (3.1%) | |
| Not eligible (n = 95) | 19 (9.9%) | 76 (39.6%) | Not eligible (n = 17) | 11 (4.8%) | 6 (2.6%) | |||
| high t-Tau:Aβ ratio | Eligible (n = 27) | 17 (8.9%) | 10 (5.2%) | high t-Tau:Aβ ratio | Eligible (n = 193) | 192 (84.2%) | 1 (0.4%) | |
| Not Eligible (n = 165) | 22 (11.5%) | 143 (74.5%) | Not Eligible (n = 35) | 23 (10.1%) | 12 (5.3%) | |||
| high t-tau | Eligible (n = 14) | 5 (2.6%) | 9 (4.7%) | high t-tau | Eligible (n = 118) | 118 (51.8%) | 0 (0%) | |
| Not eligible (n = 178) | 34 (17.7%) | 144 (75.0%) | Not eligible (n = 110) | 97 (42.5%) | 13 (5.7%) | |||
Effect of varying CSF criteria on trial populations.
Compared to a trial enrolling only those with low CSF Aβ, including patients meeting low Aβ or high p-Tau:Aβ ratio slightly lowered the overall proportion of APOE ε4 carriers and raised the group mean CSF Aβ level (Table 5). No qualitative differences were apparent, however, for sex, age, race, or performance at baseline on the MMSE, ADAS13, or CDRSB. Trials enrolling only those meeting both Aβ and t-Tau criteria demonstrated the highest proportion of APOE ε4 carriers. These criteria also resulted in the worst mean baseline performance on the ADAS13 and CDRSB, the highest mean levels of p-Tau and t-Tau, and the lowest mean levels of Aβ.
Table 5.
Baseline description of trial samples
| Characteristic | Aβ | p-Tau | t-Tau | p-Tau:Aβ ratio | t-Tau:Aβ ratio | Aβ or p-Tau:Aβ ratio | Aβ or t-Tau:Aβ ratio | Aβ and p-Tau | Aβ and t-Tau |
|---|---|---|---|---|---|---|---|---|---|
| N (%) | 403 (64.7) | 452 (72.6) | 222 (35.6) | 483 (77.5) | 360 (58.1) | 498 (79.9) | 417 (66.9) | 358 (57.5) | 210 (33.7) |
| Age, mean ± SD | 73.29 (7.12) | 72.64 (7.5) | 73.52 (7.4) | 72.66 (7.5) | 73.54 (7.2) | 72.61 (7.5) | 73.39 (7.2) | 73.31 (7.1) | 73.31 (7.3) |
| Female, n (%) | 159 (39.5) | 192 (42.5) | 105 (47.3%) | 201 (41.6) | 147 (40.8) | 204 (41.0) | 167 (40.1) | 145 (40.5) | 98 (46.7) |
| White race, n (%) | 386 (95.8) | 427 (94.5) | 214 (96.4) | 457 (94.6) | 347 (96.4) | 472 (94.8) | 399 (95.7) | 343 (95.8) | 203 (96.7) |
| Education, mean ± SD | 16.00 (2.8) | 16.04 (2.8) | 15.88 (2.8) | 16.00 (2.8) | 15.99 (2.8) | 16.01 (2.8) | 16.02 (2.8) | 16.01 (2.8) | 15.85 (2.8) |
| ApoE ε4 copies, n (%) 0 1 2 |
142 (35.2) 197 (48.9) 64 (15.9) |
188 (41.6) 206 (45.6) 58 (12.8) |
67 (30.2) 121 (54.5) 34 (15.3) |
202 (41.8) 218 (45.1) 63 (13.0) |
114 (31.7) 185 (51.4) 61 (17.0) |
211 (42.4) 223 (44.8) 64 (12.9) |
151 (36.2) 202 (48.4) 64 (15.4) |
117 (32.7) 183 (51.1) 58 (16.2) |
58 (27.6) 118 (56.2) 34 (16.2) |
| MMSE, mean ± SD | 27.42 (1.9) | 27.52 (1.9) | 27.2 (1.9) | 27.54 (1.9) | 27.3 (1.9) | 27.57 (1.9) | 27.42 (1.9) | 27.33 (1.9) | 27.16 (1.9) |
| CDR-SB, mean ± SD | 1.61 (0.9) | 1.53 (0.9) | 1.71 (0.9) | 1.53 (0.9) | 1.62 (0.9) | 1.53 (0.9) | 1.59 (0.9) | 1.62 (0.9) | 1.75 (0.9) |
| ADAS13, mean ± SD | 17.88 (6.8) | 17.26 (6.9) | 18.77 (6.4) | 17.14 (6.9) | 18.47 (6.8) | 17.05 (6.9) | 17.82 (6.8) | 18.29 (6.7) | 18.89 (6.3) |
| CSF Aβ, mean ± SD | 137.7 (24.7) | 156.09 (46.0) | 139.27 (32.7) | 154.09 (43.3) | 137.34 (28.9) | 154.73 (42.9) | 140.47 (29.0) | 135.97 (23.7) | 133.44 (21.0) |
| CSF t-tau, mean ± SD | 108.98 (58.0) | 106.84 (55.6) | 147.80 (52.4) | 103.22 (55.7) | 119.64 (55.0) | 101.16 (56.1) | 109.28 (57.4) | 116.69 (56.7) | 149.01 (53.0) |
| CSF p-tau, mean ± SD | 46.89 (23.0) | 47.26 (21.0) | 56.16 (22.5) | 45.42 (21.5) | 50.00 (22.2) | 44.49 (21.8) | 46.59 (22.7) | 50.55 (21.7) | 57.34 (22.4) |
Trial variance and power.
