

Abstract
This study explored the applicability, cellular efficacy, and osteogenic activities of porous nano-hydroxyapatite/Poly (glycerol sebacate)-grafted maleic anhydride (n-HA/PGS-g-M) composite scaffolds. Nuclear magnetic resonance (NMR) analyses indicated that approximately 43% of the hydroxide radicals in PGS were displaced by maleic anhydride. Resonance bands at 1036 cm−1 occurred in scaffolds containing nHA powders, and peak areas increased when n-HA weight increased in PGS-M-n-HA-0.4, PGS-M-n-HA-0.5, and PGS-M-n-HA-0.6 scaffolds. The n-HA/PGS-g-M composite scaffolds exhibited porous microstructure with average pore size of 150–300 µm in scanning electron microscopy (SEM) analysis. Differential scanning calorimetry (DSC) identified the glass transition temperature (Tg) as −25–30 °C, indicative of quality resilience. The modulus of compressibility increased when n-HA content increased. Interestingly, viability of human adipose-derived stem cells (hADSCs) in vitro and expression of the osteogenic related genes RUNX2, OCN, and COL1A1 was enhanced in the n-HA/PGS-g-M composite scaffolds compared to those factors observed in PGS-g-M scaffolds. Finally, simulated body fluid (SBF) tests indicated more apatite deposits on the surface of n-HA/PGS-g-M scaffolds compared to PGS-g-M scaffolds. Overall, porous n-HA/PGS-g-M composite scaffolds possessed acceptable biocompatibility and mechanical properties, and they stimulated hADSC cell proliferation and differentiation. Given these qualities, the composite scaffolds have potential applications in bone tissue engineering.
Subject terms: Stem-cell biotechnology, Tissue engineering and regenerative medicine
Introduction
A majority of skull defects are caused by disease or accidents, and these pose a major threat to human health. Over the years, the treatment of bone defects has mainly involved autologous graft or allograft; however, there are obvious deficiencies in these traditional methods, including a limitation in donor bone stocks for autologous grafts and immune rejection1. Recently, a number of biomaterials have become available for use in bone regeneration and remodelling, including three-dimensional scaffolds and membranes2,3. Bone regeneration through tissue bioengineering is a complex and advanced medical procedure. An ideal biomaterial for tissue bioengineering should possess certain features such as sufficient mechanical strength, desirable biocompatibility, biodegradability, good bone conduction performance, and high permeability3,4. Nanotechnology also plays an important role in the synthesis of scaffolds. Researchers have prepared biomaterial for tissue bioengineering using silk fibroin, collagen, biocompatible and biodegradable polymers, and hydroxyapatite (HA) using electrospinning5, phase separation6, or self-assembly methods7.
Poly glycerol sebacate (PGS), a strong biodegradable and biocompatible polyester applied in the context of tissue engineering, was first reported by Wang et al. in 2002, and this material possesses thermoset elastomeric properties8. Additionally, PGS exhibits a low elastic modulus (0.3 MPa) and resorption (under 4 months)9. To increase the mechanical strength of polymers, nano-and micro-sized particulates have been widely used in nanocomposite materials10,11. HA has typically been used for the induction of osteoconductivity, as a dermal filler for volume enhancement, and for the promotion of vascularization12–14. Bone mineral is a modified form of HA and composes up to 50% by volume and 70% by weight the teeth and bones within the human body. Therefore, it is used as a filler or coating to replace defective bone or to promote bone ingrowth in prosthetic implants15. The bioresorbability of nano-HA (nHA) is preferred over micro-HA due to a size of less than 100 nm. In hard tissue engineering, HA can induce bone regeneration and fixation through its bone conduction performance and desirable biocompatibility properties. The application of this material, however, is limited due to its brittle characteristics. Polymer materials remedy this limitation by increasing toughness and resilience. Synthetic polymer materials, such as poly(lactic-co-glycolic acid (PLGA) and poly- d, l-lactide-co-polyethylene glycol (PLA-PEG), possess desirable mechanical properties and are degraded directly in the body16,17.
PGS fumarate and nHA (PGSF-nHA) nanocomposites that are biocompatible and possess strong mechanical properties can promote cell adhesion, proliferation, and differentiation of osteoblasts in a rat calvarial bone defect model18. Bioresorbable and shape-memory nanocomposites will replace non-resorbable implants for non-healing soft tissue reconstruction. Natural compounds possessing a maleic anhydride (MAH) structure are a class of substances produced by certain microorganisms, and most of these compounds have been reported to possess desirable biological activity, particularly bactericidal activity19. Based on these reports, MAH was commonly used to improve the performance of composites such as polylactic acid, nano calcium carbonate, and others20–22. The main purpose underlying the use of MAH was to introduce reactive groups such as carboxyl groups, hydroxyl groups, and double bonds to alter cell adhesion, proliferation, and differentiation23,24. In this study, PGS grafted maleic anhydride (PGS-g-M) polymers were synthesized, and then n-HA/PGS-g-M composite scaffolds containing various weight to weight ratios (W/W) of n-HA and PGS-M (4:6, 5:5, and 6:4) were prepared and characterized by Fourier Transform Infra-Red spectroscopy (FTIR), 1H NMR, scanning electron microscopy (SEM), differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), and a universal testing machine. Subsequently, biomedical applications of these materials in the context of bone regeneration were assessed in vitro.
