

Abstract
Herein we disclose an efficient method for the conversion of carboxylic acids to trifluoromethyl groups via the combination of photoredox and copper catalysis. This transformation tolerates a wide range of functionality including heterocycles, olefins, alcohols, and strained ring systems. To demonstrate the broad potential of this new methodology for late-stage functionalization, we successfully converted a diverse array of carboxylic acid-bearing natural products and medicinal agents to the corresponding trifluoromethyl analogues.
In drug discovery, a commonly used strategy to protect against in vivo metabolism involves the incorporation of a trifluoromethyl group (CF3) into medicinal candidates. Moreover, a variety of studies have shown that CF3 groups can increase the efficacy of a drug candidate by modulating protein—ligand interactions and improving membrane permeance, among other benefits.1 While significant advancements have been made in the development of general, catalytic protocols that access aryl—CF3 adducts, methods for the formation of aliphatic Csp3—CF3 bonds remain limited.2 Conventional approaches toward Csp3—CF3 installation include (i) difunctionalization of olefins via CF3 radical addition,3 (ii) nucleophilic substitution of alkyl halides or triflates with stoichiometric [CuCF3] species,4 and (iii) oxidative coupling of alkyl boronic acids with [CuCF3] reagents.5 These elegant methods nonetheless require the pre-activation of the coupling partner, a step that can often limit their potential utility in late-stage functionalization and other diversification steps. As such, the discovery of a catalytic trifluoromethylation mechanism that allows the conversion of native, abundant functional groups into Csp3—CF3 motifs should be of considerable value to medicinal chemists and the synthetic community as a whole.
The merger of transition metal and photoredox catalysis, termed metallaphotoredox catalysis, has enabled the discovery of a range of synthetically valuable and hitherto elusive transformations.6 In particular, carboxylic acids, a naturally abundant class of compounds, can efficiently undergo decarboxylative cross-coupling via this dual catalysis platform to install a myriad of valuable functionalities including Csp3—aryl, —alkyl, —vinyl, and —acyl groups.7 Indeed, the capacity of carboxylic acids to act as an adaptive functionality renders them ideal starting materials for both early- and late-stage bond formation. Moreover, these methodologies generally employ mild conditions (i.e., visible light, room temperature) and exhibit broad functional group tolerance, features that allow for their use with a diverse array of complex, biologically relevant molecules.8 Recently, we questioned if a metallaphotoredox platform might enable a catalytic decarboxylative trifluoromethylation. As a key design element, we envisioned the use of copper as the key cross-coupling catalyst, effectively taking advantage of CuIII’s propensity to undergo facile reductive elimination from its high valency oxidation state.9 At the same time, we hoped that Csp3—carboxylic acid bonds could be activated via photoredox to generate alkyl radicals that would rapidly engage in a copper catalytic cycle. Indeed, it is well-established that aliphatic open-shell intermediates will undergo capture by copper species at rates that approach rates of diffusion.10 This concept was recently employed by our laboratory in a C—N heteroaryl bond-forming reaction wherein aliphatic carboxylic acids underwent decarboxylative coupling with many classes of N-nucleophiles.11 Herein, we report the successful translation of this new strategy to C—C bond formation and describe the first copper-catalyzed decarboxylative trifluoromethylation of aliphatic carboxylic acids.