Table 6 provides the estimated 2-year variances incorporating differing CSF inclusion criteria and employing the CDRSB or ADAS13 as primary outcome measures. No major differences in variance were observed in the trial scenarios. The lowest observed variance for the CDRSB was in trials using p-Tau:Aβ ratio and trials using p-Tau:Aβ ratio or Aβ as inclusion criteria. Trials using the combination of low Aβ and high t-Tau demonstrated the lowest variance for ADAS13. The correlation coefficients of the varying trial scenarios were largely equivalent for each outcome measure. Compared to trials enrolling only those meeting Aβ criteria, none of the more inclusive scenarios sacrificed power (Figures 1B, 1E, 1H, and 1K). In fact, power was increased for the inclusive scenarios (e.g., it is estimated that a trial enrolling low Aβ or high p-Tau:Aβ ratio would require a 12% smaller treatment effect on the ADAS13 to achieve 90% power than would a trial enrolling only those with low Aβ). This impact is attributable to the larger sample size afforded by the broader inclusion criteria in the finite population pools being considered. Trials employing more restrictive criteria generally required larger treatment effects to account for smaller sample sizes (e.g., it is estimated that compared to a trial using the ADAS13 as an outcome enrolling only participants with low Aβ, a trial enrolling patients meeting both low Aβ and high t-Tau criteria would require a 42% higher treatment effect [Table 6; Figure 1C, 1F, 1I, and 1L]). As expected given the minimal impact that the differing inclusion criteria had on outcome variation and within-subject correlation in the outcome, the smallest minimum detectable difference required for 90% power was afforded by the largest trials, those enrolling patients meeting either low Aβ or high p-Tau criteria or low Aβ or high p-Tau:Aβ ratio criteria.
Table 6.
Variance and power associated with trial design choices.
| CDRSB | ||||
|---|---|---|---|---|
| Enrollment criteria | N | Variance | ρ∗ (95% CI) | Minimum detectable treatment effect, points |
| Aβ | 404 | 5.0 | 0.48 (0.44, 0.52) | 0.74 (0.71, 0.76) |
| t-Tau | 222 | 5.0 | 0.46 (0.39, 0.51) | 1.02 (0.97, 1.1) |
| p-Tau | 452 | 4.7 | 0.50 (0.46, 0.54) | 0.67 (0.64, 0.70) |