Results
Chemical structure of PGS-g-M
The chemical structure of PGS-g-M was identified by FT-IR and 1H-NMR spectrograms. As shown in Fig. 1A, the strong peaks of a1-a3 at 1.32 ppm, 1.59 ppm, and 2.34 ppm were attributed to the methylene protons of SA in the PGS segment of PGS-M. The weak signals of b and c at 3.69–5.39 ppm indicated the protons of glycerol units in PGS. The sharp peak of d at 6.86 ppm was attributed to the protons of C=C in the MAH units of PGS-g-M. The percent grafting was approximately 43% according to the results of 1H-NMR, suggesting that 43% of the hydroxide radicals in PGS were displaced by MAH. FT-IR spectroscopy displayed peaks at 2930 cm−1 and 2855 cm−1 that were attributed to alkane groups of PGS (Fig. 1B). The strong peak at 1740 cm−1 indicated the stretching band of C=O, and the peak at 1164 cm−1 was attributed to the stretching band of C-O, indicating ester linkages.
Figure 1.
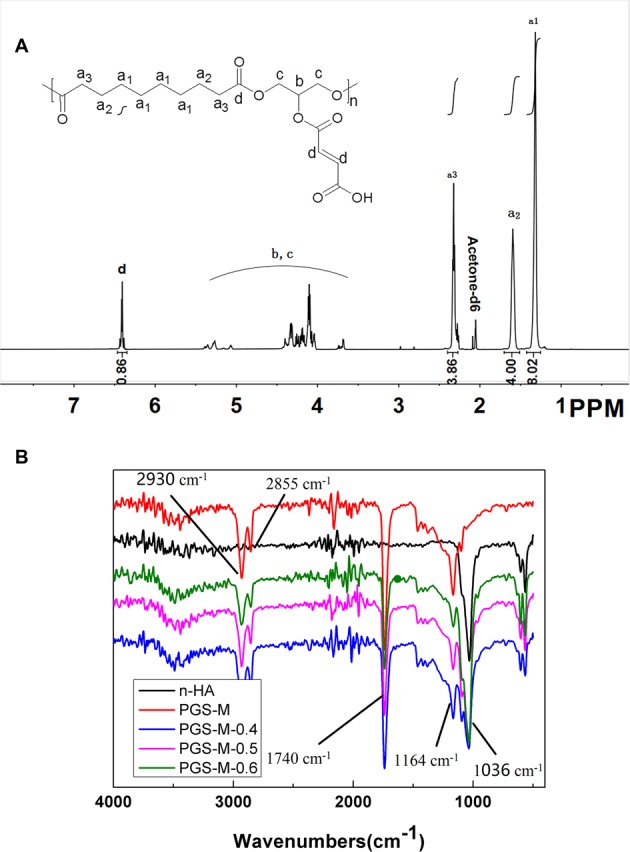
Chemical characterizations of PGS-g-M scaffolds and PGS-M-n-HA-0.4, PGS-M-n-HA-0.5, and PGS-M-n-HA-0.6 composite scaffolds. (A) The 1H-NMR spectrogram of PGS-g-M. (B) The FT-IR spectrogram of n-HA powder, PGS-g-M scaffolds, and PGS-M-n-HA-0.4, PGS-M-n-HA-0.5, and PGS-M-n-HA-0.6 composite scaffolds.
Characterizations of scaffolds
As shown in Fig. 1B, the resonance bands at 1036 cm−1, which indicated the C-O of alcohol group, could not be observed in the blank PGS-g-M scaffolds but were observed in other scaffolds with nHA powders. The peak area increased when the weight of n-HA increased in PGS-M-n-HA-0.4, PGS-M-n-HA-0.5, and PGS-M-n-HA-0.6 scaffolds.
The SEM images indicated that the n-HA/PGS-g-M composite scaffolds exhibited a porous microstructure with an average pore size of 150–300 µm (Fig. 2). When the SEM magnified the composite scaffolds two hundred times or five hundred times, an interconnected pore structure was observed. At a magnification of 1500×, many small pore structures in the sidewall of the different scaffolds due to solvent evaporation were observed, which provided further evidence that the scaffolds possess an interconnected pore structure. Additionally, the average pore size is negatively related to the W/W of n-HA/PGS-g-M. The porosities of the blank PGS-g-M, PGS-M-n-HA-0.4, PGS-M-n-HA-0.5, and PGS-M-n-HA-0.6 scaffolds were 90.34 ± 1.02%, 90.13 ± 0.24%, 88.21 ± 0.77%, and 84.56 ± 1.54%, respectively. These quantitative results were similar to the results observed in the SEM images. These results indicated that the W/W of n-HA/PGS-g-M significantly altered the porosities of scaffolds.
Figure 2.

SEM images of the PGS-g-M scaffolds (A) and the PGS-M-n-HA-0.4 (B), PGS-M-n-HA-0.5 (C), and PGS-M-n-HA-0.6 composite scaffolds (D).
The thermal properties of the scaffolds are indicated in Fig. 3. The Tg values of these scaffolds were between −25 °C and −30 °C, indicating good resilience. Additionally, crystalline peaks and melting peaks were not observed from −50 °C to 150 °C, suggesting that these scaffolds were not crystalline at 25 °C or at 37 °C (Fig. 3A). For the blank PGS-g-M scaffold, the quality change was close to 100% at 600 °C, while the quality change was only less than 5% for n-HA powder at 600 °C. The quality change was higher when the W/W of n-HA/PGS-g-M was lower (Fig. 3B). The TGA curve indicated that the W/W of n-HA/PGS-g-M was consistent.
Figure 3.

Physical characterizations of PGS-g-M scaffolds and PGS-M-n-HA-0.4, PGS-M-n-HA-0.5, and PGS-M-n-HA-0.6 composite scaffolds. (A) Differential scanning calorimetry (DSC) curves; (B) thermogravimetric analysis (TGA) curves; (C) the stress-strain curve; (D) the modulus of compressibility graphs.
