

Abstract
Nav1.6 (SCN8A) is a major voltage-gated sodium channel in the mammalian CNS, and is highly concentrated at the axon initial segment (AIS). As previously demonstrated, the microtubule associated protein MAP1B binds the cytoplasmic N terminus of Nav1.6, and this interaction is disrupted by the mutation p.VAVP(77–80)AAAA. We now demonstrate that this mutation results in WT expression levels on the somatic surface but reduced surface expression at the AIS of cultured rat embryonic hippocampal neurons from both sexes. The mutation of the MAP1B binding domain did not impair vesicular trafficking and preferential delivery of Nav1.6 to the AIS; nor was the diffusion of AIS inserted channels altered relative to WT. However, the reduced AIS surface expression of the MAP1B mutant was restored to WT levels by inhibiting endocytosis with Dynasore, indicating that compartment-specific endocytosis was responsible for the lack of AIS accumulation. Interestingly, the lack of AIS targeting resulted in an elevated percentage of persistent current, suggesting that this late current originates predominantly in the soma. No differences in the voltage dependence of activation or inactivation were detected in the MAP1B binding mutant relative to WT channel. We hypothesize that MAP1B binding to the WT Nav1.6 masks an endocytic motif, thus allowing long-term stability on the AIS surface. This work identifies a critical and important new role for MAP1B in the regulation of neuronal excitability and adds to our understanding of AIS maintenance and plasticity, in addition to identifying new target residues for pathogenic mutations of SCN8A.
SIGNIFICANCE STATEMENT Nav1.6 is a major voltage-gated sodium channel in human brain, where it regulates neuronal activity due to its localization at the axon initial segment (AIS). Nav1.6 mutations cause epilepsy, intellectual disability, and movement disorders. In the present work, we show that loss of interaction with MAP1B within the Nav1.6 N terminus reduces the steady-state abundance of Nav1.6 at the AIS. The effect is due to increased Nav1.6 endocytosis at this neuronal compartment rather than a failure of forward trafficking to the AIS. This work confirms a new biological role of MAP1B in the regulation of sodium channel localization and will contribute to future analysis of patient mutations in the cytoplasmic N terminus of Nav1.6.
Keywords: axon initial segment, endocytosis, localization, MAP1B, Nav1.6, trafficking
Introduction
Voltage-gated sodium channels (Nav) generate the depolarizing phase of most action potentials. The ion-conducting Nav α subunits are encoded by nine genes, with Nav1.1, Nav1.2, Nav1.3, and Nav1.6 predominant in the CNS (Catterall, 2012). Nav1.6, encoded by SCN8A, is one of the most abundant Nav channels and localizes to specific nanodomains on the neuronal soma (Akin et al., 2016), axon initial segment (AIS) and nodes of Ranvier (Schaller et al., 1995; Whitaker et al., 1999; Tzoumaka et al., 2000). Maintenance of the structure and composition of the AIS is essential for proper neuronal activity, with the AIS demonstrating plasticity by changing both its composition and location in response to neuronal activity and injury (Grubb and Burrone, 2010; Kuba et al., 2010; Evans et al., 2013; Hatch et al., 2017). Within the distal AIS, Nav1.6 is highly concentrated via an ankyrinG-dependent mechanism (Gasser et al., 2012; Akin et al., 2015). At the AIS, Nav1.6 plays a key role in the initiation of the action potential (Hu et al., 2009) and is a potent regulator of neocortical and hippocampal glutamatergic excitation (Kole et al., 2008).
Mutations in Nav1.6 generate neurological disorders, such as tremor, dystonia, ataxia, and premature death in mice, and epileptic encephalopathy and intellectual disability in humans (Burgess et al., 1995; Meisler et al., 2016; Wagnon et al., 2017). While most pathological sodium channel mutations are located in transmembrane or C-terminal domains and alter channel function, evidence is emerging that mutations within the cytoplasmic N terminus can cause disease, and this region may play a central role in channel delivery to the cell surface. For example, the N-terminal mutation S21P in mouse Nav1.6 causes ataxia due to retention of channels in the Golgi and impaired trafficking to the cell surface (Sharkey et al., 2009). A 29 aa motif in the N terminus of Nav1.8 is necessary for Annexin II binding, which facilitates forward channel trafficking (Okuse et al., 2002; Poon et al., 2004). The N-terminal mutation R101Q in human Nav1.1 is associated with borderline severe myoclonic epilepsy in infancy (Fukuma et al., 2004).
Yeast two-hybrid screening using the Nav1.6 N terminus as bait, and coimmunoprecipitation from mouse brain membranes, demonstrated that the MAP1B light chain binds to Nav1.6 (O'Brien et al., 2012). MAP1B is a microtubule-associated protein that plays classical roles in microtubule stabilization, axonal elongation, and neurogenesis (Yang et al., 2012; Villarroel-Campos and Gonzalez-Billault, 2014). The VAVP motif at aa 77–80 of Nav1.6 was required for MAP1B binding. Mutation of this MAP1B binding motif prevented the generation of voltage-dependent Na+ currents in transfected ND7/23 cells, and coexpression of MAP1B increased wild-type (WT) Nav1.6 current density by 50% (O'Brien et al., 2012). These data, together with previous findings that MAP1B regulates the trafficking of neurotransmitter receptors (Hanley et al., 1999; Eriksson et al., 2010; Kim et al., 2014), suggested that MAP1B binding is required for delivery of Nav1.6 to the cell surface.
Our current work analyzes the function, localization, and trafficking of the Nav1.6 mutant p.VAVP(77–80)AAAA (hereafter, MAPM) that is incapable of binding MAP1B. In primary rat hippocampal neurons, MAPM does not concentrate at the AIS, although it localizes efficiently to the soma membrane. Interestingly, MAPM vesicular delivery to the AIS and postdelivery diffusion were unaltered. Unexpectedly, the low AIS expression level of MAPM is due to rapid endocytosis at this critical neuronal compartment. Thus, lack of a functional MAP1B binding domain results in enhanced compartment-specific Nav1.6 endocytosis, rather than reduced forward trafficking as originally suggested (O'Brien et al., 2012). Furthermore, our work suggests that there is a correlation between Nav1.6 channel function and its location. The MAPM channel, as well as the Nav1.6 AnkyrinG binding domain mutant that also prevents AIS localization, exhibits an ∼5-fold greater percentage of persistent current than WT channel, suggesting that Nav1.6 persistent current is regulated by subcellular location.
Materials and Methods
Plasmids constructs.
hNav1.6-GFP, hNav1.6-LoopBAD-GFP, hNav1.6ABM-GFP, and hNav1.6ABM-LoopBAD-GFP were previously described (Akin et al., 2015, 2016). The 77/80 VAVP-AAAA mutation was incorporated by site-directed mutagenesis (Wagnon et al., 2016) into hNav1.6-GFP and hNav1.6-LoopBAD-GFP. WT Nav1.6-mCherry and WT Nav1.6-LoopBAD-mCherry were generated by replacing the GFP with mCherry derived from pmCherry-N1 (Clontech). When indicated, cells were cotransfected with Ruby2-Kv2.1 to localize the AIS (Johnson et al., 2018).
Cell culture and transfections.
Rat hippocampal neuronal cultures (rHNs) were used as previously described (Akin et al., 2015, 2016; Fox et al., 2015; Johnson et al., 2018). Briefly, dissociated rat E18 hippocampal neurons from both sexes were seeded onto poly-L-lysine (Sigma-Aldrich, catalog #RNBG0769) (dilution 1:2 in 0.15 m borate buffer, pH 8.4) coated glass-bottom 35 mm dishes (Matsunami, catalog #D11130H) and cultured in Neurobasal media (Invitrogen, catalog #21103-049) supplemented with Glutamax (Sigma-Aldrich, catalog #350350-061), penicillin/streptomycin (Cellgro, catalog #SV30010), and SM1 neuronal supplement (Stemcell Technologies, catalog #05711), per the manufacturer's instructions. Animal use was according to protocols approved by the Institutional Animal Care and Use Committee of Colorado State University (Animal Welfare Assurance number: A3572-01). This Institutional Animal Care and Use Committee specifically approved this study. Neuronal cultures were transfected after 5 d in culture (DIV 5) with Lipofectamine 2000 (Invitrogen, catalog #11668027) and 1 μg of the Nav1.6 cDNA (WT or mutant), 0.5 μg of NavBeta1 subunit, 0.5 μg of NavBeta2 subunit and, in some experiments, 0.5 μg of pSec-BirA (bacterial biotin ligase) (Akin et al., 2016). Neuronal cultures were used at DIV 6–10 as indicated.
Electrophysiology.
Transfected rHNs were analyzed by whole-cell voltage-clamp 1–2 d after transfection (DIV 6–7) in a neuronal external solution consisting of the following (in mm): 112 NaCl, 20 TEA-Cl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 0.5 CaCl2, 11.1 glucose, and 10 HEPES, pH 7.4 (adjusted with NaOH), 300 mOsm; 300 nm Tetrodotoxin (TTX) and 300 μm Ni2+ were added to the bath to block endogenous Nav channels and T-type calcium channels, respectively (Lee et al., 1999; Herzog et al., 2003). When lower external Na+ concentration was desired for better control of transient peak currents, half the Na+ was replaced with N-methyl-d-glucamine (NMDG), using an Na+/NMDG neuronal external solution consisting of the following (in mm): 56 NaCl, 56 NMDG, 20 TEA-Cl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 0.5 CaCl2, 11.1 glucose, and 10 HEPES, pH 7.4 (adjusted with HCl), 300 mOsm. In all the cases, the pipette solution contained the following (in mm): 131 CsCl, 4 NaCl, 1.5 MgCl2, 10 HEPES, 0.5 EGTA, pH 7.4 (adjusted with CsOH), 280 mOsm. Pipettes had resistances of 1.3–2.5 mΩ, and Ra was ≤5 mΩ. Whole-cell Na+ currents were recorded at room temperature using an Axopatch 200B amplifier. Ionic currents were capacitance and series resistance compensated by 70%–90%, sampled at 10 kHz (Digidata 1440), filtered at 2 kHz, and leak subtracted online using the P/4 method in pClamp10.6. Cells were held at −70 mV, stepped to −100 mV for 50 ms to release channels from inactivation, then depolarized to potentials between −70 and 75 mV for 100 ms in 5 mV steps, and stepped back to −70 mV. We did not include data from cells that exhibited delayed inward current at negative voltages, a known space-clamp artifact. For the analysis of the persistent current, only the 0 mV trace was used, and the amount of current remaining during the last 2 ms of the 100 ms voltage-step at 0 mV was normalized to the peak current. Activation curves were plotted by converting peak ionic currents (I) to conductance (G) using the relationship: G = I/(V − ENa). ENa was calculated to be 85 mV for normal external Na+. Steady-state inactivation was determined using a 100 ms prepulse ranging from −140 to 90 mV (10 mV increments), then stepping to a potential of −10 mV for 40 ms to measure the noninactivated channels. Peak inward currents were then normalized to the maximal peak current (Imax).
Live-cell surface labeling.