| t-Tau:Aβ ratio | 360 | 5.0 | 0.48 (0.43, 0.53) | 0.79 (0.75, 0.82) |
| p-Tau:Aβ ratio | 484 | 4.6 | 0.50 (0.45, 0.54) | 0.64 (0.61, 0.66) |
| Aβ or t-Tau | 416 | 4.9 | 0.49 (0.44, 0.53) | 0.72 (0.69, 0.75) |
| Aβ or p-Tau | 498 | 4.7 | 0.50 (0.47, 0.54) | 0.63 (0.61, 0.65) |
| Aβ or t-Tau:Aβ ratio | 418 | 4.9 | 0.49 (0.44, 0.53) | 0.72 (0.69, 0.75) |
| Aβ or p-Tau:Aβ ratio | 498 | 4.6 | 0.50 (0.46, 0.54) | 0.63 (0.61, 0.65) |
| Aβ and t-Tau | 210 | 4.9 | 0.45 (0.39, 0.50) | 1.05 (1.0, 1.1) |
| Aβ and p-Tau | 358 | 5.0 | 0.47 (0.42, 0.51) | 0.79 (0.76, 0.83) |
| Aβ and t-Tau:Aβ ratio | 346 | 5.0 | 0.47 (0.42, 0.52) | 0.81 (0.77, 0.84) |
| Aβ and p-Tau:Aβ ratio | 388 | 4.9 | 0.48 (0.44, 0.52) | 0.75 (0.72, 0.78) |
| ADAS13 | ||||
| Enrollment criteria | N | Variance | ρ∗ (95% CI) | Minimum detectable treatment effect, points |
| Aβ | 404 | 104.7 | 0.63 (0.58, 0.67) | 2.9 (2.7, 3.0) |
| t-Tau | 222 | 93.0 | 0.57 (0.51, 0.63) | 3.9 (3.6, 4.2) |
| p-Tau | 452 | 104.6 | 0.65 (0.61, 0.69) | 2.6 (2.5, 2.8) |
| t-Tau:Aβ ratio | 360 | 104.3 | 0.59 (0.53, 0.65) | 3.2 (2.9, 3.4) |
| p-Tau:Aβ ratio | 484 | 104.9 | 0.64 (0.58, 0.68) | 2.6 (2.4, 2.8) |
| Aβ or t-Tau | 416 | 103.7 | 0.63 (0.59, 0.67) | 2.8 (2.6, 3.0) |
| Aβ or p-Tau | 498 | 103.2 | 0.65 (0.61, 0.69) | 2.5 (2.3, 2.6) |
| Aβ or t-Tau:Aβ ratio | 418 | 103.9 | 0.61 (0.55, 0.66) | 2.9 (2.7, 3.1) |
| Aβ or p-Tau:Aβ ratio | 498 | 103.7 | 0.64 (0.58, 0.68) | 2.5 (2.4, 2.7) |
| Aβ and t-Tau | 210 | 92.5 | 0.56 (0.49, 0.62) | 4.1 (3.8, 4.3) |
| Aβ and p-Tau | 358 | 104.8 | 0.62 (0.57, 0.67) | 3.1 (2.9, 3.3) |
| Aβ and t-Tau:Aβ ratio | 346 | 104.5 | 0.61 (0.56, 0.66) | 3.2 (3.0, 3.4) |
| Aβ and p-Tau:Aβ ratio | 388 | 105.8 | 0.63 (0.58, 0.67) | 2.9 (2.8, 3.1) |
Figure 1.
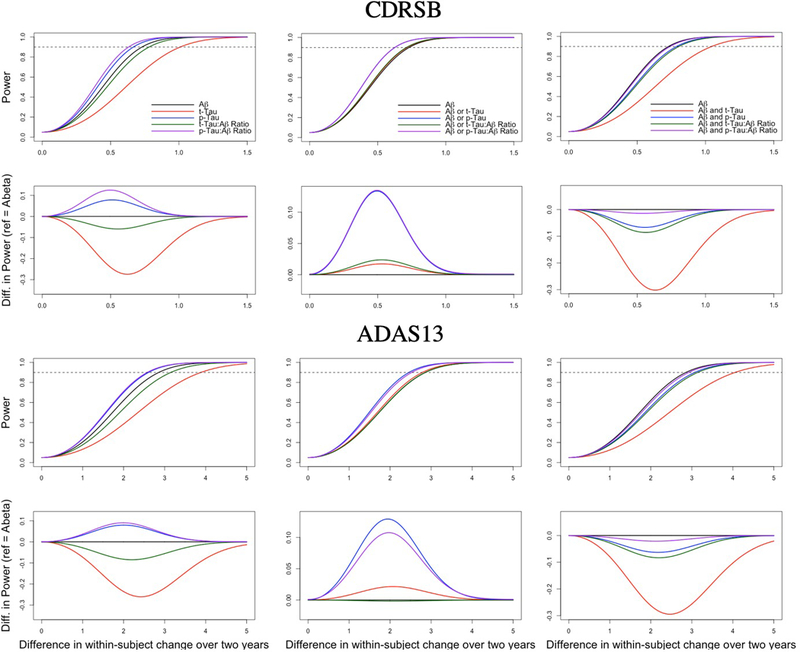
Power curves for trials incorporating the CDRSB (A-F) and ADAS13 (G-L) as primary outcomes. In the rows 1 (A-C) and 3 (G-I), power is plotted for trials using indicated biomarker inclusion criteria (and the associated sample sizes based on ADNI) for varying estimated treatment effects. The dotted horizontal line indicates 90% power. In rows 2 (D-F) and 4 (J-L), relative power is plotted, using trials enrolling based only on CSF Aβ as a reference.