Compressive measurements were used to evaluate the mechanical properties of n-HA/PGS-g-M composite scaffolds. In Fig. 3C, the compressive strengths of scaffolds increased when the content of n-HA increased. Similarly, the modulus of compressibility increased when the content of n-HA increased (Fig. 3D). Together, these results indicated that n-HA/PGS-g-M composite scaffolds possessing interconnected pore structure were successfully prepared.
In vitro cell proliferation of scaffolds with different W/W rate of n-HA/PGS-g-M
To investigate the effects of these scaffold properties on cell proliferation, hADSC cells were seeded onto the scaffolds. The cell viability significantly increased at 7 days compared with that at 1 day when scaffolds with various W/W ratios of n-HA/PGS-g-M were used, with the exception of PGS-M scaffolds (all P < 0.05, Fig. 4A). At 7 days, cell viability significantly increased in the blank PGS-g-M, PGS-M-n-HA-0.4, PGS-M-n-HA-0.5, and PGS-M-n-HA-0.6 scaffolds when compared to cell viability of the blank cells (all P < 0.05).
Figure 4.

The biomedical applications of PGS-M-n-HA-0.4, PGS-M-n-HA-0.5, and PGS-M-n-HA-0.6 composite scaffolds in bone regeneration in vitro. (A) the effects of these scaffolds on cell proliferation in human adipose-derived stem cells (hADSCs); (B) the expression levels of proinflammatory factor IL-6, and the expression levels of genes associated with osteoblast differentiation by qRT-PCR; The levels of RUNX2, OCN proteins by western blot (C) and immunohistochemical staining (D).
Inflammation response and osteoblast differentiation of hADSCs on the composite scaffolds
The inflammation response played a key role in tissue bioengineering. In this study, the expression levels of proinflammatory factor IL-6 were reduced in PGS-M-n-HA-0.4, PGS-M-n-HA-0.5, and PGS-M-n-HA-0.6 composite scaffolds compared to those observed in PGS-g-M scaffolds (P < 0.01, Fig. 4B). When compared to PGS-g-M scaffolds, PGS-M-n-HA-0.4 and PGS-M-n-HA-0.5 composite scaffolds significantly induced the expression levels of genes associated with osteoblast differentiation, including RUNX2 (P < 0.05) and COL1A1 (P < 0.01). PGS-M-n-HA-0.6 composite scaffolds induced increased expression levels of RUNX2, OCN (both P < 0.05), and COL1A1 (P < 0.01) (Fig. 4B). The levels of RUNX2 and OCN proteins were also increased in these composite scaffolds (Fig. 4C and Supplementary Fig. 1) and the immunohistochemical results were consistent with the other observations (Supplementary Fig. 2).
Calcium phosphorus (Ca–P) precipitation on the surface of scaffolds
Figure 5 shows the surface SEM morphology of the apatite layer on each scaffold when immersed in SBF for 4 weeks. Numerous smaller apatite deposits were observed on the surface of n-HA/PGS-g-M scaffolds when compared to PGS-g-M scaffolds; however, the number of apatite deposits on the scaffolds was not significantly different when nHA content increased in the composite scaffolds. Additionally, the results of EDS analysis indicate that a Ca-P chemical element was detected on the surface and porous layer of the four scaffolds, indicating that PO43− and Ca2+ ions can pass through the porous layers and form apatite structure on the surfaces (Supplementary Fig. 3).
Figure 5.

SEM images of scaffolds in simulated body fluid (SBF) from 1 week to 4 weeks. The upper left corner: magnification of SEM images.
Discussion
PGS has been used in a large number of biomedical applications including tissue engineering and drug release due to its strength and to its biodegradable and thermoset elastomeric properties25. The purpose of our present study was to explore the structural characterization, cellular efficacy, and osteogenic activities of porous n-HA/PGS-g-M composite scaffolds. Wang et al. reported that the intense C=O stretch in ester bonds of PGS in FT-IR spectroscopy was located at 1740 cm−1, while the C-O stretching bond was present at 1164 cm−1. These findings indicated that PGS is a polyester8. Similarly, our results revealed peaks of alkane groups at 2930 cm−1 and 2855 cm−1, the stretching band of C=O at 1740 cm−1, and the stretching band of C-O at 1164 cm−1, which indicated ester linkages in the PGS polymer. Additionally, 1H-NMR spectra of PGS displayed methylene peaks at 1.30, 1.62, and 2.35 ppm that were attributed to the methylene protons of SA in PGS, while peaks observed at 4.05–5.30 ppm indicated the protons of the glycerol units in PGS26. These findings are similar to our results. Additionally, the sharp peak of d at 6.86 ppm was attributed to the protons of C=C in the MAH units of PGS-g-M. Taken together, our observations of FT-IR and 1H-NMR spectrum indicated that PGS-g-M scaffolds were successfully prepared.