For live-cell surface labeling in conjunction with either total internal reflection fluorescence (TIRF) or spinning-disk confocal imaging, rHN cultures were washed quickly with neuronal imaging saline (NIS) (126 mm NaCl, 4.7 mm KCl, 2.5 mCaCl2, 0.6 mm MgSO4 0.15 mm), and incubated for 10 min with 1.2 nm streptavidin-conjugated fluorophores (SA-CF640, Biotium, catalog #29041; or SA-A594, Life Science Technologies, catalog #1005935) to label biotinylated surface channels as previously described (Akin et al., 2015). When an AIS marker was required, neurons were simultaneously labeled for 10 min with a monoclonal antibody-anti-Neurofascin-186 (NF-186) (dilution 1:2000) (Neuromab, catalog #75-172, RRID:AB_2282826) and a secondary antibody goat-anti-mouse-Alexa-594 (or Alexa-647) (dilution 1:2000) (Life Science Technologies, catalog #A11032 and A21235, RRID:AB_141672 and RRID:AB_141693, respectively). Cells were washed three times over 5 min after labeling and immediately imaged without fixation.
Immunocytochemistry.
Briefly, rHN cultures were washed quickly with NIS and immediately fixed with 4% PFA (diluted in NIS) for 15 min at 37°C. After 3 washes of 5 min each with PBS, cells were incubated in PBS with 0.1% Triton X-100 and 10% goat serum for 30 min at room temperature. After 3 washes with PBS 0.05% Triton X-100, cells were incubated with PanNav antibody (Neuromab, catalog #N419/40, RRID:AB_2750795; dilution 1:1000) overnight at 4°C. Then, after 3 washes with PBS 0.05% Triton X-100, secondary antibody goat anti-mouse AlexaFluor-647 (Life Science Technologies, catalog #A21235, RRID:AB_141693) was applied (dilution 1:1000) at room temperature for 30 min. After 3 washes with PBS, cells were mounted with AquaPoly/Mount (Polysciences, catalog #18606) and imaged with our spinning disk microscope.
Microscopy.
Most experiments used TIRF imaging with an Eclipse Ti fluorescence microscope (Nikon) equipped with 405, 488, 561, and 633 nm diode lasers, 100 mW each, split evenly between TIRF and photoactivation unit pathways. Images were collected using an iXon EMCCD DU-897 camera (Andor) through a Plan Apo 100×, NA 1.49, TIRF objective. Both the objective and dish were temperature controlled, and z drift was mitigated through the use of the Perfect-Focus system (Nikon).
Vesicle analysis and fluorescence recovery after photobleaching (FRAP) experiments were performed using a spinning disk confocal microscope based on a Yokogawa CSUX1 system built on an Olympus IX83 inverted stand coupled to an Andor laser launch containing 405, 488, 561, and 637 nm diode lasers, 100–150 mW each. Images were collected using either 100× Plan Apo, 1.4 NA or a 60× Plan Apo N 1.4 NA objective and two iXon EMCCD cameras (DU-897, Andor). This system is equipped with the ZDC constant focus system and a Tokai Hit chamber and objective heater. Fixed immunocytochemistry samples were also imaged on the spinning disk confocal microscope using a 100× Plan Apo, 1.4 NA. z stacks were acquired (one slice every 0.25 μm), and z maximal projections were processed and analyzed with Fiji software.
In all cases, appropriate use of sequential excitation with the appropriate dichroic and bandpass filters permitted fluorophore separation. No images were acquired under saturating conditions. Additional details regarding our microscopy have been previously described (Akin et al., 2015, 2016; Johnson et al., 2018). For all imaging experiments, the stage and objective were heated to 37°C.
Vesicular traffic analysis.
To visualize vesicles moving along the axon (identified with NF-186 immunolabeling), selected regions (which ranged from 40 × 30 to 59 × 30 pixels) were photobleached using the FRAPPA Scanning Laser Active Illumination System (Andor) attached to the spinning-disk microscope described above. Cells were imaged at 2 Hz for ∼3.5 min (400 frames) with 100 ms exposures. Manual identification/counting of vesicles was performed in each cell imaged. Kymograph analysis of vesicle speed and number of stops were created and analyzed with the Kymograph tool from MetaMorph Software.
Dual cargo-carrying vesicles were analyzed in a similar way, but using a simultaneous dual-acquisition mode (two EMCCD DU-897 cameras) and here the bleached regions ranged from 30 × 25 to 32 × 65 pixels. A 60× Plan Apo N 1.40 NA objective was also used to minimize chromatic aberrations.
Insertion event experiments.
Detection of newly inserted channels to the neuronal surface was performed as previously described in detail (Akin et al., 2015). In brief, biotinylated Nav1.6-BAD-GFP channels (WT or mutant) on the neuronal surface 18–36 h after transfection were blocked with 1 μm NeutrAvidin (Invitrogen, catalog #A2666) for 5 min; then unbound NeutrAvidin was removed with 5–6 NIS washes. Cells were immediately placed on the TIRF microscope stage; and once a transfected cell was located, during TIRF imaging at 2.5 Hz (100 ms exposure), 1.2 nm SA-CF640 was added to the NIS and imaging continued for ∼30 min. For these experiments, to avoid possible stearic problems with antibodies preventing SA-CF40 from binding to the newly inserted channels, live cell NF-186 immunolabeling was not performed, but axonal processes were identified by cotransfection with Ruby2-Kv2.1 that concentrates at the AIS (Sarmiere et al., 2008).
FRAP experiments.
Photobleaching was performed using the FRAPPA system (Andor). Images were acquired with a 100× Plan Apo, 1.4 NA objective, every 30 s during 30 min, and the recovery of SA-594 fluorescence within the bleached region (35 × 30 pixels) was quantitated, background subtracted, and normalized to the initial prephotobleach signal.
Nav1.6 single molecule tracking.
To achieve the low surface labeling density required to track single Nav1.6 channels, neurons were incubated with 0.06–0.09 pm SA-CF640 for only 3 min and immediately washed with NIS three times. Transfected cells (identified by the GFP signal) were located under TIRF imaging conditions, and cells were imaged for ∼1 min at 20 Hz (50 ms exposure). Images were processed and the U-Track algorithm was applied to track single channels diffusing on the AIS surface as previously described (Akin et al., 2016; Sikora et al., 2017; Weron et al., 2017). Diffusion coefficients were calculated from the ensemble average of the time-averaged mean square displacements.
Endocytosis inhibition.
DIV 7 rHNs were incubated with 100 μm Dynasore (Tocris Bioscience, catalog #2897) (0.1% DMSO in supplemented Neurobasal media) for 2 h. After treatment, cells were labeled with SA-CF640 and NF-186 antibody for 10 min in Dynasore-containing NIS. After 3 rinses, cells were imaged under TIRF conditions, in NIS. Because inhibition of endocytosis by Dynasore is reversible (Macia et al., 2006), 50 μm Dynasore was present throughout the experiment, including during surface labeling, washes, and imaging.
Image presentation and data analysis.
No images were acquired under saturating conditions, and all analysis was performed using raw images. Images shown were processed a posteriori only to the extent necessary to appreciate the quantitated differences. Image processing (pseudo-coloring, background subtraction, cropping, adjustment of brightness and contrast) was performed with Fiji. All panels shown in a given figure were processed and contrasted in the same way. Volocity 6.1.1 analysis software was used to create the kymograph in Figure 6 (insertion events). MetaMorph 7.8.0.0 was used to create the kymographs shown in Figure 4 (vesicles). Origin 2018 was used to analyze and fit numerical data. Custom MATLAB and LabView codes were used for mean square displacement calculation and U-track algorithms (Akin et al., 2015, 2016; Sikora et al., 2017; Weron et al., 2017).
Figure 6.

Nav1.6 WT and MAPM channels are both preferentially delivered to the AIS. DIV 7 rHNs expressing either the WT or MAPM Nav1.6-LoopBAD-GFP tagged channels were incubated with NeutrAvidin for 5 min to saturate biotinylated Nav1.6 channels already on the cell surface, rinsed to remove unbound NeutrAvidin, and then imaged under TIRF conditions in the presence of SA-CF640 to visualize the real-time insertion of nascent Nav1.6-LoopBAD-GFP WT or Nav1.6-LoopBAD-GFP MAPM to the membrane. A, GFP TIRF footprint of transfected neurons. Left, WT. Right, MAPM. Shape of the cell is outlined in white. Orange lines indicate the line segments used to generate the kymographs shown in B. Scale bars, 10 μm. B, Kymographs illustrating SA-CF640 fluorescence indicative of channel insertion. Orange lines indicate line scans starting in the soma (solid line) and continuing through the AIS (dashed line). Scale bars: x: 125 s; y: 29 and 30 μm for WT and MAPM, respectively. C, Average number of insertion events/μm2 in the somatic and AIS compartments. No significant differences were detected between WT (dark gray) and MAPM (light gray). However, in both cases, channels were preferentially inserted in the AIS compartment. For the insertion events/μm2 into the soma: 0.032 ± 0.009 and 0.053 ± 0.013 for the WT and MAPM, respectively. For the insertion events/μm2 into the AIS: 0.100 ± 0.026 and 0.163 ± 0.035 for the WT and MAPM, respectively. Error bars indicate SEM. *p ≤ 0.05, **p ≤ 0.01, ns, nonsignificant.
Figure 4.

The lack of MAPM targeting to the AIS is not due to a defect in vesicular packaging or transport to the AIS. Small regions of the AIS in Nav1.6-GFP WT, MAPM, or ABM expressing DIV 6 rHNs were photobleached to visualize vesicles traveling along the axon. A, Axonal trafficking vesicle detection. Shown are different frames of the same movie, where the AIS was bleached (white squares in second column). White arrows point to retrograde vesicles (moving to the left). Yellow arrow points to an anterograde vesicle (moving to the right, from the soma). Scale bars, 5 μm. B, Kymographs from the same processes shown in A. y axis represents time (200 s); the bleach does not happen until 5 s (10 frames) after the movie was initiated. x axis represents position along the AIS. Scale bars, 5 μm. Anterograde vesicles (moving from the soma to the process) are depicted by lines going to the right, and retrograde (trafficking toward the soma) vesicles are depicted by lines moving to the left. C, Average number of vesicles/cell (left graph), distribution of anterograde and retrograde vesicles per cell (middle graph), and average of vesicular transport rate (right graph). No significant differences were found between constructs (4.0 ± 0.46, 3.31 ± 0.65, and 3.56 ± 0.39 vesicles/cell for the WT, MAPM, and ABM constructs). Likewise, there were no differences in transport direction: 60.6 ± 7.7%, 68.9 ± 8.4%, and 77.5 ± 6.6% retrograde vesicles for the WT, MAPM, and ABM, respectively. There were also no differences in the vesicle's velocity between conditions. Error bars indicate SEM. D, MAPM and ABM constructs coexpressed with Nav1.6 WT-mCherry. Images of the GFP (cyan) and the mCherry (magenta) fluorescence at prebleach, 5 s after bleach, and different time points are shown. White arrows point to vesicles carrying WT-mCherry and either WT-GFP (top), MAPM-GFP (middle), or ABM-GFP (bottom). t1 and t2 correspond to different frames 5.5, 42, and 3.5 s apart for WT, MAPM, and ABM coexpression, respectively. Scale bars, 5 μm.