For trials testing a therapy designed to slow disease progression, trial power will be impacted by the rate of change in the control arm. To explore how specific participants meeting varying criteria differ in disease progression, we plotted change over time for subpopulations of the most inclusive modeled trial, one enrolling either low Aβ or high p-Tau:Aβ ratio. Figure 2 demonstrates the individual and group changes for the outcome measures specifically among those participants meeting only high p-Tau:Aβ ratio, only low Aβ, or both criteria. Those meeting both low Aβ and high p-Tau:Aβ ratio criteria demonstrated greater change over 24 months for the ADAS13, compared to those meeting only low Aβ or only high p-Tau:Aβ ratio criteria. For each additional month, ADAS13 increased by 0.153 (95% CI: 0.130, 0.177), on average, for subjects satisfying both low Aβ and high p-Tau ratio. The estimates were 0.040 (95% CI: −0.069, 0.150) and −0.003 (95% CI: −0.057, 0.051) for the low Aβ only and the high p-Tau:Aβ ratio only groups, respectively. For CDRSB, those meeting only low Aβ demonstrated changes equivalent to those meeting both low Aβ and high p-Tau:Aβ ratio criteria; both groups demonstrated greater change than those meeting only high p-Tau:Aβ ratio criteria. For each additional month, CDRSB increased by approximately 0.038 (95% CI: −0.007, 0.082) for the subjects meeting both criteria. The corresponding estimates were 0.057 (95% CI: 0.051, 0.062) for the low Aβ only group and 0.008 (95% CI: −0.002, 0.018) for the high p-Tau:Aβ ratio only group.
Figure 2.

Twenty-four month changes in the CDRSB (A) and ADAS13 (B) plotted based on CSF enrollment criteria. Data for individual MCI patients meeting only high p-Tau:Aβ ratio (blue), only low Aβ (green), or both low Aβ and high p-Tau:Aβ ratio criteria (red) are shown, along with smoothed best fit lines for each group.
Using our definition of progression, 148 subjects converted to dementia out of 601 who had at least one visit after baseline. Subjects who satisfied low Aβ and p-Tau:Aβ ratio were at higher risk for progression (OR=5.12; 95% CI: 2.49, 10.53), compared to subjects with a similar age, education, gender, and APOE status who did not satisfy either criteria. The odds of progression were also higher for those who only satisfied low Aβ (OR=3.7; 95% CI: 1.10, 12.54) but lower for subjects who only satisfied p-Tau:Aβ ratio eligibility (OR=0.61; 95% CI: 0.19, 2.01), compared to subjects satisfying neither criteria.
DISCUSSION
Clinical trial inclusion criteria should ensure participant safety, maximize the probability of trial success, and reflect those patients likely to use and benefit from the investigational intervention if it is efficacious. Overly restrictive criteria may limit generalizability of results and reduce trial feasibility. Recruitment to prodromal AD trials is challenging, due in part to high screen failure rates.8,23 Additional barriers to successful recruitment to prodromal AD trials include that the clinical window of eligibility—a current diagnosis of MCI—is temporary,24 limiting the total time a given patient may be trial eligible. Patients must also agree to biomarker testing, as well as randomization to placebo or a drug that may have side effects.10 Thus, optimizing the probability that patients who are aware, willing, and meet clinical criteria to enroll in prodromal AD trials will also meet biomarker criteria is critical to expediting drug development in this diagnostic category. Here, we found that incorporating a minimum p-Tau level or a minimum p-Tau:Aβ ratio resulted in eligibility of an additional 15% of MCI participants (a potential 25% boost in enrollment), compared to trials using Aβ criteria alone. Incorporating additional eligibility based on CSF t-Tau or t-Tau:Aβ ratio had minimal effects on eligibility rates.