Our examination of the porosity of the scaffolds revealed that the porosity was greatly decreased by the introduction of nHA into the polymer. These results are consistent with those of SEM images. This phenomenon was also consistent with a study by Huang et al.27. All of the scaffolds possessed a high porosity of >84%, and this aided in cell adhesion and proliferation27,28. In the present study, the compressive strengths of scaffolds increased when the content of n-HA increased in the n-HA/PGS-g-M composite scaffolds. As mentioned above, PGS is an elastomeric polymer, and HA exhibits bone conduction performance and desirable biocompatibility properties. Thus, the addition of HA reduced the elastomer of PGS and increased the strength. The results were consistent with previous studies that that demonstrated that increasing HA improved the compressive strength29,30. The mechanical properties were consistent with the structural observations that the elasticity for PGS-M-n-HA-0.6 and −0.4 were lower than that of the PGS-M. This phenomenon may result from the presence of the stiff hydroxyl caused by the higher incorporation of nHA groups that prevent C-O bending. The copolymers with high mechanical strength enhanced the applicability for hard tissue engineering. Additionally, the compressive modulus of n-HA/PGS-g-M composite scaffolds is enhanced when HA contents are increased. In a previous study, the compressive modulus of PGS was demonstrated to be similar (0.024 MPa) to that of PGS-g-M31. Hence, n-HA/PGS-g-M composite scaffolds may prove useful for hard tissue engineering.
n-HA/PGS-g-M composite scaffolds also enhanced cell viability and promoted the expression of osteogenic related genes in vitro. The expression of osteogenic related genes detected by qRT-PCR in bone tissue engineering has been reported by some researchers. RUNX2 has been demonstrated to strongly influence the differentiation process of human mesenchymal stem cells into osteogenesis in the early stage of development32. The relative expression of the osteogenic marker RUNX2 significantly increased during osteogenic differentiation when human mesenchymal stem cells were seeded into organic/inorganic hybrid scaffolds that contained functional rhBMP-233. Similarly, PGS-M-n-HA-0.4 and PGS-M-n-HA-0.5 composite scaffolds significantly induced the expression levels of genes associated with osteoblast differentiation such as RUNX2 and COL1A1. mPCL-CaP composite scaffolds up-regulated OCN and OPN expression34. On the biphasic Ca-P scaffolds coated with HA-PCL composites, overexpression of RUNX2 and COL1A1 indicated that these scaffolds induced the differentiation of primary human bone-derived cells35. Therefore, PGS-M-n-HA-0.4 and PGS-M-n-HA-0.5 composite scaffolds may prove useful for bone tissue engineering.
To investigate the response process and biological activity of scaffolds, SBF was used in this study, as it is a quick and easy assay possesses ion concentrations and a pH that are similar to those encountered under physiological conditions36. SBF has been used to evaluate the potential mineralization abilities of implant material for bone tissue engineering by previous researchers37,38. The formation of apatite on synthetic materials is induced, as functional groups present a negative charge and form apatite nuclei through the formation of amorphous Ca-P39. It has been reported that a greater number of apatite nuclei form on chitosan/nHA composite scaffolds than that of chitosan-only scaffolds due to the ability of nHA particles to act as nucleation sites37. The Ca and P ions present in the SBF can be spontaneously assembled into the surrounding apatite core once the apatite core is formed. Additionally, Blaker et al. reported that there was significant HA layer formation on the surface of PDLLA/Bioglass bone scaffolds after immersion in SBF solution for 70 days40. Similarly, apatite layers were found on the surface SEM morphology of the porous n-HA/PGS-g-M composite scaffolds when immersed in SBF for 4 weeks, suggesting that the formation of apatite on n-HA/PGS-g-M composite scaffolds could provide a marker for bioactivity. Additionally, more apatite deposits were observed on the surface of n-HA/PGS-g-M scaffolds when compared to those observed on PGS-g-M scaffolds. It should be noted that the number of apatite deposits was not significantly different when nHA content was increased in the composite scaffolds. Therefore, n-HA/PGS-g-M scaffolds were more effective in imparting bioactivity to the material than was the control scaffold.
In the present study, porous n-HA/PGS-g-M composite scaffolds with desirable biocompatibility, strong mechanical properties, and bioactivity have been successfully fabricated. Compared to PGS-g-M scaffolds, n-HA/PGS-g-M composite scaffolds induced elevated cell proliferation and expression levels of osteogenic related genes while causing little inflammatory response. Therefore, these n-HA/PGS-g-M composite scaffolds may be potential candidates for application in bone regeneration and remodelling.
Materials and Methods
Chemicals
Sebacic acid (SA) and anhydrous N,N-Dimethylformamide (DMF) were both purchased from J&K Scientific Ltd. (Beijing, China). Glycerinum was purchased from Sigma-Aldrich Chemical Co. (St. Louis, Missouri, USA), and nHA (H97%, <100 nm) was purchased from Aladdin (Shanghai, China). Acetone for HPLC was purchased from Yonghua Chemical Co. (Changshu, Jiangsu, China). Maleic anhydride (MAH) was purchased from TCI (Shanghai, China).
Synthesis of PGS
The synthesis of PGS was modified based on a previous procedure8. Briefly, 20.2574 g of SA and 9.2037 g of glycerol were placed in a three-necked flask and melted at 135 °C. Then, the mixture was stirred with nitrogen for 24 h, and this was followed by vacuuming for 48 h. After the reaction system was cooled to room temperature, PGS was obtained.
Preparation and characterization of PGS-g-M
The reaction scheme for the synthesis of PGS-g-M is shown in Fig. 6. Mixtures of PGS (2.6668 g), MAH (2.0263 g), and anhydrous DMF (2.7 mL) were reacted at 110 °C under N2 for 50 min before 150 mL of deionized water was used to obtain the precipitate. Then, the precipitate was dissolved into acetone and washed again with 150 mL of deionized water. The above procedure was repeated twice. The faint yellow sticky solid appeared under a vacuum air-removed system (4 mbar) after 24 h. PGS-M was identified by 1H NMR (Bruke, 600 MHz, Germany) in deuterated acetone solution and by Fourier Transform Infra-Red spectroscopy (FTIR, Nicolet 6700, Madison, WI). The range of FTIR analysis was 400–4000 cm−1. The resolution of the instrument and the number of scans were 4 cm and 32, respectively.