Experimental design and statistical analysis.
All experiments were performed in rat cultured hippocampal neurons at DIV 6–10 (derived from E18 embryos of both sexes). As early as DIV 4, these neurons have a developing AIS, and our previous studies indicate that at DIV 6 there is direct vesicular transport of Nav1.6 to the AIS surface (Akin et al., 2015). The endogenous Nav1.6 does not appear consistently in our neuronal cultures until DIV 10 (Akin et al., 2015). Our primary concern with respect to experimental design focused on overexpression issues because high-level expression could induce altered Nav1.6 trafficking and localization. The levels of transfected Nav1.6 in the neurons are only 1.3-fold higher than the level of the endogenous channel as determined by comparing the immunostaining of transfected cells with that against the endogenous channel (Akin et al., 2015). In addition, endogenous inward currents in our DIV 6 neurons were 214.3 ± 64.9 pA/pF (Akin et al., 2015), whereas transfected Nav1.6 generated (TTX resistant) currents of 141.10 ± 24.40 pA/pF. High expression levels were not necessary given the sensitivity of our TIRF and spinning disk microscopes, which are both capable of imaging single molecules.
For the majority of statistical analysis, one-way ANOVA with ad hoc Tukey's tests with significance levels of 0.05 were performed using Origin 2018b software. Where appropriate, unpaired Student's t test were performed, directly comparing one condition with another, with significance levels of 0.05. Statistical details, number of ROIs and cells examined, degrees of freedom (df), and p values are indicated in detail in each Results section. Data are presented as mean ± SEM unless otherwise stated.
Code/software.
MATLAB codes used for the analysis of single-particle diffusion were used as previously described (Akin et al., 2016; Sikora et al., 2017; Weron et al., 2017) and are available upon request. No new codes were written for the present analysis.
Results
MAPM Nav1.6 demonstrates reduced surface expression at the AIS but normal somatic surface expression
Previous studies determined that aa 77–80 (VAVP) within the Nav1.6 N terminus are necessary for both MAP1B binding and functional expression on the surface of ND7/23 cells (O'Brien et al., 2012). ND7/23 is an immortalized cell line, and proteins expressed in this heterologous system may behave very differently than when expressed in more native systems, such as primary neuronal cultures (Yin et al., 2016). Our previous imaging studies demonstrated that our rat hippocampal neuronal cultures have well-developed AIS and somatic compartments with respect to Nav1.6 localization (Akin et al., 2015) and that these cells are a useful system with which to study Nav1.6 trafficking. Therefore, we used rHNs to examine the trafficking of the mutant Nav1.6 channel MAPM [p.VAVP (77–80)AAAA] that is incapable of binding MAP1B. To visualize the cell surface expression of MAPM, a biotin acceptor domain (BAD) that does not affect the function or trafficking of Nav1.6 or Kv2.1 channels (Deutsch et al., 2012; Akin et al., 2015) was introduced into an extracellular loop (Fig. 1), referred to as LoopBAD. We and others have previously shown that the AnkyrinG binding motif mutant of Nav1.6, designated ABM, is localized only to the soma and not to the AIS due to its inability to bind AnkyrinG (Gasser et al., 2012; Akin et al., 2015). Therefore, ABM served as a control with known AIS trafficking defects. The MAPM and ABM mutations are located in two different cytoplasmic domains, the N terminus and loop 2, respectively (Fig. 1). Coexpression of the LoopBAD constructs with the bacterial biotin ligase BirA resulted in biotinylated surface channels that were detected by labeling live cells with cell impermeable CF640-conjugated streptavidin (SA-CF640).
Figure 1.
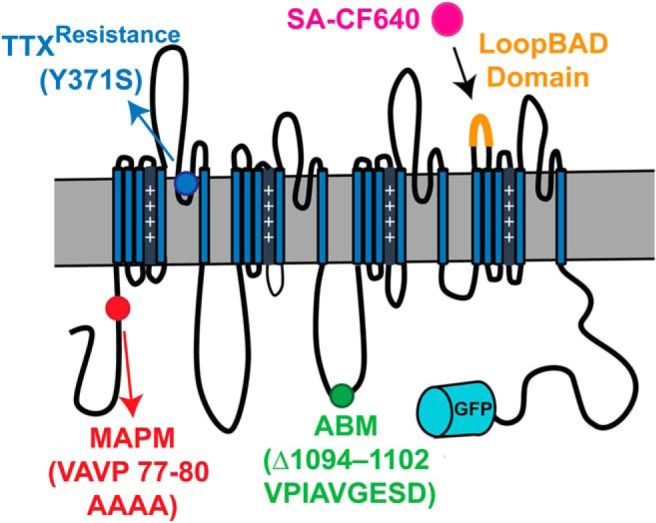
Schematic of the Nav1.6-LoopBAD-GFP construct with mutations used for this study. Diagram depicting the locations of the MAP1B (MAPM, red) and Ankyrin binding motif mutations (ABM, green), the location of the GFP (cyan barrel), and the biotin acceptor domain (LoopBAD orange). SA-CF640 binding (magenta) is used to visualize the surface channels. All our Nav1.6 constructs carry the Y371S mutation (blue) that confers TTX resistance, allowing us to record currents from only the transfected channels in the presence of TTX.
Figure 2A shows representative TIRF images of rHNs transfected with Nav1.6 constructs containing the extracellular biotinylation domain as well as a C-terminal GFP. Transfected cells were identified by GFP fluorescence, immunodetection of the AIS was with anti-neurofascin-186 (NF-186) antibodies, and specific labeling of the Nav1.6 channel at the cell surface used the SA-CF640. As shown in the top row, Nav1.6 WT clearly targets to the AIS, whereas ABM (bottom row) does not, as previously described (Akin et al., 2015). Interestingly, MAPM was not concentrated at the AIS (Fig. 2A, middle row; for a better appreciation of AIS localization, see enlarged insets). In all cases, the previously described (Akin et al., 2016) somatic nanoclusters are noticeable (white arrows) in the surface labeling inset panels corresponding to an enlargement of the white square within the somatic compartment. Quantitative analysis of all the acquired images (n = 48, n = 32, and n = 28 for WT, MAPM, and ABM Nav1.6-LoopBAD-GFP transfected cells, respectively) demonstrated that both ABM and MAPM mutants showed significantly less AIS surface labeling than WT Nav1.6 (Fig. 2B, top left) (for the SA-CF640 AIS signal: 1792.1 ± 186.0, 702.1 ± 205.7, and 120.7 ± 18.7 Arbitrary Units (AU) for the WT, MAPM, and ABM, respectively; one-way ANOVA significant F(2,105) = 23.77, p = 3.05 × 10−9, with post hoc pairwise Tukey's tests, p = 0.00006 and 0 for WT-MAPM and WT-ABM comparisons, respectively). There was no significant difference in somatic surface labeling among the three constructs (for the SA-CF640 somatic signal: 49.7 ± 7.7, 38.8.4 ± 10.3, and 46.8 ± 10.1 AU for the WT, MAPM, and ABM, respectively; one-way ANOVA, not significant, F(2,94) = 0.374, p = 0.689) (Fig. 2B, top right). Similar trends were observed in the GFP signal (predominantly intracellular channel) as illustrated in the two bottom graphs (for the GFP AIS signal: 2526.2 ± 239.1, 947.4 ± 279.3, and 195.3 ± 34.7 AU for the WT, MAPM, and ABM, respectively; one-way ANOVA significant F(2,105) = 27.297, p = 2.84 × 10−10, with post hoc pairwise Tukey's tests p = 0.000009 and 0 for WT-MAPM and WT-ABM comparisons, respectively; for the GFP somatic signal: 835.8 ± 224.6, 880.5 ± 163.5, and 486.2 ± 93.2 AU for the WT, MAPM, and ABM, respectively; one-way ANOVA, not significant, F(2,94) = 0.902, p = 0.409). In all cases n = 48, n = 32, and n = 28 cells for WT, MAPM, and ABM, respectively. This analysis was performed with nonsaturated images. All images shown in Figure 2A were identically processed and contrasted after analysis to allow for an accurate comparison. Together, these results indicate that MAPM is efficiently synthesized and trafficked to the somatic surface but shows impaired AIS localization.
Figure 2.

Nav1.6 MAPM is not concentrated at the AIS but effectively localizes to the soma. A, Representative TIRF images of rHNs (DIV 10) expressing Nav1.6-LoopBAD-GFP WT, MAPM, or ABM (top, middle, or bottom row, respectively). The GFP fluorescence is shown in the first column (cyan), anti-neurofascin (NF)-186 immunostaining in the second column (yellow), and surface labeling in the third column (magenta). Insets in the third column represent enlargements of the somatic surface expression of Nav1.6 corresponding to the white squares. White arrows point to somatic nanoclusters which are stable domains containing multiple Nav1.6 channels (Akin et al., 2016). Two right-hand panels represent enlargements of the NF-186 immunolabeling and surface Nav1.6 expression in AIS (yellow rectangles). Scale bars: Full-size, 10 μm; Enlargements, 5 μm. B, Scatter plots summarizing mean surface labeling signal (SA-CF640, top row, magenta) or mean GFP fluorescence (bottom row, cyan) in the AIS (left) or somatic compartment (right). Lines indicate mean values ± SEM. Nav1.6 MAPM presented significantly less AIS surface labeling and AIS GFP signal than WT, similar to the ABM. There were no significant differences in the somatic compartment, with both mutants showing expression levels similar to the WT. For the GFP AIS signal: 2526.2 ± 239.1 AU, 947.4 ± 279.3 AU, and 195.3 ± 34.7 AU for the WT, MAPM, and ABM, respectively. For the GFP somatic signal: 835.8 ± 224.6 AU, 880.5 ± 163.5 AU, and 486.2 ± 93.2 AU for the WT, MAPM, and ABM, respectively. For the SA-CF640 AIS signal: 1792.1 ± 186.0 AU, 702.1 ± 205.7 AU, and 120.7 ± 18.7 AU for the WT, MAPM, and ABM, respectively. For the SA-CF640 somatic signal: 49.7 ± 7.7 AU, 38.8.4 ± 10.3 AU, and 46.8 ± 10.1 AU for the WT, MAPM, and ABM, respectively. C, Averaged line profile of SA-CF640 intensity along AIS (NF-186 staining was used as a guideline) for WT (black) and MAPM (red) channels. MAPM presents a distribution similar to WT, with smaller values of intensity due to its decreased abundance at the AIS. Line indicates mean values ± SEM. Inset, Normalized SA-CF640 maximum intensity along the AIS, for WT (black) and MAPM (red) channels. Each individual AIS line profile was normalized to its maximal value, and then all line profiles (WT, n = 49; or MAPM, n = 34) were averaged. Because each individual line profile had its maximum value at different position, the averaged normalized line shown does not reach 1 in any specific point.