Most prodromal AD trials incorporate a biomarker of Aβ as an inclusion criterion (Table 1). This practice is due to the stance that Aβ is an earlier and more specific marker of AD than is tau2 and observations in mild-to-moderate AD trials, in which ~20% of participants failed to demonstrate elevated brain Aβ.25,26 A proposed biological framework specifies that a marker of abnormal Aβ is necessary for a diagnosis of AD.7 Individuals with adequately high p-Tau:Aβ ratio but subthreshold Aβ would not be considered AD within this categorization scheme. Recent studies reveal that some individuals with subthreshold Aβ are at increased risk for cognitive decline and neurofibrillary tangle deposition27,28 and more than 75% of patients eligible based only on p-Tau:Aβ ratios in this study had CSF Aβ measures within 1 SD of the eligibility criterion cutoff for that protein. Determining the optimal threshold for AD biomarker criteria will be a critical area of research to identify those most likely to demonstrate disease progression and to benefit from effective AD therapies.
As a group, amyloid negative symptomatic patients do not demonstrate similar disease progression, compared to amyloid positive patients.29 Using a practical model of trial power, one contingent upon a limited pool of MCI patients who can be recruited to trials, we showed that power to demonstrate drug effect is increased in trials incorporating broader inclusion criteria (Figure 1), primarily as a result of the increased sample size gained through increased eligibility. The specific individuals eligible based on high p-Tau:Aβ ratios (i.e., who were not eligible based on low Aβ), however, demonstrated less change over time on trial outcomes compared to those with high p-Tau:Aβ ratio and low Aβ (Figure 2) and were less likely to progress to dementia.
Thus, the current results illustrate the challenges faced by investigators designing prodromal AD trials. More inclusive biomarker criteria (e.g., permitting participation for those meeting either low Aβ or an adequate ratio) will expedite enrollment by reducing screen failure rates. From a standpoint of instructing future clinical practice, broader inclusion in prodromal AD clinical trials, with pre-specified secondary analyses in biomarker-specific groups, could provide critical information related to drug safety as well as mechanistic pathways for efficacy. Alternatively, more restrictive criteria (e.g., enrolling only those with low Aβ and high CSF tau) may afford smaller trials due to higher rates of progression but bring more challenging accrual.
Other factors influence trial enrollment criteria selection. Specific drug mechanism of action may mandate more stringent criteria (e.g., ensuring fibrillar brain Aβ is present in participants when testing specific amyloid-lowering therapies or ensuring multiple pathologies are present in combination trials). Trials now most frequently incorporate amyloid PET imaging as biomarker evidence supportive of prodromal AD. Amyloid PET offers some advantages compared to CSF biomarker criteria, including avoiding the reluctance of many patients to undergo lumbar puncture.10,30 The current data indicate that using amyloid PET to determine prodromal AD trial eligibility, much like using low CSF Aβ alone as a criterion, may exclude otherwise eligible participants, here including 15% of those with low CSF Aβ (Table 4).31–33 This finding may represent Aβ changing earlier in CSF than can be observed by amyloid PET34 or deposition of diffuse but not neuritic plaques in these MCI patients.35 Additionally, a third of participants eligible based on p-Tau:Aβ ratio (and 92% of those with high p-Tau:Aβ ratio but ineligible based on CSF Aβ) were ineligible based on amyloid PET. Future research should focus on which biomarker enrollment criteria are associated with greater costs, including the costs of recruitment, time to accrual, and the biomarkers themselves.
Beyond study power and ease of enrollment, there may be ethical implications of these findings. Specifically, if excluded individuals are likely use the intervention under study were it found to be efficacious (e.g., off-label use), then the controlled trial setting would be the only place in which a pure assessment of safety and efficacy in these sub-populations could be performed.
Limitations.
Minor differences among the ADNI samples were not controlled for in our analyses. ADNI-2 enrolled early and late stage MCI; we did not incorporate these diagnostic distinctions into our analyses, reflecting current trial practices. Amyloid PET was available for only a subset of participants with CSF data, potentially limiting the generalizability of these results. ADNI is performed nearly exclusively at academic medical centers and may not entirely recapitulate observations from actual trials, which often include a mix of academic and commercial site types.36 We did not apply additional trial exclusion criteria beyond those related to biomarker cutoffs, though ADNI is largely designed to recapitulate trial criteria.37 Moreover, we examined only one cutoff for each assessed biomarker and no definitive consensus as yet exists for where these thresholds should be placed.38,39 It is possible that setting different cutoffs could reduce or increase the observed discrepancies in eligibility estimates. Finally, there is an imperative to increase the diversity of trial samples40 and limited information is available for whether biomarker criteria could differentially affect eligibility among unique racial or ethnic groups.41,42 This study lacked adequate diversity to assess for such potential differences.