Figure 6.

The reaction scheme for the synthesis of PGS-g-M. PGS-g-M, poly (glycerol sebacate)-graft-Maleic anhydride.
Preparation of n-HA/PGS-g-M composite scaffold
Firstly, a cylindrical mould (inner diameter of 4 cm) was prepared by a strong magnet. Then, sodium chloride particles with a diameter of 150–300 µm were spread over the entire mould. The mould was transferred to a relative humidity environment for 1.5 h. Third, the sample was vacuum dried at 120 °C, and the mould was removed to prepare a salt mould. Fourth, PGS-M and acetone were mixed to prepare a 280 g/L solution, and this solution was divided into 4 equal portions. Fifth, an appropriate amount of n-HA was added to the each acetone solution, bringing the final n-HA/PGS-M weight to weight ratio (W/W) to 0 (control: PGS-M), 4:6 (PGS-M-n-HA-0.4), 5:5 (PGS-M-n-HA-0.5), or 6:4 (PGS-Mn-HA-0.6). Sixth, after ultrasonic treatment for 20 min, the salt mould was soaked into the mixed solution. After evaporating, it was placed in a vacuum oven and crosslinked at 150 °C under a pressure of 1 Torr for 24 hours. Finally, the mould was washed with distilled water, and a porous tissue engineering scaffold was obtained by freeze drying for 48 h. The chemical characteristics of the scaffolds were determined by FTIR.
Surface morphological characteristics and porosity of the scaffolds
The surface morphology and microstructure of n-HA/PGS-M composite scaffolds were both observed by a SEM (Hitachi, SU8010, Tokyo, Japan). Fluid-discharge therapy41 and the liquid displacement method were both used to calculate the porosity of the scaffolds, was used. Briefly, a known amount of ethanol (volume V1) was decanted into a graduated cylinder. Then, the n-HA/PGS-M composite scaffold was also placed in the same graduated cylinder, and the ethanol volume (V2) was recorded after the scaffold was completely infiltrated. Subsequently, the scaffold sample saturated with ethanol was removed, and the volume of ethanol in the graduated cylinder was defined as V3. The porosity of the scaffolds was calculated according to the following formula:
Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA)
The thermal properties of the scaffolds were characterized by a DSC (204F1, Netzsch, Germany). The temperature program heated at a ratio of 10 °C/min from room temperature to 150 °C and cooled to −50 °C, and then again heated back to 150 °C. Thus, the glass transition temperature (Tg) was automatically detected. Additionally, thermo-gravimetric analyses were performed using a Q5000IR (TA, USA) under air atmosphere at a rate of 10 °C/min from 40 °C to 600 °C.
Mechanical test
A universal testing machine (E42, MTS, USA) possessing a 25N sensor was used to test the mechanical properties of PGS-M and PGS-M-n-HA scaffolds. Each sample was compressed to 40% at a rate of 1.00 mm/min using a preload force of 0.01N. The compressive modulus was calculated from the instrument software.
Cell Counting Kit-8 (CCK-8) assay
Human adipose-derived stem cells (hADSCs) were cultured in Minimum Essential Medium (MEM). The blank PGS-g-M, PGS-M-n-HA-0.4, PGS-M-n-HA-0.5, and PGS-M-n-HA-0.6 scaffolds (all 1 mm × 5 mm) were sterilized and co-cultured with MEM overnight. After the MEM was removed, these scaffolds were seeded with hADSCs (2 × 104/ well) by pipetting of the cell suspension onto the scaffolds, and they were cultured in 24-well plates with 1 mL of MEM under conditions of small amplitude vibration. The MEM was changed every 3 days, and hADSCs treated without any scaffolds were defined as the control group. After 1, 4, and 7 days, 100 µL of 5-mg/mL CCK-8 solutions were added into each well and incubated for 2 h in the dark. The optical density (OD) value of each well was measured at 450 nm using a microplate reader. The cell viability was evaluated using the following equation:
Quantitative real-time quantitative PCR (qRT-PCR) assay
The hADSCs (1 × 105 cells) were seeded onto these scaffolds in 6-well plates for 14 days. Then, qRT-PCR was performed to estimate the expression levels of genes associated with osteoblast differentiation (e.g. RUNX2, OCN, COL1A1, and BMP2) and inflammation-associated genes (e.g. IL-6). Briefly, the total RNA was extracted. Then, the single-stranded cDNA was synthesized using 4 µL of 5× PrimeScript RT Master Mix (TaKaRa Biotechnology, Dalian, China), 0.5 µg of total RNA, and RNase-free water based on the product instructions. Subsequently, RT-PCR was performed using SYBR Premix EX Taq and an ABI 7500 Real Time PCR System (7900HT FAST, Applied Biosystems) under 50 °C for 3 min, 95 °C for 3 min, and 40 cycles of 95 °C for 10 s and 60 °C for 30 s. The primers specific to each gene are shown in Table 1. Amplifications were performed in triplicate, and the relative expression levels of genes were calculated according to 2−ΔΔCT method42.
Table 1.