Because some MAPM-transfected cells seemed to present a possibly more proximal distribution within the AIS (Fig. 2A), further analysis of Nav1.6 WT and MAPM channel localization was performed. Line profiles (2 pixels wide) were drawn along the AIS (using NF-186 labeling as a guideline for where the AIS was located) and the intensity of the surface-labeled Nav1.6 channels measured (Fig. 2C). No significant differences were detected regarding the length of the AIS (26.17 ± 1.55 μm, n = 49; 25.92 ± 2.12 μm, n = 34; and 27.67 ± 1.72 μm, n = 28; for WT, MAPM, and ABM, respectively; one-way ANOVA F(2,108) = 0.230, p = 0.795). As shown in Figure 2C (inset), the normalized intensities along the AIS for both WT and MAPM channels are superimposable, demonstrating that both channels present the same distribution along the AIS. Together, although fewer MAPM channels localize at the AIS, their overall distribution resembles that of WT channels.
Functional effects of Nav1.6 mutations
Because the function of the MAP1B binding mutant had not been previously characterized due to insufficient expression in ND7/23 cells (O'Brien et al., 2012), we performed voltage-clamp experiments on rat hippocampal cultures transfected with the appropriate Nav1.6 plasmid. All transfected Nav1.6 channels carried a point mutation (Y371S) imparting TTX resistance as illustrated in Figure 1, thus permitting separation of the transfected and endogenous Nav currents as previously described (Herzog et al., 2003; Akin et al., 2015).
As illustrated in Figure 3A (top right), and in contrast to the previous studies in ND7/23 cells (O'Brien et al., 2012), Nav1.6 MAPM generated inward currents in rHNs with a magnitude of −1901 ± 575 pA at 0 mV (n = 9). However, consistent with a localization or trafficking defect, this peak current was significantly smaller than that produced by the WT Nav1.6, −4088 ± 679 pA (n = 7). Interestingly, the ABM Nav1.6 mutant, which is incapable of binding Ankyrin and completely failed to localize to the AIS (Gasser et al., 2012; Akin et al., 2015), generated peak inward currents of −1820 ± 509 pA (n = 12), which are similar in magnitude to the MAPM mutant (one-way ANOVA, significant, F(2,25) = 4.23, p = 0.0263, with post hoc pairwise Tukey's tests, p = 0.053 and p = 0.994 for WT-MAPM and MAPM-ABM, respectively). Figure 3A (bar graph, bottom right) summarizes the average peak current density (pA/pF) observed with the WT, MAPM, and ABM channels. Average peak current densities of ABM and MAPM were significantly smaller than WT (145.14 ± 27.13 pA/pF, n = 7 cells; −59.59 ± 22.07 pA/pF, n = 9 cells; and −62.84 ± 17.72 pA/pF, n = 12 cells; for WT, MAPM, and ABM, respectively; one-way ANOVA, F(2,25) = 4.28, p = 0.0251, with post hoc pairwise Tukey's tests p = 0.0405 and 0.0363 for WT-MAPM and WT-ABM comparisons, respectively). While the activation midpoints were statistically indistinguishable between MAPM and ABM, the ABM midpoint was significantly different from that of the WT channel (Fig. 3B, left). In contrast, no significant differences were detected regarding the voltage dependence of inactivation between the two mutants versus WT (Fig. 3B, right). Voltage mid-activation points: −19.44 ± 2.39 mV (n = 7 cells), −14.27 ± 1.49 mV (n = 9 cells), and −12.01 ± 1.05 mV (n = 12 cells), for WT, MAPM, and ABM, respectively (one-way ANOVA, significant, F(2,25) = 6.195, p = 0.00653, with post hoc pairwise Tukey's tests, p = 0.067, p = 0.005, and p = 0.527 for WT-MAPM, WT-ABM, and ABM-WT comparisons, respectively). Voltage mid-inactivation points: −50.00 ± 1.80 mV (n = 7 cells), −46.21 ± 1.34 mV (n = 7 cells), and −48.03 ± 1.26 mV (n = 7 cells) for the WT, MAPM, and ABM, respectively (one-way ANOVA not significant, F(2,18) = 1.627, p = 0.2241).
Figure 3.

Functional characterization of Nav1.6 MAPM in rat hippocampal neurons. A, Representative current traces (at −70, −50, −30, −10, 0, 10, and 30 mV) from whole-cell voltage-clamp recordings in rHNs (DIV 6–7) expressing WT (top left), MAPM (top right), or ABM (bottom left) Nav1.6-GFP channels. Bottom right, Bar graph with the average peak current density values normalized to whole-cell capacitance (pA/pF) for WT, MAPM, and ABM transfected rHN cells. Error bars indicate mean ± SEM. Both mutants generated significantly smaller peak current densities than WT Nav1.6 (−145.14 ± 27.13 pA/pF, −59.59 ± 22.07 pA/pF, and −62.84 ± 17.72 pA/pF for WT, MAPM, and ABM, respectively). B, Voltage dependence of activation (left) and inactivation (right) for Nav1.6 WT (black filled squares), MAPM (gray filled triangles), and ABM (gray inverted triangles). Error bars indicate mean ± SEM. ABM showed a small but significant shift to more depolarized values of activation midpoint, compared with WT and MAPM Nav1.6 (−19.44 ± 2.39 mV, −14.27 ± 1.49 mV, and −12.01 ± 1.05 mV for WT, MAPM, and ABM, respectively). Voltage midpoints were −50.00 ± 1.80 mV, −46.21 ± 1.34 mV, and −48.03 ± 1.26 mV for the WT, MAPM, and ABM, respectively. C, Comparison of persistent current magnitude. Left, Current traces for Nav1.6 WT (black), MAPM (dark gray), and ABM (light gray) are shown, after normalization to the peak current. Inset, Enlargement of the region boxed with the dashed line. Right, Average percentage persistent current 100 ms after initial depolarization for WT, MAPM, and ABM transfected rHNs. Error bars indicate SEM. Both mutants consistently presented a significantly greater percentage of persistent current than WT (1.64 ± 0.45%, 11.06 ± 3.07%, and 11.13 ± 3.62% for WT, MAPM, and ABM, respectively. D, Comparison of WT currents in normal (black) and low (white) external Na+ concentration. Left, Average WT transient peak currents (pA). Right, Average percentage persistent current of WT transfected rHN. WT peak currents at 0 mV were of −2309 ± 477 pA in low Na+ concentration (56 mm) compared with −4088 ± 679 pA in normal external solution (112 mm). Remaining current after 100 ms of depolarization with the 56 mm Na+/NDMG solution was 2.4 ± 0.6% of peak current at 0 mV for WT compared with 1.6 ± 0.5% in normal Na+ (112 mm). Error bars indicate SEM. *p ≤ 0.05, n.s., nonsignificant.
One characteristic of Nav1.6 channels is a persistent current during prolonged depolarization, which can be up to 5% of the peak current (Rush et al., 2005). This prolonged current is thought to be due to reopening of Nav1.6 channels from the inactivated state (Chatelier et al., 2010). Interestingly, the MAPM and ABM mutants presented a significantly greater percentage of persistent current than WT, as measured by the ratio of the current remaining after 100 ms of depolarization to the peak current (Fig. 3C) (average percentage persistent current 100 ms after initial depolarization: 1.64 ± 0.45%, n = 10 cells, 11.06 ± 3.07%, n = 6 cells, and 11.13 ± 3.62%, n = 7 cells, for WT, MAPM, and ABM, respectively; one-way ANOVA was significant, F(2,20) = 5.95, p = 0.00938, with post hoc pairwise Tukey's tests p = 0.0286 and 0.0205 for WT-MAPM and WT-ABM comparisons, respectively). This difference was due to a decreased max peak current, without a change in the actual value of remaining current (remaining current after 100 ms of depolarization: −103.78 ± 39.70 pA, n = 10, −196.63 ± 79.66 pA, n = 6, and −141.10 ± 24.40 pA for WT, MAPM, and ABM; one-way ANOVA, not significant, F(2,20) = 0.906, p = 0.420). The increased percentage of persistent current and lack of AIS accumulation observed for the ABM and MAPM mutants suggest a link between location and function, with AIS and somatic Nav1.6 channels having distinct functional properties, with the persistent current being primarily generated in the somatic compartment. Together, the imaging and voltage-clamp data presented in Figures 2 and 3 indicate that almost two-thirds of WT whole-cell transient current is derived from channels localized within the AIS compartment in this expression system.
Transient WT peak currents are much larger than MAPM and could be poorly controlled relative to the smaller persistent current at 100 ms. In order to ensure that the relative difference in the percentage persistent current was not due to incomplete voltage control within the AIS, we performed voltage-clamp experiments with reduced external Na+ concentration, to decrease the transient peak currents of the WT. With the reduced external Na+ concentration (56 mm), we observed reduced WT peak currents with magnitudes similar to the MAPM average peak currents under normal external solutions (Fig. 3D, left). Importantly, there was no significant difference in the percentage persistent current (Fig. 3D, right). WT peak currents with low Na+ at 0 mV averaged −2309 ± 477 pA (n = 13) compared with −4088 ± 679 pA in normal external solution (Student's t test, p = 0.04, df = 18) and demonstrated 2.4 ± 0.6% persistent current at 0 mV compared with the 1.6 ± 0.5% persistent current observed in normal external Na+ (Student's t test, p = 0.39, df = 21). This result supports the idea that persistent current is generated in the soma rather than in the AIS and that our detected increased in percentage persistent current between MAPM versus WT (with normal external solution) is not due to an artifact induced by improper voltage control. In addition, the persistent current should be under voltage control even in normal external Na+ due to its small size and time separation from the voltage step. Therefore, the finding that its absolute magnitude is the same between neurons with and without AIS targeted channels, where the AIS localization represents two-thirds of total surface Nav, supports the proposed relationship between persistent current and somatic localization.
Vesicular trafficking of MAPM displays WT characteristics
Nav1.6 interaction with MAP1B implies microtubule-based trafficking of the channel to the cell surface; indeed, we have previously reported direct vesicular delivery of Nav1.6 channels to the AIS (Akin et al., 2015). Therefore, we determined whether impaired vesicular packaging and/or transport of MAPM could account for the reduction in AIS localization. rHN cultures were transfected with WT, MAPM, or ABM Nav1.6-GFP constructs and imaged 15–20 h later. After photobleaching small portions of the AIS to remove the fluorescence of channels already inserted into the plasma membrane or accumulated intracellularly, we observed vesicles traveling along the AIS in both anterograde and retrograde directions relative to the somatic compartment (Fig. 4A). Kymographs from the same regions are shown in Figure 4B. The number of vesicles per cell (each cell imaged for the same time) did not differ between constructs (Fig. 4C, left histogram). There was no difference in vesicle direction between WT, MAPM, and ABM channels (Fig. 4C, middle), or on the vesicle transport rates (Fig. 4C, right), which were characteristic of kinesin-based transport (Maday et al., 2014) and showed no statistically significant differences between the three channel constructs: number of vesicles/cell: 4.0 ± 0.46 (n = 21 cells), 3.31 ± 0.65 (n = 16 cells), and 3.56 ± 0.39 (n = 16 cells) vesicles/cell for the WT, MAPM, and ABM constructs (one-way ANOVA, not significant, F(2,50) = 0.500, p = 0.609); percentage of retrograde vesicles: 60.6 ± 7.7% (n = 21 cells), 68.9 ± 8.4% (n = 16 cells), and 77.5 ± 6.6% (n = 16 cells) for the WT, MAPM, and ABM, respectively (one-way ANOVA, not significant, F(2,50) = 1.255, p = 0.294); anterograde vesicle's velocity (in μm/s): 0.57 ± 0.05 (n = 38 vesicles), 0.46 ± 0.07 (n = 17 vesicles), and 0.52 ± 0.10 (n = 11 vesicles) for WT, MAPM, and ABM, respectively (one-way ANOVA, not significant, F(2,63) = 0.804, p = 0.452); retrograde vesicle's velocity (in μm/s): 0.60 ± 0.04 (n = 60 vesicles), 0.49 ± 0.05 (n = 34 vesicles), and 0.58 ± 0.05 (n = 46 vesicles) for WT, MAPM, and ABM, respectively (one-way ANOVA, not significant, F(2,137) = 1.454, p = 0.237).