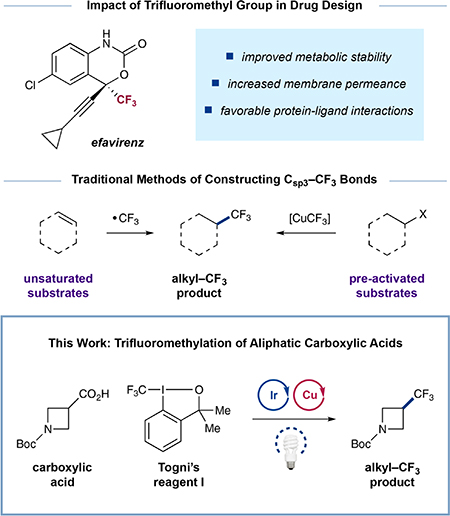
Details of the proposed dual copper-photoredox cycle are outlined in Scheme 1. We envisioned that photoexcitation of the IrIII photocatalyst Ir[dF(CF3)ppy]2(4,4′-dCF3bpy)PF6 (1) with visible light would generate the highly oxidizing excited-state vs SCE in MeCN).12 At the same time, the carboxylate anion derived from carboxylic acid 3 and base would ligate the CuII catalyst. Single-electron transfer (SET) from copper carboxylate 4 to highly oxidizing excited-state *IrIII 2 would then deliver CuIII carboxylate 5, or in the dissociated form, a carboxyl radical and CuII complex (6), along with reduced IrII photocatalyst 7. The resulting carboxyl radical would extrude CO2 to generate an alkyl radical and CuII (8), which would rapidly recombine to form CuIII species 9. At this point, we hypothesize that a single-electron transfer would then occur between 9 and the reduced IrII state of photocatalyst 7 ( = −0.79 V vs SCE in MeCN) to close the photoredox catalytic cycle and produce an alkylcopper(II) species (10). At this stage, we hoped that 10 would engage Togni’s reagent I (11) to deliver alkyl—CF3 product 12, presumably via reductive elimination from a transiently formed CuIII intermediate.13 This is in accord with previous stoichiometric studies in which an arylcopper(II) intermediate was trifluoromethylated.14 The resulting complex 13 would then undergo ligand exchange with substrate acid 3 to complete this dual-catalytic cycle.
Scheme 1.

Pathway of Decarboxylative Trifluoromethylation
Our investigation into this new decarboxylative trifluoromethylation began with exposure of 1-benzoylpiperidine-4-carboxylic acid (3) to various electrophilic trifluoromethyl sources13,15 in the presence of photocatalyst 1, CuCl2, 3,4,7,8-tetramethyl-1,10-phenanthroline, and Barton’s base (2-tert-butyl-1,1,3,3-tetramethylguanidine, BTMG) in ethyl acetate. To our delight, product formation was observed in 39% yield when Togni’s reagent I (11, Ep[11/11•−] = −1.51 V vs SCE in MeCN)16 was used (Table 1, entry 3). More oxidizing CF3 sources such as Umemoto reagent (14, Ep[14+/14•] = −0.32 V vs SCE in MeCN, entry 1)16 and Togni’s reagent II (15, Ep[15/15•−] = −0.79 V vs SCE in MeCN, entry 2)16 were not as efficient, presumably due to their unproductive reduction by the reduced photocatalyst (7, V vs SCE in MeCN). Further evaluation revealed that the use of CuCN as the copper source, bathophenanthroline as the ligand, and addition of 30 equiv of water further improved the reaction efficiency (entry 5, 78% isolated yield). It is worth noting that both CuI and CuII salts are capable precatalysts for this transformation. When CuI sources such as CuCN are used, a significant change in color can be observed upon addition of Togni’s reagent I, potentially due to a change in oxidation state of the copper species. This hypothesis is supported by in situ 19F NMR analysis, in which an equivalent of Togni’s reagent is consumed upon mixing with a ligated CuI species (see Supporting Information (SI) for details). It should be noted that sub-stoichiometric base (0.5 equiv, cf. Table 1, entry 6) is critical to the success of the reaction. Carboxylates are prone to form chelates with copper and deactivate the Cu catalyst.17 The tertiary alcohol byproduct originating from Togni’s reagent I after transfer of the CF3 moiety ultimately serves as stoichiometric base (cf. 13 to 4). Control experiments revealed that photocatalyst, copper catalyst, and light were all essential for the success of this new decarboxylative trifluoromethylation protocol (Table 1, entries 7–9). With respect to the proposed mechanism, it is important to note that [CuCF3] was not detected by 19F NMR analysis (<1 mol%). This mechanism contrasts with the recent studies of the Li group, whereby stoichiometric Cu(CF3)x was used to capture alkyl radicals.18 Moreover, mechanistic experiments reveal a copper-mediated decarboxylation is likely operative (see SI for details).
Table 1.