The primers in real-time quantitative PCR assay.
| Primers | Sequence (5′-3′) |
|---|---|
| human-GAPDHF | AGAAGGCTGGGGCTCATTTG |
| human-GAPDHR | AGGGGCCATCCACAGTCTTC |
| IL-6-hF | ATGAGGAGACTTGCCTGGTG |
| IL-6-hR | GCATTTGTGGTTGGGTCAGG |
| RUNX2-hF | CCGCCTCAGTGATTTAGGGC |
| RUNX2-hR | GGGTCTGTAATCTGACTCTGTCC |
| BMP2-hF | GAACGGACATTCGGTCCTTG |
| BMP2-hR | GCAACGCTAGAAGACAGCG |
| COL1A1-hF | CTGGCCTCCCTGGAATGAAG |
| COL1A1-hR | GGCAGCACCAGTAGCACC |
| OCN-hF | GGATGACCCCCAAATAGCCC |
| OCN-hR | CTTGGACACAAAGGCTGCAC |
hF, human forward primer; hR, human reversed primer.
Western blot
After the hADSCs (3 × 105 cells) were seeded onto scaffolds in 6-well plates for 14 days. Briefly, total protein was extracted and the protein samples were mixed in a 5× loading buffer and loaded into SDS-PAGE following by transferring to polyvinylidene difluoride (PVDF) membranes, blocking with 5% skim milk at 37 °C for 1–2 h, and washing six times (5 min per time) with 1× PBS-T buffer (1000 mL of 1× PBS and 1 mL of Tween-20). Subsequently, the membranes were incubated with the corresponding primary antibodies at 4 °C overnight, and this was followed by incubation with secondary antibody labelled with horseradish peroxidase at room temperature for 2 h. Chemiluminescence was captured by a Millipore ECL system.
Immunofluorescence
The hADSCs (3 × 105 cells) were seeded onto scaffolds in 6-well plates for 14 days, and the expression of RUNX2 and OCN in hADSCs cells was visualized by immunofluorescent staining. The cell samples were fixed with 4% paraformaldehyde and immersed into PBS three times (3 min/time), and then they were transferred into a 0.5% Triton X-100 PBS solution at room temperature for 20 min. After blocking with goat serum, the primary antibody against RUNX2 (1:50, mouse monoclonal antibody) and OCN (1:50, mouse monoclonal antibody) was added at 4 °C overnight. Subsequently, goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody (1:200, Alexa Fluor 594) was incubated at 20–37 °C for 1 h in the dark. Finally, the 4,6-diamidino-2-phenylindole (DAPI) was added, and this was incubated in the dark for 5 min. The images were captured by an inverted fluorescence microscope (IX73, Olympus, Japan).
Bioactivity of scaffolds in simulated body fluid (SBF)
The SBF was prepared based on the following steps43. First, 800 mL deionized water was taken in a 1000-mL beaker. Then, 11.994 g NaCl, 0.525 g NaHCO3, 0.336 g KCl, 0.342 g K2HPO4·3H2O, 0.4575 g MgCl2·6H2O, 0.417 g CaCl2, 0.1065 g Na2SO4, and 9.086 g NH4C(CH2OH)3 were added to the beaker successively. The temperature was maintained at 36.5 °C. The pH was adjusted to between 7.2–7.4. Finally, the volume was adjusted to 1000 mL. The scaffolds were sequentially immersed in SBF at 37 °C for 4 weeks. The SBF was changed every three days, and the surface morphologies of composite scaffolds were observed using SEM. Additionally, chemical analysis on the surface of each scaffold was performed by energy-dispersive X-ray spectroscopy (EDS) coupled with SEM.
Statistical analysis
Data were analysed by SPSS 19.0 software (SPSS Inc., USA) and presented as mean ± standard deviation. The differences among multiple groups were determined using one-way ANOVA followed by the Fishers least significant difference (LSD) method. Statistical significance was considered at P < 0.05.
Supplementary information
Author Contributions
J.L. designed this study. Y.W., Y.L.Z., B.Z., Z.Z., X.Z., Y.Y.Z., H.L. and Y.Z. acquired the data. Y.W., N.S. and Z.Z. analysed the data. Y.W. and N.S. conducted the statistical analysis. Y.W. drafted the manuscript. Y.L.Z. and B.Z. edited and reviewed this manuscript ensuring inclusion of important intellectual content. All authors read and approved the final manuscript.
Data Availability
The datasets used and analysed during the current study are available from the corresponding author upon reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-44478-8.