To ensure that MAPM was indeed packaged into the same vesicles as the WT channel, WT Nav1.6-mCherry was cotransfected with the GFP tagged mutant constructs. Vesicles carrying both WT Nav1.6-mCherry and MAPM-GFP or ABM-GFP channels were detected (Fig. 4D, white arrows). A total of 21, 8, and 16 vesicles were observed carrying GFP and Cherry at the same time (from a total of 31, 28, and 25 vesicles observed from 8, 11, and 12 cells), for WT, MAPM, and ABM, respectively. Thus, both mutants are packaged into the same vesicles as the WT channel. Together, the data in Figure 4 indicate that defective vesicular packaging and transport do not play a role in the lack of MAPM targeting to the AIS.
The observation of vesicles carrying both MAPM and WT channels raised the question of whether the presence of the mutant in the vesicles could affect the delivery of the WT channel to the surface, and whether the WT channel could restore MAPM mutant targeting to the AIS. When rHN cultures were transfected with WT Nav1.6-LoopBAD-mCherry in the absence or presence of the MAPM Nav1.6-GFP construct (Fig. 5A), the Nav1.6 WT AIS surface labeling was not significantly affected: 519.8 ± 106.9 AU (n = 20 cells) and 364.2 ± 45.8 AU (n = 20 cells) for WT-LoopBad-mCherry alone and WT-LoopBad-mCherry + MAPM-GFP, respectively (Student' t test, not significant, p = 0.193, df = 26). However, the somatic surface labeling of Nav1.6 WT was significantly reduced: 121.3 ± 23.5 AU (n = 20 cells) and 68.1 ± 10.0 AU (n = 20 cells) for the WT-LoopBad-mCherry alone and WT-LoopBad-mCherry + MAPM-GFP, respectively (Student's t test, p = 0.0469, df = 26) (Fig. 5B), suggesting that the number of Nav1.6 channels on the somatic surface is limited/regulated, and that both channel variants are competing for these slots. When the experiment was repeated with surface labeling of the MAPM Nav1.6-LoopBAD-GFP construct, the same competition effect was observed (Fig. 5C). The somatic surface labeling and the small amount of AIS surface labeling of MAPM were decreased by the presence of the WT (Fig. 5D): for the SA-CF640 AIS signal, 58.2 ± 8.8 AU (n = 36 cells) and 31.5 ± 4.0 AU (n = 24 cells) for MAPM Nav1.6-LoopBad-GFP alone and MAPM Nav1.6-LoopBad-GFP + WT Nav1.6-mCherry, respectively (Student's t test, p = 0.008, df = 48); for the SA-CF640 soma signal, 36.4 ± 4.9 AU (n = 36 cells) and 24.2 ± 3.3 AU (n = 24 cells) for MAPM Nav1.6-LoopBad-GFP alone and MAPM Nav1.6-LoopBad-GFP + WT Nav1.6-mCherry, respectively (Student's t test, p = 0.0436, df = 56). These results indicate that expression and delivery of the WT Nav1.6 do not rescue, but further reduce, surface expression of MAPM channels at the AIS. Together, these results suggest that there are a limited number of Nav1.6 slots on the neuronal surface, and that WT channel may be more stable than MAPM once on the AIS surface.
Figure 5.

Nav channel expression is unaltered by WT or MAPM Nav1.6 transfection. A, Representative TIRF images of a DIV 7 rHN expressing Nav1.6-LoopBAD-mCherry WT in the absence (top) or presence (bottom) of Nav1.6-GFP MAPM. mCherry fluorescence is shown in the first column (yellow), surface labeling with SA-CF640 in the second column (magenta), and GFP fluorescence in the third column (cyan). Scale bars, 10 μm. B, Average values for SA-CF640 fluorescence of WT Nav1.6 in the AIS (top) or somatic (bottom) compartments. Error bars indicate mean ± SEM. The difference in AIS labeling was not significant. For the mean SA-CF640 AIS signal: 519.8 ± 106.9 AU for and 364.2 ± 45.8 AU for WT-LoopBad-mCherry alone and WT-LoopBad-mCherry + MAPM-GFP, respectively. The decreased surface expression of the WT on the soma in the presence of MAPM was significant. For SA-CF640 soma signal: 121.3 ± 23.5 AU and 68.1 ± 10.0 AU for the WT-LoopBad-mCherry alone and WT-LoopBad-mCherry + MAPM-GFP, respectively. C, Representative TIRF images of DIV 7 rHN expressing Nav1.6-LoopBAD-GFP MAPM in absence (top) or presence (bottom) of Nav1.6-mCherry WT. The GFP fluorescence is shown in the first column (cyan), surface labeling with SA-CF640 in the second (magenta), and the mCherry fluorescence in the third column (yellow). Scale bars, 10 μm. D, Average values of SA-CF640 fluorescence in the AIS (top) or somatic (bottom) compartments. Error bars indicate mean ± SEM. The differences in surface labeling in both the AIS and soma were significant. For the SA-CF640 AIS signal: 58.2 ± 8.8 AU and 31.5 ± 4.0 AU for MAPM Nav1.6-LoopBad-GFP alone and MAPM Nav1.6-LoopBad-GFP + WT Nav1.6-mCherry, respectively. For the SA-CF640 soma signal: 36.4 ± 4.9 AU and 24.2 ± 3.3 AU for MAPM Nav1.6-LoopBad-GFP alone and MAPM Nav1.6-LoopBad-GFP + WT Nav1.6-mCherry, respectively. E, Representative AIS maximum intensity Z projections from DIV 7 rHN cultures either untransfected, that is, endogenous (left), transfected with WT Nav1.6-GFP (middle) or MAPM Nav1.6-GFP (right). GFP (cyan) and PanNav immunolabeling staining (magenta) are shown. Scale bars, 5 μm. F, Average values of AIS PanNav staining of untransfected (endogenous), WT, or MAPM transfected cells. Error bars indicate mean ± SEM. *p ≤ 0.05, **p ≤ 0.01.
Furthermore, to determine whether Nav1.6 MAPM expression affected expression of endogenous Nav channels, immunocytochemistry was performed using a PanNav antibody in untransfected DIV 7 rHN cultures and cultures transfected with Nav1.6 WT or MAPM (Fig. 5E). Similar immunostaining was observed between untransfected (endogenous) and transfected cells, with no significant differences between conditions: 627.12 ± 54.52 (n = 49 AISs), 851.57 ± 96.64 (n = 32 AISs), and 638.56 ± 84.31 (n = 28 AISs), for untransfected, WT, and MAPM transfected cells, respectively (one-way ANOVA, not significant, F(2,105) = 2.588, p = 0.080). Thus, the data summarized in Figure 5F indicate that we are not overexpressing WT or MAPM, and that MAPM expression does not impair trafficking of endogenous Nav channels to the AIS.
Delivery of Nav1.6-MAPM to the AIS is not impaired
To determine whether differences in surface delivery itself are responsible for a lack of MAPM accumulation at the AIS, membrane insertion events were detected as previously described (Akin et al., 2015). In summary, WT or MAPM LoopBAD-GFP constructs were transfected into neurons; and after 18–36 h, biotinylated surface channels were blocked with Neutravidin for 5 min. After removal of the unbound Neutravidin, neurons were imaged for 30 min at 2.5 Hz in the presence of SA-CF640 to detect newly delivered channels. GFP signal of representative neurons transfected with WT or MAPM Nav1.6 channels is shown in Figure 6A. Kymographs corresponding to the orange lines (solid lines indicate soma; dashed lines indicate axon) are shown in Figure 6B. In the kymographs, the y axis indicates distance along the dashed line, and the x axis indicates time. Multiple insertion events are visualized as the sudden appearance of fluorescence (magenta lines), with the horizontal nature representing immediate channel immobilization upon surface delivery. Quantitative analysis from 9 cells for WT Nav1.6, and 15 cells for MAPM Nav1.6 resulted in the detection of a total of 148 (WT) and 346 (MAPM) insertion events (sum of insertion events at the somatic and AIS compartments). No significant differences were observed in the number of insertion events/μm2 between WT and MAPM in either the soma or AIS (Fig. 6C) (number of insertion events/μm2 into the soma: 0.032 ± 0.009, n = 9 cells, and 0.053 ± 0.013, n = 15 cells, for the WT and MAPM, respectively; Student's t test, p = 0.184, df = 22; number of insertion events/μm2 into the AIS: 0.100 ± 0.026, n = 9 cells, and 0.163 ± 0.035, n = 15 cells, for the WT and MAPM, respectively; Student's t test, p = 0.164, df = 22). These data indicate that MAPM is still preferentially delivered to the AIS, like WT Nav1.6 (with a ratio of insertion events in AIS/soma of 3.20 ± 0.49 for WT vs 4.03 ± 0.80 for MAPM, Student's t test, p = 0.392, df = 21).
While the straight horizontal lines in the kymographs of Figure 6B indicate that newly delivered channels are anchored immediately upon surface delivery, presumably to AnkyrinG, lateral diffusion of the MAPM out of the AIS might explain its lack of accumulation in this compartment. To further assess diffusion of the WT and MAPM channels within the AIS over different time scales, and without a preference for newly inserted channels, FRAP and single-particle tracking experiments were performed. For the FRAP experiments, rHNs transfected with WT or MAPM Nav1.6-LoopBAD-GFP were first labeled with SA-A594 to visualize the surface channel. Photobleaching and fluorescence recovery at the AIS are illustrated in Figure 7A (within the white square in the second panel) where recovery of the surface-labeled channels (magenta) was followed by imaging every 30 s for 28 min. As illustrated by the still-frame time points (Fig. 7A) and the summarized time courses (Fig. 7B), we observed little recovery of surface SA-A594 fluorescence corresponding to either construct (WT in black, MAPM in red). No significant differences were detected in the recovery of SA-A594 between WT and MAPM (Fig. 7B): percentage SA-A594 recovery after 28 min, 5.3 ± 5.6% for WT (n = 12 cells) versus 8.2 ± 1.3% for MAPM (n = 11 cells) (Student's t test, p = 0.090, df = 21). These data suggest that the few MAPM channels that do reside on the AIS surface have the same low lateral mobility as the WT channels that accumulate there.