Decarboxylative Trifluoromethylation: Initial Studiesa
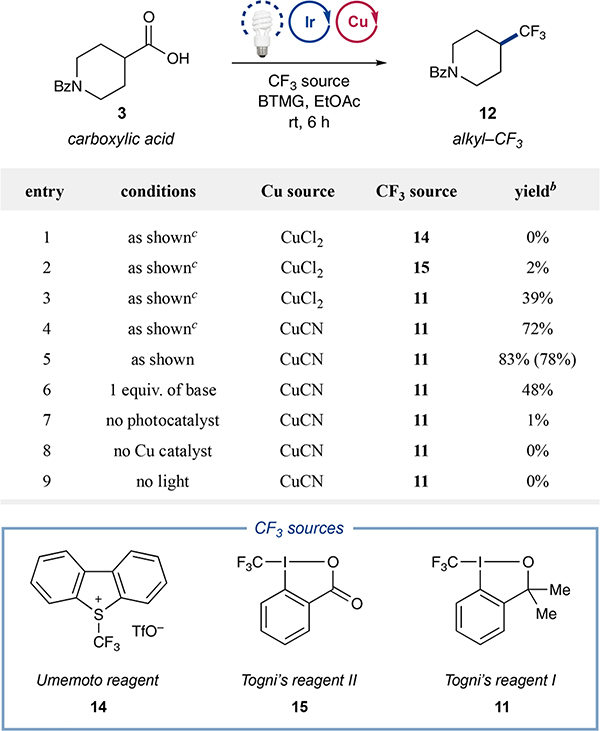 |
Performed with acid 3 (0.05 mmol), photocatalyst 1 (1 mol%), Cu source (20 mol%), bathophenanthroline (30 mol%), 2-tert-butyl-1,1,3,3-tetramethylguanidine (BTMG, 0.5 equiv), CF3 source (1.25 equiv), and H2O (30 equiv) in EtOAc (0.025 M).
Yields by 19F NMR analysis. Yields in parentheses are isolated yields.
The ligand 3,4,7,8-tetramethyl-1,10-phenanthroline was used on the Cu catalyst.
With optimized conditions in hand, we sought to evaluate the scope of the Csp3–CF3 bond-forming reaction. As shown in Table 2, a myriad of primary carboxylic acids are successful substrates, providing the corresponding CF3 analogue in good to excellent yield. Phenylacetic acids are generally competent, with a diverse range of electron-withdrawing (16–19, 70–80% yield), electron-donating (20, 74% yield), and electron-neutral substituents (21 and 22, 60 and 70% yield, respectively) tolerated on the aryl ring. Notably, the efficacy of the reaction was not hindered by ortho substitution (20, 74% yield), and useful levels of efficiency were observed for an ortho,ortho-disubstituted heterocycle (26, 30% yield). Notably, boronic esters and aryl bromides were also tolerated using this mild copper-catalyzed protocol, with no chemoselectivity issues (17 and 21, 80 and 60% yield, respectively). The reaction is also amenable to a range of non-benzylic primary acids (27–33, 31–82% yield). Moreover, substrates incorporating β,β-branching afforded the desired product in good yields (31 and 32, 63 and 71% yield, respectively) revealing that steric constraints can be overcome. Gratifyingly, an array of heterocycles, many of which include basic nitrogens, can be utilized in this transformation (23–26 and 28–30, 30–70% yield).
Table 2.