Conclusions.
Though amyloid PET predominates current prodromal AD trials as a biomarker inclusion criterion, investigators designing these trials may wish to consider additional biomarker eligibility criteria. Including patients meeting CSF Aβ criteria, and even more so for CSF p-Tau criteria, may enable enrollment of otherwise ineligible participants. Biomarker criteria selection, however, must be done with specific therapy mechanisms and trial goals in mind. Especially in early phase trials, such decisions may be critical to maximizing accrual and instructing later stage trial decisions.
ACKNOWLEDGEMENTS
JDG and DLG were supported by NIA AG016573, 1R21AG056931, AG059407, and UC Cures Alzheimer’s BRD-16-501350. JDG is currently supported by UL1 TR000153. MMN supported by the National Science Foundation Graduate Research Fellowship (grant No. DGE-1321846). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California
Footnotes
Disclosures
Dr. Grill has received research funding from Biogen Idec, Eli Lilly & Company, Genentech, the Alzheimer’s Disease Cooperative Study, and the Alzheimer’s Therapeutic Research Institute. He has consulted for CogniCiti.
Ms. Nuño has nothing to disclose.
Dr. Gillen has nothing to disclose.
References
- 1.Petersen RC, Thomas RG, Aisen PS, et al. Randomized controlled trials in mild cognitive impairment: Sources of variability. Neurology 2017;88(18):1751–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7(3):270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet neurology 2014;13(6):614–629. [DOI] [PubMed] [Google Scholar]
- 4.Cummings J Lessons Learned from Alzheimer Disease: Clinical Trials with Negative Outcomes. Clin Transl Sci 2017. [DOI] [PMC free article] [PubMed]
- 5.Grill JD, Di L, Lu PH, et al. Estimating sample sizes for predementia Alzheimer’s trials based on the Alzheimer’s Disease Neuroimaging Initiative. Neurobiol Aging 2013;34(1):62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thal LJ, Kantarci K, Reiman EM, et al. The role of biomarkers in clinical trials for Alzheimer disease. Alzheimer Dis Assoc Disord 2006;20(1):6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jack CR Jr., Bennett DA, Blennow K, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018;14(4):535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coric V, Salloway S, van Dyck CH, et al. Targeting Prodromal Alzheimer Disease With Avagacestat: A Randomized Clinical Trial. JAMA neurology 2015;72(11):1324–1333. [DOI] [PubMed] [Google Scholar]
- 9.Fargo KN, Carrillo MC, Weiner MW, Potter WZ, Khachaturian Z. The crisis in recruitment for clinical trials in Alzheimer’s and dementia: An action plan for solutions. Alzheimers Dement 2016;12(11):1113–1115. [DOI] [PubMed] [Google Scholar]
- 10.Nuno MM, Gillen DL, Dosanjh KK, et al. Attitudes toward clinical trials across the Alzheimer’s disease spectrum. Alzheimers Res Ther 2017;9(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vos SJ, Verhey F, Frolich L, et al. Prevalence and prognosis of Alzheimer’s disease at the mild cognitive impairment stage. Brain 2015;138(Pt 5):1327–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petersen RC, Aisen P, Boeve BF, et al. Mild cognitive impairment due to Alzheimer disease in the community. Ann Neurol 2013;74(2):199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petersen RC, Aisen PS, Beckett LA, et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology 2010;74(3):201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol 1999;56(3):303–308. [DOI] [PubMed] [Google Scholar]
- 15.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol 2009;65(4):403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jagust WJ, Landau SM, Koeppe RA, et al. The Alzheimer’s Disease Neuroimaging Initiative 2 PET Core: 2015. Alzheimers Dement 2015;11(7):757–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Landau SM, Mintun MA, Joshi AD, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol 2012;72(4):578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. The American journal of psychiatry 1984;141(11):1356–1364. [DOI] [PubMed] [Google Scholar]
- 19.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43(11):2412–2414. [DOI] [PubMed] [Google Scholar]
- 20.Sano M, Raman R, Emond J, et al. Adding delayed recall to the Alzheimer Disease Assessment Scale is useful in studies of mild cognitive impairment but not Alzheimer disease. Alzheimer Dis Assoc Disord 2011;25(2):122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kozauer N, Katz R. Regulatory innovation and drug development for early-stage Alzheimer’s disease. The New England journal of medicine 2013;368(13):1169–1171. [DOI] [PubMed] [Google Scholar]