Figure 7.

MAPM channels at the AIS are as immobile as WT. A, Representative spinning disk confocal images of the AIS of DIV 7 rHNs expressing either Nav1.6-LoopBAD-GFP WT or Nav1.6-LoopBAD-GFP MAPM. A small portion of the AIS was bleached (white square in second column), and recovery after photobleaching was monitored for 28 min. The SA-A594 surface labeling (magenta) before, immediately after (0 s), 60 s and 28 min after photobleaching are shown. Scale bar, 5 μm. B, Average SA-A594 surface labeling fluorescence recovery (normalized to the initial signal) of the WT (black) or MAPM (red). Images were acquired every 30 s to minimize photobleaching during the recovery. Lines indicate mean ± SEM. The percentage SA-A594 recovery after 28 min was 5.3 ± 5.6% for WT versus 8.2 ± 1.3% for MAPM. C, Representative TIRF GFP footprint images of rHNs expressing Nav1.6-LoopBAD-GFP WT (top) or Nav1.6-LoopBAD-GFP MAPM (bottom). Right panels, Images represent the area marked with white rectangles in the left panels. Top images represent the tracks obtained after tracking individual Nav1.6 channels in the AIS. Bottom images represent a maximum projection of the surface labeling using all the frames of the movie used for the tracking. A very low concentration of SA-CF640 (see Materials and Methods) was used to isolate single-channel molecules within the AIS. Scale bars: White, 10 μm; Black, 2.2 μm.
Cell surface mobility was next examined using our established single-particle tracking approaches (Akin et al., 2016; Sikora et al., 2017; Weron et al., 2017). Here, rHNs transfected with WT or MAPM channel were surface-labeled with a low concentration of SA-CF640 to achieve the low labeling density in the AIS necessary for single-particle tracking. ROIs corresponding to the AIS of transfected cells were digitally cropped, and individual channels were tracked with U-track algorithms (Akin et al., 2015, 2016; Sikora et al., 2017; Weron et al., 2017). Representative transfected cells are shown in Figure 7C (with GFP signal in cyan). Tracks of individual Nav1.6 channels derived from the ROI marked in white in the left-hand panels are shown to the right in black for WT and red for MAPM. The bottom row corresponds to a time compression of the movie of single SA-CF640 labeled surface channels imaged over 51 and 155 s, respectively. Note the perfect alignment of the compressed video image (bottom) and the single-channel tracks (top), with both showing restricted/confined diffusion of the WT and MAPM Nav1.6 channels, regardless of the ability of the channel to bind MAP1B. The diffusion coefficients obtained from the single-particle tracks were 0.0014 ± 0.0002 μm2/s (n = 569 tracks) and 0.0016 ± 0.0002 μm2/s (n = 248 tracks) for WT and MAPM, respectively (Student's t test not significant, p = 0.493, df = 815), which are one order of magnitude smaller than our previously reported Nav1.6 ABM channels' diffusion coefficient of 0.03 ± 0.03 μm2/s (Akin et al., 2015). This result agrees with the low diffusion seen in the kymographs of the insertion events (Fig. 6B) and the low recovery after photobleaching (Fig. 7A), and further indicates that both channels are stably anchored within the AIS plasma membrane over the shorter and longer time scales, and that AnkyrinG is still binding and anchoring channels at the AIS.
Inhibition of endocytosis restores stable AIS localization of MAPM
Results thus far indicate that MAPM Nav1.6 channels are delivered to, and anchored within, the AIS just as the WT channel. Nonetheless, without MAP1B binding capability, Nav1.6 does not concentrate within this critical neuronal compartment. To test whether enhanced endocytosis of MAPM at the AIS is responsible for its lack of accumulation, we examined the effect of the endocytosis inhibitor Dynasore on MAPM surface expression. rHNs transfected with the WT, MAPM, or ABM channel were treated with 100 μm Dynasore for 2 h, and surface channel was detected via SA-CF640 labeling. Immunolabeling with anti-NF-186 antibodies was used to detect the AIS. To avoid the reversible effect of the endocytosis blocker (Macia et al., 2006), 50 μm Dynasore was included during washes, incubations, and imaging on the microscope stage. Figure 8A shows representative TIRF images of rHNs transfected with Nav1.6-LoopBAD-GFP WT (left), MAPM (middle), or ABM (right) either untreated or incubated with 100 μm Dynasore. The GFP signal is shown in cyan (top row), NF-186 immunofluorescence is shown in yellow (middle row), and surface labeling with SA-CF640 signal is shown in magenta (bottom row). All images were processed and contrasted equally. The bottom row enlargements highlight the labeling within the AIS regions indicated by the white rectangles. Whereas Dynasore treatment did not affect either WT or ABM expression or localization, surface labeling of MAPM was increased by Dynasore specifically in the axonal compartment. Quantitative analysis of the fluorescence in the soma and AIS compartments with and without Dynasore treatment is summarized in Figure 8B. There were no significant effects on WT or ABM channel surface labeling in the soma and AIS. In contrast, MAPM demonstrated a much higher SA-CF640 signal in the AIS after Dynasore treatment (for the mean SA-CF640 AIS signal: 287.0 ± 49.4 AU for WTUntreated, 304.1 ± 51.2 AU for WTDynasore, 74.2 ± 17.7 AU for MAPMUntreated, 280.8 ± 58.7 AU for MAPMDynasore, 28.1 ± 3.8 AU for ABMUntreated, and 29.7 ± 4.8 AU for ABMDynasore; one-way ANOVA was significant, F(5,143) = 7.443, p = 3.11 × 10−6, with post hoc pairwise Tukey's tests, p = 0.9998, 0.0241, 9.698 × 10−4 and 0.0222, 0.0145, and 1 for WTUntreated-WTDynasore, WTUntreated-MAPMUntreated, WTDynasore-MAPMUntreated, MAPMUntreated-MAPMDynasore, WTUntreated-ABMUntreated, and ABMUntreated-ABMDynasore respectively). Most importantly, when comparing WTUntreated-MAPMDynasore, p = 1, indicating that Dynasore rescued the AIS expression of the MAPM. For the mean SA-CF640 soma signal: 60.33 ± 21.0 AU for WTUntreated, 51.8 ± 9.1 AU for WTDynasore, 41.0 ± 8.2 AU for MAPMUntreated, 41.0 ± 9.1 AU for MAPMDynasore, 39.1 ± 4.6 AU for ABMUntreated, and 30.7 ± 6.0 AU for ABMDynasore (one-way ANOVA, not significant, F(5,143) = 0.668, p = 0.648; n = 21, n = 44, n = 31, n = 24, n = 17, and n = 12 cells, respectively). These data indicate that the inhibition of endocytosis restores MAPM concentration at the AIS. Because the localization of the WT and ABM channels was not affected by Dynasore, it is unlikely that off-target actions, such as reduction of cholesterol in the plasma membrane (Preta et al., 2015), are responsible for restoring MAPM to the AIS.
Figure 8.

Nav1.6 MAPM is endocytosed from the AIS compartment. DIV 7 rHNs expressing WT, MAPM, or ABM were incubated with 100 μm Dynasore for 2 h and then surface-labeled and imaged under TIRF conditions. A, Representative images of transfected cells with Nav1.6-LoopBAD-GFP WT (left two panels), MAPM (middle two panels), or ABM (right two panels) untreated (left) or treated with Dynasore (right). Cyan represents GFP signals. Yellow represents axonal neurofascin immunolabeling. Magenta represents surface labeling with SA-CF640. Bottom row represents enlargements of the NF-186 and SA-CF640 signals corresponding to the AIS regions marked with a white rectangle. Scale bars: Full-size, 10 μm; Enlargements, 5 μm. B, Mean SA-CF640 fluorescence in AIS and somatic compartments under the different conditions. Dark color represents untreated. Light color represents Dynasore treatment. Error bars indicate mean ± SEM. *Significance. For the mean SA-CF640 AIS signal: 287.0 ± 49.4.1 AU for WTUntreated, 304.1 ± 51.2 AU for WTDynasore, 74.2 ± 17.7 AU for MAPMUntreated and 280.8 ± 58.7 AU for MAPMDynasore, 25.9 ± 5.4 AU for ABMUntreated and 33.1 ± 5.3 AU for ABMDynasore. For the mean SA-CF640 soma signal: 60.33 ± 21.0 AU for WTUntreated, 51.8 ± 9.1 AU for WTDynasore, 41.0 ± 8.2 AU for MAPMUntreated and 41.0 ± 9.1 AU for MAPMDynasore, 38.6 ± 7.2 AU for ABMUntreated, and 33.5 ± 6.9 AU for ABMDynasore.
In summary, while we originally hypothesized that MAP1B-dependent microtubule interaction would be involved in trafficking to the AIS, it now appears that Nav1.6 requires MAP1B binding to avoid internalization. Interestingly, anchoring to AnkyrinG, which limits Nav1.6 lateral diffusion on the cell surface, is not sufficient to prevent endocytosis of the Nav1.6 MAPM channel.
Discussion
The N terminus of Nav1.6 binds MAP1B, and substitution of residues 77–80 (Val-Ala-Val-Pro) by (Ala-Ala-Ala-Ala) prevents MAP1B binding. This MAP1B interaction is required for Nav1.6 cell surface expression in ND7/23 cells (O'Brien et al., 2012). The data presented here demonstrate that the Nav1.6 MAP1B mutant (MAPM) is trafficked efficiently to both AIS and somatic plasma membrane but fails to accumulate at the AIS even though it exhibits WT distribution on the somatic surface. While MAPM is delivered and inserted into the neuronal surface at the AIS (Fig. 6), it is specifically endocytosed from this compartment compared with the Nav1.6 WT channel (Fig. 8). This MAP1B-regulated, compartment-specific endocytosis results in decreased surface expression of MAPM only at the AIS (Fig. 2).
MAP1B is classically viewed as a microtubule binding protein that promotes microtubule polymerization (Takemura et al., 1992; Villarroel-Campos and Gonzalez-Billault, 2014). MAP1B has microtubule-binding domains and actin-binding domains, indicating that it can also play a structural role crosslinking microtubules and microfilaments (Pedrotti and Islam, 1996; Tögel et al., 1998). Indeed, MAP1B is implicated as a regulator of the actin cytoskeleton and dendritic spine morphology (Meixner et al., 2000; Tortosa et al., 2011; Yang et al., 2012). In addition, an increasing number of noncanonical functions of MAP1B unrelated to microtubule binding have been identified (Villarroel-Campos and Gonzalez-Billault, 2014). For example, MAP1B interacts with neurotransmitter receptors to regulate localization, function, and trafficking (Hanley et al., 1999; Eriksson et al., 2010; Gandini et al., 2014b; Kim et al., 2014). Similar to what we find with Nav1.6 at the AIS, MAP1B light chain 1 binding to 5-HT6 receptors promotes surface expression by reducing endocytosis (Kim et al., 2014). In addition, light chain 2 promotes the stabilization of Cav2.2 channels at the presynaptic membrane (Leenders et al., 2008), whereas light chain 1 promotes Cav2.2 proteasomal degradation (Gandini et al., 2014a,b).