References
- 1.Betz RR. Limitations of autograft and allograft: new synthetic solutions. Orthopedics. 2002;25:561–570. doi: 10.3928/0147-7447-20020502-04. [DOI] [PubMed] [Google Scholar]
- 2.Pobloth AM, et al. Mechanobiologically optimized 3D titanium-mesh scaffolds enhance bone regeneration in critical segmental defects in sheep. Science Translational Medicine. 2018;10:eaam8828. doi: 10.1126/scitranslmed.aam8828. [DOI] [PubMed] [Google Scholar]
- 3.Omar O, Dahlin A, Gasser A, Dahlin C. Tissue dynamics and regenerative outcome in two resorbable non‐cross‐linked collagen membranes for guided bone regeneration: A preclinical molecular and histological study in vivo. Clinical oral implants research. 2018;29:7–19. doi: 10.1111/clr.13032. [DOI] [PubMed] [Google Scholar]
- 4.Ando K, et al. Effects of a self-assembling peptide as a scaffold on bone formation in a defect. PloS one. 2018;13:e0190833. doi: 10.1371/journal.pone.0190833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshimoto H, Shin Y, Terai H, Vacanti J. A biodegradable nanofiber scaffold by electrospinning and its potential for bone tissue engineering. Biomaterials. 2003;24:2077–2082. doi: 10.1016/S0142-9612(02)00635-X. [DOI] [PubMed] [Google Scholar]
- 6.Hua FJ, Park TG, Lee DS. A facile preparation of highly interconnected macroporous poly (d, l-lactic acid-co-glycolic acid)(PLGA) scaffolds by liquid–liquid phase separation of a PLGA–dioxane–water ternary system. Polymer. 2003;44:1911–1920. doi: 10.1016/S0032-3861(03)00025-9. [DOI] [Google Scholar]
- 7.Gelain F, Horii A, Zhang S. Designer self‐assembling peptide scaffolds for 3‐D tissue cell cultures and regenerative medicine. Macromolecular Bioscience. 2007;7:544–551. doi: 10.1002/mabi.200700033. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Ameer GA, Sheppard BJ, Langer R. A tough biodegradable elastomer. Nature Biotechnology. 2002;20:602–606. doi: 10.1038/nbt0602-602. [DOI] [PubMed] [Google Scholar]
- 9.Rosenbalm TN, et al. Structural and mechanical characterization of bioresorbable, elastomeric nanocomposites from poly(glycerol sebacate)/nanohydroxyapatite for tissue transport applications. J Biomed Mater Res B Appl Biomater. 2016;104:1366–1373. doi: 10.1002/jbm.b.33467. [DOI] [PubMed] [Google Scholar]
- 10.Blondeel PN, et al. Evaluation of a New Skin Closure Device in Surgical Incisions Associated With Breast Procedures. Annals of Plastic Surgery. 2013;73:631. doi: 10.1097/SAP.0b013e3182858781. [DOI] [PubMed] [Google Scholar]
- 11.Sariibrahimoglu K, et al. Development of porous polyurethane/strontium-substituted hydroxyapatite composites for bone regeneration. Journal of Biomedical Materials Research Part A. 2015;103:1930–1939. doi: 10.1002/jbm.a.35327. [DOI] [PubMed] [Google Scholar]
- 12.Alves CD, Jansen JA, Leeuwenburgh SC. Synthesis and application of nanostructured calcium phosphate ceramics for bone regeneration. Journal of Biomedical Materials Research Part B Applied Biomaterials. 2012;100B:2316–2326. doi: 10.1002/jbm.b.32794. [DOI] [PubMed] [Google Scholar]
- 13.Oh KJ, Ko YB, Jaiswal S, Whang IC. Comparison of osteoconductivity and absorbability of beta-tricalcium phosphate and hydroxyapatite in clinical scenario of opening wedge high tibial osteotomy. Journal of Materials Science Materials in Medicine. 2016;27:179. doi: 10.1007/s10856-016-5795-1. [DOI] [PubMed] [Google Scholar]
- 14.Kang IG, et al. Hydroxyapatite Microspheres as an Additive to Enhance Radiopacity, Biocompatibility, and Osteoconductivity of Poly(methyl methacrylate) Bone Cement. Materials. 2018;11:258. doi: 10.3390/ma11020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodenas-Rochina J, Vidaurre A, Cortázar IC, Lebourg M. Effects of hydroxyapatite filler on long-term hydrolytic degradation of PLLA/PCL porous scaffolds. Polymer Degradation and Stability. 2015;119:121–131. doi: 10.1016/j.polymdegradstab.2015.04.015. [DOI] [Google Scholar]
- 16.Bhaskar B, et al. Composite porous scaffold of PEG/PLA support improved bone matrix deposition in vitro compared to PLA‐only scaffolds. Journal of Biomedical Materials Research Part A. 2018;106:1334–1340. doi: 10.1002/jbm.a.36336. [DOI] [PubMed] [Google Scholar]
- 17.Gentile P, Chiono V, Carmagnola I, Hatton PV. An overview of poly (lactic-co-glycolic) acid (PLGA)-based biomaterials for bone tissue engineering. International journal of molecular sciences. 2014;15:3640–3659. doi: 10.3390/ijms15033640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bodakhe S, et al. Injectable photocrosslinkable nanocomposite based on poly (glycerol sebacate) fumarate and hydroxyapatite: Development, biocompatibility and bone regeneration in a rat calvarial bone defect model. Nanomedicine. 2013;8:1777–1795. doi: 10.2217/nnm.12.192. [DOI] [PubMed] [Google Scholar]
- 19.Xiaolong C, Yuguo Z, Yinchu S. Natural products with maleic anhydride structure: nonadrides, tautomycin, chaetomellic anhydride, and other compounds. Chemical Reviews. 2007;107:1777. doi: 10.1021/cr050029r. [DOI] [PubMed] [Google Scholar]
- 20.Zhang J-F, Sun X. Mechanical properties of poly (lactic acid)/starch composites compatibilized by maleic anhydride. Biomacromolecules. 2004;5:1446–1451. doi: 10.1021/bm0400022. [DOI] [PubMed] [Google Scholar]
- 21.Yao M, et al. Modification of poly (lactic acid)/poly (propylene carbonate) blends through melt compounding with maleic anhydride. Express Polym Lett. 2011;5:937–949. doi: 10.3144/expresspolymlett.2011.92. [DOI] [Google Scholar]