- 22.Fitzmaurice GM, Laird NM, Ware JH. Applied longitudinal analysis Hoboken, NJ: John Wiley & Sons; 2012. [Google Scholar]
- 23.Ostrowitzki S, Lasser RA, Dorflinger E, et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther 2017;9(1):95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fleisher AS, Sowell BB, Taylor C, Gamst AC, Petersen RC, Thal LJ. Clinical predictors of progression to Alzheimer disease in amnestic mild cognitive impairment. Neurology 2007;68(19):1588–1595. [DOI] [PubMed] [Google Scholar]
- 25.Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. The New England journal of medicine 2014;370(4):322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siemers ER, Sundell KL, Carlson C, et al. Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement 2016;12(2):110–120. [DOI] [PubMed] [Google Scholar]
- 27.Landau SM, Horng A, Jagust WJ, Alzheimer’s Disease Neuroimaging I. Memory decline accompanies subthreshold amyloid accumulation. Neurology 2018;90(17):e1452–e1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leal SL, Lockhart SN, Maass A, Bell RK, Jagust WJ. Subthreshold amyloid predicts tau deposition in aging. J Neurosci 2018. [DOI] [PMC free article] [PubMed]
- 29.Landau SM, Horng A, Fero A, Jagust WJ, Alzheimer’s Disease Neuroimaging I. Amyloid negativity in patients with clinically diagnosed Alzheimer disease and MCI. Neurology 2016;86(15):1377–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marcantonio ER, Aneja J, Jones RN, et al. Maximizing clinical research participation in vulnerable older persons: identification of barriers and motivators. Journal of the American Geriatrics Society 2008;56(8):1522–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Landau SM, Lu M, Joshi AD, et al. Comparing positron emission tomography imaging and cerebrospinal fluid measurements of beta-amyloid. Ann Neurol 2013;74(6):826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattsson N, Insel PS, Donohue M, et al. Independent information from cerebrospinal fluid amyloid-beta and florbetapir imaging in Alzheimer’s disease. Brain 2015;138(Pt 3):772–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oh M, Seo M, Oh SY, et al. Clinical significance of visually equivocal amyloid PET findings from the Alzheimer’s Disease Neuroimaging Initiative cohort. Neuroreport 2018;29(7):553–558. [DOI] [PubMed] [Google Scholar]
- 34.Palmqvist S, Mattsson N, Hansson O, Alzheimer’s Disease Neuroimaging I. Cerebrospinal fluid analysis detects cerebral amyloid-beta accumulation earlier than positron emission tomography. Brain 2016;139(Pt 4):1226–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cairns NJ, Ikonomovic MD, Benzinger T, et al. Absence of Pittsburgh compound B detection of cerebral amyloid beta in a patient with clinical, cognitive, and cerebrospinal fluid markers of Alzheimer disease: a case report. Arch Neurol 2009;66(12):1557–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edland SD, Emond JA, Aisen PS, Petersen RC. NIA-funded Alzheimer centers are more efficient than commercial clinical recruitment sites for conducting secondary prevention trials of dementia. Alzheimer Dis Assoc Disord 2010;24(2):159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weiner MW, Veitch DP, Aisen PS, et al. The Alzheimer’s Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement 2012;8(1 Suppl):S1–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jack CR Jr., Bennett DA, Blennow K, et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016;87(5):539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jack CR Jr., Knopman DS, Chetelat G, et al. Suspected non-Alzheimer disease pathophysiology--concept and controversy. Nature reviews Neurology 2016;12(2):117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watson JL, Ryan L, Silverberg N, Cahan V, Bernard MA. Obstacles and opportunities in Alzheimer’s clinical trial recruitment. Health affairs (Project Hope) 2014;33(4):574–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Howell JC, Watts KD, Parker MW, et al. Race modifies the relationship between cognition and Alzheimer’s disease cerebrospinal fluid biomarkers. Alzheimers Res Ther 2017;9(1):88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morris JC, Schindler SE, McCue LM, et al. Assessment of Racial Disparities in Biomarkers for Alzheimer Disease. JAMA neurology 2019. [DOI] [PMC free article] [PubMed]