Although there are no reports demonstrating endocytosis of Nav1.6 at the AIS, endocytosis of Nav channels has been investigated. Using CD4 chimeras containing Nav1.2 sequence Garrido et al. (2001) reported that an AP-2 binding dileucine motif (LI 1839–1840) in the cytoplasmic C terminus played a role in axonal Nav1.2 compartmentalization and its elimination from the somatodendritic compartment. AP-2 is one of the four members of a family of adaptor clathrin complexes. AP-2 specifically binds to either [D/E]xxxL[L/I] acidic dileucine or to Yxxφ motifs located in membrane proteins subject to clathrin-mediated endocytosis (Bonifacino and Traub, 2003). Interestingly, although such a dileucine motif is also present in Nav1.1 and Nav1.6, it does not play a role in the trafficking of CD4-Nav1.1 or CD4-Nav1.6 chimeras (Garrido et al., 2001). This difference was proposed to be due to different C terminus structures masking the exposure of the dileucine motif. Later, the same laboratory proposed that a CD4-Nav1.2 chimera was preferentially delivered to the somatodendritic compartment only to be removed by endocytosis, with a 19 aa motif in the cytoplasmic II-III loop required for internalization (residues 1010–1030) (Fache et al., 2004). It was proposed that AnkyrinG, which binds the nearby residues 1094–1102 (Gasser et al., 2012), traps the chimera at the AIS, blocking endocytosis and allowing AIS accumulation. In contrast, the MAPM Nav1.6 in the present study binds AnkyrinG and shows low diffusive behavior as the WT channel, but is internalized, indicating that AnkyrinG binding in cytoplasmic loop 2 is insufficient to block endocytosis at the AIS. While MAP1B binding to WT Nav1.6 may protect the channel from being endocytosed at the AIS, the molecular mechanism linking MAP1B binding and endocytosis is unknown. It is possible that MAP1B binding masks specific Nav1.6 domains required for internalization. For example, there is an unconfirmed AP-2 binding site, YGDI (Yxxφ motif), only 5 aa proximal to the VAVP motif. This AP-2 binding site (residues 69–72) may be blocked by MAP1B binding to residues 77–80. Another potential mechanism is that MAP1B binding blocks a ubiquitination site, which, if unmasked, leads to ubiquitination and removal of Nav1.6 from the neuronal surface followed by degradation in the proteasome. Interestingly, Nav1.6 contains two Nedd-4 ubiquitin ligase binding motifs, 551-PGS-553 and 1941-PSY-1945, which may play a role in modulation of Nav1.6 by p38 MAPK (Fotia et al., 2004; Gasser et al., 2010). Because of the lack of inhibitors specific to either of these pathways, and the lack of a 3D structure of the intracellular domains of mammalian Nav channels, it is difficult to predict whether MAP1B binding at the 77–80 position would mask either the AP-2 or Nedd-4 motifs and/or any specific ubiquitination site. It is also possible that the MAPM mutation simply induces a conformational change in the N-terminal domain (due to removal of proline) that is responsible for enhanced endocytosis due to the exposure of previously masked motifs. Without structural information on the Nav intracellular domains, it is difficult to distinguish between these or other mechanisms. However, the fact that MAP1B overexpression in ND7/23 cells enhances WT Nav1.6 current (O'Brien et al., 2012) supports the regulation of surface expression by a specific channel-MAP1B interaction.
Our data show that, although MAPM does not present a global trafficking defect in rHNs, it has a mislocalization phenotype due to enhanced AIS-specific endocytosis. This loss of AIS localization is linked to a functional change in the whole-cell current. Both MAPM and ABM mutants, which fail to localize at the AIS through different mechanisms, exhibit increased persistent current. While some publications have suggested that persistent current originates in the AIS (Astman et al., 2006), our data suggest that most of Nav1.6-specific persistent current is generated in the soma. Interestingly, direct AnkyrinG binding to Nav1.6 has been suggested to decrease Nav1.6 persistent current in tSA cells (Shirahata et al., 2006). Our data are consistent with AnkyrinG regulation of persistent current, for although MAPM is able to bind to AnkyrinG, most of the surface expressed channel is located on the soma, where there is little AnkyrinG (Kordeli et al., 1995). Interestingly, while we see a small, but significant, shift in the voltage dependence activation for ABM to more depolarized values relative to WT, no shift was observed for MAPM. Because a single aa AnkyrinG binding Nav1.6 mutant (E1100A) expressed in rHNs also showed no shift in the activation midpoint (Gasser et al., 2012), it appears that AIS localization does not have a major impact on voltage sensitivity. Rather, only the magnitude of persistent current is linked to neuronal compartment localization.
The AIS is an organized structure, characterized by enrichment of Kv and Nav channels, scaffolding proteins, cell adhesion molecules, and a highly ordered cytoskeleton (Huang and Rasband, 2018). Although it is a complex structure, AIS plasticity (changes in length and distance from the somatic compartment, which lead to change in neuronal excitability) has been observed following prolonged depolarization in certain pathological conditions and animal models of disease (Grubb and Burrone, 2010; Kuba et al., 2010; Yamada and Kuba, 2016; Hatch et al., 2017). AIS plasticity has been suggested to function as a negative feedback mechanism to maintain homeostasis of the neuronal circuit. This would require tight regulation of component assembly in the AIS. Key factors involved in upstream signaling leading to AIS remodeling include increased [Ca]i through L-type channels and activation of either calcineurin phosphatase or CDK5 kinase (Evans et al., 2013; Chand et al., 2015). Interestingly, MAP1B interactions are highly regulated via phosphorylation by casein kinase II, GSK3, and CDK5 (Villarroel-Campos and Gonzalez-Billault, 2014), whereas AnkyrinG-Nav channel interaction is modulated by casein kinase II activity (Bréchet et al., 2008). The AIS also controls neuronal polarity by regulating trafficking vesicle entry into the axon. Not all vesicles are allowed to enter the AIS, and MAP2 seems to play a role in defining the preaxonal filtering zone, regulating KIF1- versus KIF5-dependent cargo transport (Gumy et al., 2017). We now propose that MAP1B is an important regulator of both the organization and plasticity of the highly specialized AIS compartment.
The Nav1.6 motif required for MAP1B binding (VAVP) is not conserved within the Nav family. Only Nav1.6 contains this sequence, although all nine channels share a proline at the corresponding position. Nav1.1, with the sequence VSEP, did not bind MAP1B in the yeast two-hybrid experiments (O'Brien et al., 2012), suggesting that the negative charge in the third position blocks MAP1B binding. Nav1.3, Nav1.5, Nav1.7, and Nav1.8 also have glutamate at this position. Interestingly, the Nav1.2 channel sequence is VSVP. The data suggest that MAP1B binding and regulation of endocytosis could contribute to the localization of both Nav1.2 and Nav1.6 within the AIS (Hu et al., 2009), but direct evaluation of the interaction of MAP1B with Nav1.2 will be required to test this hypothesis.
Although Nav channels have been the focus of research for >50 years (Catterall, 2012), the molecular mechanisms involved in their distinctive subcellular localizations remain elusive. While MAP1B is most appreciated for its regulation of microtubule structure, recent studies indicate a major role in regulation of synaptic signaling proteins. Our study expands the noncanonical functions of MAP1B by linking the MAP1B binding domain of Nav1.6 to the maintenance of AIS structure and neuronal excitability.
Footnotes
This work was supported by National Institutes of Health Grant RO1 NS085142 to M.M.T., and Grant R01 NS034509 to M.H.M. We thank Ashley Leek, Emily Maverick, and Ben Johnson for critical review of the manuscript; and Dr. Diego Krapf for developing algorithms and codes used for single-particle tracking.
The authors declare no competing financial interests.