Scope of Decarboxylative Trifluoromethylation of Aliphatic Carboxylic Acids via Copper and Photoredox Catalysisa
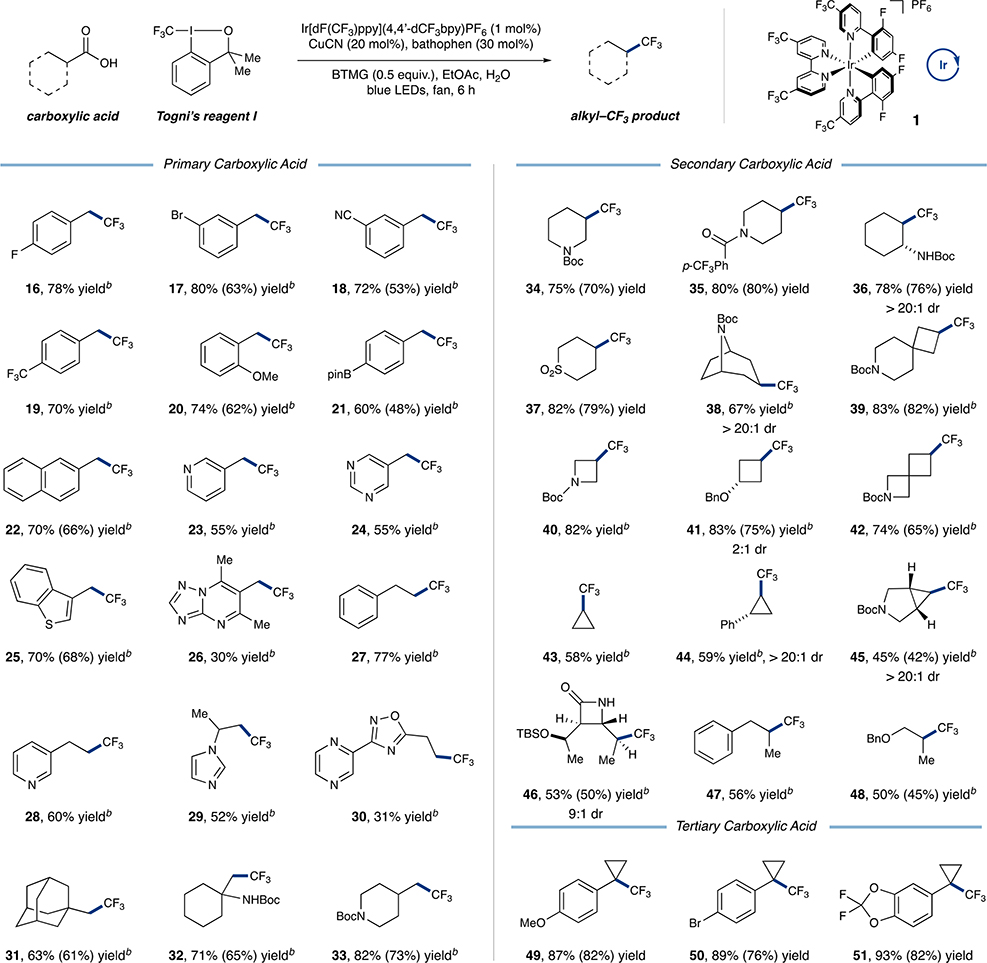 |
Performed on a 0.5 mmol scale. Due to volatility of products, yields were obtained by 19F NMR analysis of the crude reaction mixtures using an internal standard. Isolated yields are provided in parentheses. See SI for isolation conditions and yields.
Reaction performed under modified conditions. See SI for details.
We next turned our attention to secondary and tertiary carboxylic acids (34–51, 45–93% yield). We were pleased to find that cyclic carboxylic acids, including strained 3- and 4-membered rings, participate in this trifluoromethylation in moderate to excellent yield (39–45, 45–83% yield). Several bicyclic and spirocyclic compounds (38, 39, 42, and 45, 45–83% yield) formed the desired bond with good efficiency. This is a significant finding given that many of these bicyclic systems serve as aryl ring bioisosteres in drug design.19 Moreover, acyclic systems also produced the desired CF3 product in good efficiency (46–48, 50–56% yield). Notably, the compatibility of this reaction with medicinally relevant structures was demonstrated with a β-lactam (46, 53% yield). Last, a number of tertiary carboxylic acids were also successfully converted to Csp3-CF3 bonds in excellent yield (49–51, 87–93% yield).
To further demonstrate the utility of this decarboxylative trifluoromethylation protocol, we performed a series of late-stage modifications on medicinal agents and natural products (Table 3). To our delight, a large variety of biologically relevant systems that incorporate olefins, carbonyls, alcohols, and heterocycles underwent chemoselective installation of a CF3 moiety (52–63, 30–71% yield). Remarkably, mupirocin was transformed in good yield, a notable finding given that this natural product incorporates an epoxide, a diol, and a Michael acceptor (63, 45% yield).