- 22.Huang H, et al. Crystallization kinetics of polypropylene composites filled with nano calcium carbonate modified with maleic anhydride. Journal of Applied Polymer Science. 2011;119:1516–1527. doi: 10.1002/app.32842. [DOI] [Google Scholar]
- 23.Zhou C, et al. Electrospun bio-nanocomposite scaffolds for bone tissue engineering by cellulose nanocrystals reinforcing maleic anhydride grafted PLA. ACS applied materials & interfaces. 2013;5:3847–3854. doi: 10.1021/am4005072. [DOI] [PubMed] [Google Scholar]
- 24.Xiang Y, et al. [Effects of maleic anhydride-modified poly(D,L-lactic acid) on the adhesion, proliferation and differentiation of osteoblasts] Journal of Biomedical Engineering. 2011;28:753–757. [PubMed] [Google Scholar]
- 25.Rai R, Tallawi M, Grigore A, Boccaccini AR. Synthesis, properties and biomedical applications of poly(glycerol sebacate) (PGS): A review. Progress in Polymer Science. 2012;37:1051–1078. doi: 10.1016/j.progpolymsci.2012.02.001. [DOI] [Google Scholar]
- 26.Patel A, et al. Highly elastomeric poly(glycerol sebacate)-co-poly(ethylene glycol) amphiphilic block copolymers. Biomaterials. 2013;34:3970–3983. doi: 10.1016/j.biomaterials.2013.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Y, et al. Preparation and properties of poly (lactide-co-glycolide)(PLGA)/nano-hydroxyapatite (NHA) scaffolds by thermally induced phase separation and rabbit MSCs culture on scaffolds. Journal of biomaterials applications. 2008;22:409–432. doi: 10.1177/0885328207077632. [DOI] [PubMed] [Google Scholar]
- 28.Hayati AN, Rezaie H, Hosseinalipour S. Preparation of poly (3-hydroxybutyrate)/nano-hydroxyapatite composite scaffolds for bone tissue engineering. Materials Letters. 2011;65:736–739. doi: 10.1016/j.matlet.2010.11.004. [DOI] [Google Scholar]
- 29.Rosenbalm TN, et al. Structural and mechanical characterization of bioresorbable, elastomeric nanocomposites from poly (glycerol sebacate)/nanohydroxyapatite for tissue transport applications. Journal of Biomedical Materials Research Part B: Applied Biomaterials. 2016;104:1366–1373. doi: 10.1002/jbm.b.33467. [DOI] [PubMed] [Google Scholar]
- 30.Du M, et al. Fabrication and biological application of nano-hydroxyapatite (nHA)/alginate (ALG) hydrogel as scaffolds. Journal of Materials Chemistry. 2011;21:2228–2236. doi: 10.1039/C0JM02869J. [DOI] [Google Scholar]
- 31.Pomerantseva I, et al. Degradation behavior of poly(glycerol sebacate) Journal of Biomedical Materials Research Part A. 2009;91A:1038–1047. doi: 10.1002/jbm.a.32327. [DOI] [PubMed] [Google Scholar]
- 32.Alessandro P, et al. Osteoinduction of Human Mesenchymal Stem Cells by Bioactive Composite Scaffolds without Supplemental Osteogenic Growth Factors. PloS one. 2011;6:e26211. doi: 10.1371/journal.pone.0026211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chatzinikolaidou M, et al. Recombinant human bone morphogenetic protein 2 (rhBMP-2) immobilized on laser-fabricated 3D scaffolds enhance osteogenesis. Colloids & Surfaces B Biointerfaces. 2016;149:233–242. doi: 10.1016/j.colsurfb.2016.10.027. [DOI] [PubMed] [Google Scholar]
- 34.Zhou Y, et al. Combined marrow stromal cell-sheet techniques and high-strength biodegradable composite scaffolds for engineered functional bone grafts. Biomaterials. 2007;28:814–824. doi: 10.1016/j.biomaterials.2006.09.032. [DOI] [PubMed] [Google Scholar]
- 35.Roohani-Esfahani SI, et al. The influence hydroxyapatite nanoparticle shape and size on the properties of biphasic calcium phosphate scaffolds coated with hydroxyapatite-PCL composites. Biomaterials. 2010;31:5498. doi: 10.1016/j.biomaterials.2010.03.058. [DOI] [PubMed] [Google Scholar]
- 36.Rezwan K, Chen QZ, Blaker JJ, Boccaccini AR. Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering. Biomaterials. 2006;27:3413–3431. doi: 10.1016/j.biomaterials.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 37.Kong L, et al. A study on the bioactivity of chitosan/nano-hydroxyapatite composite scaffolds for bone tissue engineering. European Polymer Journal. 2006;42:3171–3179. doi: 10.1016/j.eurpolymj.2006.08.009. [DOI] [Google Scholar]
- 38.Sari, N. K., Indrani, D. J., Johan, C. & Corputty, J. E. M. Evaluation of chitosan-hydroxyapatite-collagen composite strength as scaffold material by immersion in simulated body fluid. 884(1), 012116 (2017).
- 39.Weng J, et al. Formation and characteristics of the apatite layer on plasma-sprayed hydroxyapatite coatings in simulated body fluid. Biomaterials. 1997;18:1027–35. doi: 10.1016/S0142-9612(97)00022-7. [DOI] [PubMed] [Google Scholar]
- 40.Blaker JJ, Nazhat SN, Maquet V, Boccaccini AR. Long-term in vitro degradation of PDLLA/Bioglass® bone scaffolds in acellular simulated body fluid. Acta biomaterialia. 2011;7:829–840. doi: 10.1016/j.actbio.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 41.Yang J, et al. Fabrication and surface modification of macroporous poly(L-lactic acid) and poly(L-lactic-co-glycolic acid) (70/30) cell scaffolds for human skin fibroblast cell culture. Journal of Biomedical Materials Research Part B Applied Biomaterials. 2002;62:438. doi: 10.1002/jbm.10318. [DOI] [PubMed] [Google Scholar]
- 42.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 43.Peng X, et al. Surface bioactivity studies of nano-hydroxyapatite/polyamide 66 composites in simulated body fluids (SBF) High-tech communication. 2003;13:38–42. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and analysed during the current study are available from the corresponding author upon reasonable request.