Table 3.
Late-Stage Decarboxylative Trifluoromethylation of Natural Products and Medicinal Agents Containing Carboxylic Acidsa
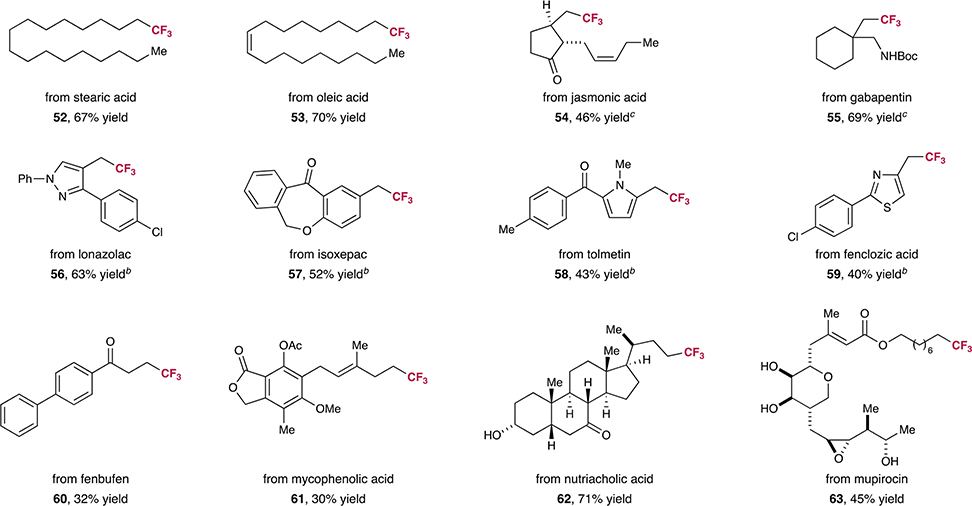 |
All yields are isolated. Performed with acid (0.5 mmol), Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1 mol%), CuCl2 (20 mol%), 3,4,7,8-tetramethyl-1,10-phenanthroline (Me4phen, 25 mol%), 2-tert-butyl-1,1,3,3-tetramethylguanidine (BTMG, 0.5 equiv), Togni’s reagent I (1.25 equiv), and H2O (30 equiv) in EtOAc (0.025 M).
Performed with di(2-pyridyl) ketone and 1,1,3,3-tetramethylguanidine (TMG) in lieu of Me4phen and BTMG, and no water was added.
Supplementary Material
ACKNOWLEDGMENTS
Financial support was provided by the NIHGMS (RO1 GM103558-05), and kind gifts from Merck, BMS, Pfizer, Janssen, and Eli Lilly are acknowledged. J.A.K. thanks the NSF GRFP (DGE 1656466) for funding.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b02650.
General experimental protocols, procedure for optimization studies, general procedure for decarboxylative trifluoromethylation, mechanistic studies, characterization data for all key compounds, and spectral data (PDF)
REFERENCES
- (1).(a) Hagmann WK J. Med. Chem 2008, 51, 4359. [DOI] [PubMed] [Google Scholar]; (b) Purser S; Moore PR; Swallow S; Gouverneur V Chem. Soc. Rev 2008, 37, 320. [DOI] [PubMed] [Google Scholar]; (c) Müller K; Faeh C; Diederich F Science 2007, 317, 1881. [DOI] [PubMed] [Google Scholar]; (d) Bégué JP; Bonnet-Delpon D Bioorganic and Medicinal Chemistry of Fluorine; J. Wiley & Sons: Hoboken, NJ, 2008. [Google Scholar]
- (2).Alonso C; de Marigorta EM; Rubiales G; Palacios F Chem. Rev 2015, 115, 1847. [DOI] [PubMed] [Google Scholar]
- (3).(a) Merino E; Nevado C Chem. Soc. Rev 2014, 43, 6598. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) He Z; Tan P; Hu J Org. Lett 2016, 18, 72. [DOI] [PubMed] [Google Scholar]; (c) Xu X; Chen H; He J; Xu H Chin. J. Chem 2017, 35, 1665. [Google Scholar]
- (4).(a) Chen Q-Y; Wu S-WJ Chem. Soc., Chem. Commun 1989, 705. [Google Scholar]; (b) Chen Q-Y; Duan J-X Tetrahedron Lett. 1993, 34, 4241. [Google Scholar]; (c) Novak P; Lishchynskyi A; Grushin VV J. Am. Chem. Soc 2012, 134, 16167. [DOI] [PubMed] [Google Scholar]; (d) Tyrra W; Naumann D; Quadt S; Buslei S; Yagupolskii YL; Kremlev MM J. Fluorine Chem 2007, 128, 813. [Google Scholar]; (e) Sevenard DDV; Kirsch P; Roschenthaler GV; Movchun VN; Kolomeitsev AA Synlett 2001, 2001, 379. [Google Scholar]
- (5).Xu J; Xiao B; Xie CQ; Luo DF; Liu L; Fu Y Angew. Chem., Int. Ed 2012, 51, 12551. [DOI] [PubMed] [Google Scholar]
- (6).Twilton J; Le C; Zhang P; Shaw MH; Evans RW; MacMillan DWC Nature Rev 2017, 1, 0052. [Google Scholar]
- (7).Jin Y; Fu H Asian J. Org. Chem 2017, 6, 368. [Google Scholar]
- (8).Bloom S; Liu C; Kolmel DK; Qiao JX; Zhang Y; Poss MA; Ewing WR; MacMillan DWC Nat. Chem 2018, 10, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Liu L; Zhu M; Yu H-T; Zhang W-X; Xi ZJ Am. Chem. Soc 2017, 139, 13688. [DOI] [PubMed] [Google Scholar]
- (10).Freiberg M; Meyerstein DJ Chem. Soc., Faraday Trans. 1 1980, 76, 1825. [Google Scholar]
- (11).Liang Y; Zhang X; MacMillan DWC ChemRxiv 2018, DOI: 10.26434/chemrxiv.5877868.v1. [DOI] [Google Scholar]
- (12).Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Nature 2016, 539, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Charpentier J; Fruh N; Togni A Chem. Rev 2015, 115, 650. [DOI] [PubMed] [Google Scholar]
- (14).Huang Y; Fang X; Lin X; Li H; He W; Huang K-W; Yuan Y; Weng Z Tetrahedron 2012, 68, 9949. [Google Scholar]
- (15).(a) Umemoto T Chem. Rev 1996, 96, 1757. [DOI] [PubMed] [Google Scholar]; (b) Shibata N; Matsnev A; Cahard D Beilstein J. Org. Chem 2010, 6, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Charpentier J; Früh N; Togni A Chem. Rev 2015, 115, 650. [DOI] [PubMed] [Google Scholar]; (d) Chachignon H; Cahard D Chin. J. Chem 2016, 34, 445. [Google Scholar]
- (16).Mizuta S; Verhoog S; Wang X; Shibata N; Gouverneur V; Médebielle MJ Fluorine Chem. 2013, 155, 124. [Google Scholar]
- (17).Vantourout JC; Miras HN; Isidro-Llobet A; Sproules S; Watson AJB J. Am. Chem. Soc 2017, 139, 4769. [DOI] [PubMed] [Google Scholar]
- (18).(a) Shen H; Liu Z; Zhang P; Tan X; Zhang Z; Li CJ Am. Chem. Soc 2017, 139, 9843. [DOI] [PubMed] [Google Scholar]; (b) Tan X; Liu Z; Shen H; Zhang P; Zhang Z; Li CJ Am. Chem. Soc 2017, 139, 12430. [DOI] [PubMed] [Google Scholar]
- (19).(a) Patani GA; LaVoie EJ Chem. Rev 1996, 96, 3147. [DOI] [PubMed] [Google Scholar]; (b) Meanwell NA J. Med. Chem 2011, 54, 2529. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.