References
- Akin EJ, Solé L, Dib-Hajj SD, Waxman SG, Tamkun MM (2015) Preferential targeting of Nav1.6 voltage-gated Na+ channels to the axon initial segment during development. PLoS One 10:e0124397. 10.1371/journal.pone.0124397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akin EJ, Solé L, Johnson B, Beheiry ME, Masson JB, Krapf D, Tamkun MM (2016) Single-molecule imaging of Nav1.6 on the surface of hippocampal neurons reveals somatic nanoclusters. Biophys J 111:1235–1247. 10.1016/j.bpj.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astman N, Gutnick MJ, Fleidervish IA (2006) Persistent sodium current in layer 5 neocortical neurons is primarily generated in the proximal axon. J Neurosci 26:3465–3473. 10.1523/JNEUROSCI.4907-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem 72:395–447. 10.1146/annurev.biochem.72.121801.161800 [DOI] [PubMed] [Google Scholar]
- Bréchet A, Fache MP, Brachet A, Ferracci G, Baude A, Irondelle M, Pereira S, Leterrier C, Dargent B (2008) Protein kinase CK2 contributes to the organization of sodium channels in axonal membranes by regulating their interactions with ankyrin G. J Cell Biol 183:1101–1114. 10.1083/jcb.200805169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH (1995) Mutation of a new sodium channel gene, Scn8a, in the mouse mutant 'motor endplate disease.' Nat Genet 10:461–465. 10.1038/ng0895-461 [DOI] [PubMed] [Google Scholar]
- Catterall WA. (2012) Voltage-gated sodium channels at 60: structure, function and pathophysiology. J Physiol 590:2577–2589. 10.1113/jphysiol.2011.224204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chand AN, Galliano E, Chesters RA, Grubb MS (2015) A distinct subtype of dopaminergic interneuron displays inverted structural plasticity at the axon initial segment. J Neurosci 35:1573–1590. 10.1523/JNEUROSCI.3515-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatelier A, Zhao J, Bois P, Chahine M (2010) Biophysical characterisation of the persistent sodium current of the Nav1.6 neuronal sodium channel: a single-channel analysis. Pflugers Arch 460:77–86. 10.1007/s00424-010-0801-9 [DOI] [PubMed] [Google Scholar]
- Deutsch E, Weigel AV, Akin EJ, Fox P, Hansen G, Haberkorn CJ, Loftus R, Krapf D, Tamkun MM (2012) Kv2.1 cell surface clusters are insertion platforms for ion channel delivery to the plasma membrane. Mol Biol Cell 23:2917–2929. 10.1091/mbc.e12-01-0047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson M, Samuelsson H, Björklund S, Tortosa E, Avila J, Samuelsson EB, Benedikz E, Sundström E (2010) MAP1B binds to the NMDA receptor subunit NR3A and affects NR3A protein concentrations. Neurosci Lett 475:33–37. 10.1016/j.neulet.2010.03.039 [DOI] [PubMed] [Google Scholar]
- Evans MD, Sammons RP, Lebron S, Dumitrescu AS, Watkins TB, Uebele VN, Renger JJ, Grubb MS (2013) Calcineurin signaling mediates activity-dependent relocation of the axon initial segment. J Neurosci 33:6950–6963. 10.1523/JNEUROSCI.0277-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fache MP, Moussif A, Fernandes F, Giraud P, Garrido JJ, Dargent B (2004) Endocytotic elimination and domain-selective tethering constitute a potential mechanism of protein segregation at the axonal initial segment. J Cell Biol 166:571–578. 10.1083/jcb.200312155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotia AB, Ekberg J, Adams DJ, Cook DI, Poronnik P, Kumar S (2004) Regulation of neuronal voltage-gated sodium channels by the ubiquitin-protein ligases Nedd4 and Nedd4–2. J Biol Chem 279:28930–28935. 10.1074/jbc.M402820200 [DOI] [PubMed] [Google Scholar]
- Fox PD, Haberkorn CJ, Akin EJ, Seel PJ, Krapf D, Tamkun MM (2015) Induction of stable endoplasmic reticulum/plasma membrane junctions by Kv2.1 potassium channels. J Cell Sci 128:2096–2105. 10.1242/jcs.166009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuma G, Oguni H, Shirasaka Y, Watanabe K, Miyajima T, Yasumoto S, Ohfu M, Inoue T, Watanachai A, Kira R, Matsuo M, Muranaka H, Sofue F, Zhang B, Kaneko S, Mitsudome A, Hirose S (2004) Mutations of neuronal voltage-gated Na+ channel α1 subunit gene SCN1A in core severe myoclonic epilepsy in infancy (SMEI) and in borderline SMEI (SMEB). Epilepsia 45:140–148. 10.1111/j.0013-9580.2004.15103.x [DOI] [PubMed] [Google Scholar]
- Gandini MA, Henríquez DR, Grimaldo L, Sandoval A, Altier C, Zamponi GW, Felix R, González-Billault C (2014a) CaV2.2 channel cell surface expression is regulated by the light chain 1 (LC1) of the microtubule-associated protein B (MAP1B) via UBE2L3-mediated ubiquitination and degradation. Pflugers Arch 466:2113–2126. 10.1007/s00424-014-1476-4 [DOI] [PubMed] [Google Scholar]
- Gandini MA, Sandoval A, Zamponi GW, Felix R (2014b) The MAP1B-LC1/UBE2L3 complex catalyzes degradation of cell surface CaV2.2 channels. Channels 8:452–457. 10.4161/19336950.2014.949162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido JJ, Fernandes F, Giraud P, Mouret I, Pasqualini E, Fache MP, Jullien F, Dargent B (2001) Identification of an axonal determinant in the C-terminus of the sodium channel Nav1.2. EMBO J 20:5950–5961. 10.1093/emboj/20.21.5950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser A, Cheng X, Gilmore ES, Tyrrell L, Waxman SG, Dib-Hajj SD (2010) Two Nedd4-binding motifs underlie modulation of sodium channel Nav1.6 by p38 MAPK. J Biol Chem 285:26149–26161. 10.1074/jbc.M109.098681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser A, Ho TS, Cheng X, Chang KJ, Waxman SG, Rasband MN, Dib-Hajj SD (2012) An AnkyrinG-binding motif is necessary and sufficient for targeting Nav1.6 sodium channels to axon initial segments and nodes of Ranvier. J Neurosci 32:7232–7243. 10.1523/JNEUROSCI.5434-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Burrone J (2010) Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature 465:1070–1074. 10.1038/nature09160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumy LF, Katrukha EA, Grigoriev I, Jaarsma D, Kapitein LC, Akhmanova A, Hoogenraad CC (2017) MAP2 defines a pre-axonal filtering zone to regulate KIF1- versus KIF5-dependent cargo transport in sensory neurons. Neuron 94:347–362.e7. 10.1016/j.neuron.2017.03.046 [DOI] [PubMed] [Google Scholar]
- Hanley JG, Koulen P, Bedford F, Gordon-Weeks PR, Moss SJ (1999) The protein MAP-1b links GABA(C) receptors to the cytoskeleton at retinal synapses. Nature 397:66–69. 10.1038/16258 [DOI] [PubMed] [Google Scholar]
- Hatch RJ, Wei Y, Xia D, Götz J (2017) Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta Neuropathol 133:717–730. 10.1007/s00401-017-1674-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog RI, Cummins TR, Ghassemi F, Dib-Hajj SD, Waxman SG (2003) Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J Physiol 551:741–750. 10.1113/jphysiol.2003.047357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, Rasband MN (2018) Axon initial segments: structure, function, and disease. Ann N Y Acad Sci 1420:46–61. 10.1111/nyas.13718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Tian C, Li T, Yang M, Hou H, Shu Y (2009) Distinct contributions of Nav1.6 and Nav1.2 in action potential initiation and backpropagation. Nat Neurosci 12:996–1002. 10.1038/nn.2359 [DOI] [PubMed] [Google Scholar]
- Johnson B, Leek AN, Solé L, Maverick EE, Levine TP, Tamkun MM (2018) Kv2 potassium channels form endoplasmic reticulum/plasma membrane junctions via interaction with VAPA and VAPB. Proc Natl Acad Sci U S A 115:E7331–E7340. 10.1073/pnas.1805757115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Kim DH, Lee KH, Im SK, Hur EM, Chung KC, Rhim H (2014) Direct interaction and functional coupling between human 5-HT6 receptor and the light chain 1 subunit of the microtubule-associated protein 1B (MAP1B-LC1). PLoS One 9:e91402. 10.1371/journal.pone.0091402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH, Ilschner SU, Kampa BM, Williams SR, Ruben PC, Stuart GJ (2008) Action potential generation requires a high sodium channel density in the axon initial segment. Nat Neurosci 11:178–186. 10.1038/nn2040 [DOI] [PubMed] [Google Scholar]
- Kordeli E, Lambert S, Bennett V (1995) Ankyrin(G): a new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem 270:2352–2359. 10.1074/jbc.270.5.2352 [DOI] [PubMed] [Google Scholar]
- Kuba H, Oichi Y, Ohmori H (2010) Presynaptic activity regulates Na+ channel distribution at the axon initial segment. Nature 465:1075–1078. 10.1038/nature09087 [DOI] [PubMed] [Google Scholar]
- Lee JH, Gomora JC, Cribbs LL, Perez-Reyes E (1999) Nickel block of three cloned T-type calcium channels: low concentrations selectively block α1H. Biophys J 77:3034–3042. 10.1016/S0006-3495(99)77134-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenders AG, Lin L, Huang LD, Gerwin C, Lu PH, Sheng ZH (2008) The role of MAP1A light chain 2 in synaptic surface retention of CaV2.2 channels in hippocampal neurons. J Neurosci 28:11333–11346. 10.1523/JNEUROSCI.3078-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T (2006) Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 10:839–850. 10.1016/j.devcel.2006.04.002 [DOI] [PubMed] [Google Scholar]
- Maday S, Twelvetrees AE, Moughamian AJ, Holzbaur EL (2014) Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 84:292–309. 10.1016/j.neuron.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler MH, Helman G, Hammer MF, Fureman BE, Gaillard WD, Goldin AL, Hirose S, Ishii A, Kroner BL, Lossin C, Mefford HC, Parent JM, Patel M, Schreiber J, Stewart R, Whittemore V, Wilcox K, Wagnon JL, Pearl PL, Vanderver A, et al. (2016) SCN8A encephalopathy: research progress and prospects. Epilepsia 57:1027–1035. 10.1111/epi.13422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meixner A, Haverkamp S, Wässle H, Führer S, Thalhammer J, Kropf N, Bittner RE, Lassmann H, Wiche G, Propst F (2000) MAP1B is required for axon guidance and is involved in the development of the central and peripheral nervous system. J Cell Biol 151:1169–1178. 10.1083/jcb.151.6.1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien JE, Sharkey LM, Vallianatos CN, Han C, Blossom JC, Yu T, Waxman SG, Dib-Hajj SD, Meisler MH (2012) Interaction of voltage-gated sodium channel Nav1.6 (SCN8A) with microtubule-associated protein Map1b. J Biol Chem 287:18459–18466. 10.1074/jbc.M111.336024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuse K, Malik-Hall M, Baker MD, Poon WY, Kong H, Chao MV, Wood JN (2002) Annexin II light chain regulates sensory neuron-specific sodium channel expression. Nature 417:653–656. 10.1038/nature00781 [DOI] [PubMed] [Google Scholar]
- Pedrotti B, Islam K (1996) Dephosphorylated but not phosphorylated microtubule associated protein MAP1B binds to microfilaments. FEBS Lett 388:131–133. 10.1016/0014-5793(96)00520-0 [DOI] [PubMed] [Google Scholar]
- Poon WY, Malik-Hall M, Wood JN, Okuse K (2004) Identification of binding domains in the sodium channel NaV1.8 intracellular N-terminal region and annexin II light chain p11. FEBS Lett 558:114–118. 10.1016/S0014-5793(03)01512-6 [DOI] [PubMed] [Google Scholar]
- Preta G, Cronin JG, Sheldon IM (2015) Dynasore: not just a dynamin inhibitor. Cell Commun Signal 13:24. 10.1186/s12964-015-0102-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Dib-Hajj SD, Waxman SG (2005) Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. J Physiol 564:803–815. 10.1113/jphysiol.2005.083089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmiere PD, Weigle CM, Tamkun MM (2008) The Kv2.1 K+ channel targets to the axon initial segment of hippocampal and cortical neurons in culture and in situ. BMC Neurosci 9:112. 10.1186/1471-2202-9-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller KL, Krzemien DM, Yarowsky PJ, Krueger BK, Caldwell JH (1995) A novel, abundant sodium channel expressed in neurons and glia. J Neurosci 15:3231–3242. 10.1523/JNEUROSCI.15-05-03231.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharkey LM, Cheng X, Drews V, Buchner DA, Jones JM, Justice MJ, Waxman SG, Dib-Hajj SD, Meisler MH (2009) The ataxia3 mutation in the N-terminal cytoplasmic domain of sodium channel Nav1.6 disrupts intracellular trafficking. J Neurosci 29:2733–2741. 10.1523/JNEUROSCI.6026-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirahata E, Iwasaki H, Takagi M, Lin C, Bennett V, Okamura Y, Hayasaka K (2006) Ankyrin-G regulates inactivation gating of the neuronal sodium channel, Nav1.6. J Neurophysiol 96:1347–1357. 10.1152/jn.01264.2005 [DOI] [PubMed] [Google Scholar]
- Sikora G, Wylomanska A, Gajda J, Solé L, Akin EJ, Tamkun MM, Krapf D (2017) Elucidating distinct ion channel populations on the surface of hippocampal neurons via single-particle tracking recurrence analysis. Phys Rev E 6:062404. 10.1103/PhysRevE.96.062404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura R, Okabe S, Umeyama T, Kanai Y, Cowan NJ, Hirokawa N (1992) Increased microtubule stability and alpha tubulin acetylation in cells transfected with microtubule-associated proteins MAP1B, MAP2 or tau. J Cell Sci 103:953–964. [DOI] [PubMed] [Google Scholar]
- Tögel M, Wiche G, Propst F (1998) Novel features of the light chain of microtubule-associated protein MAP1B: microtubule stabilization, self interaction, actin filament binding, and regulation by the heavy chain. J Cell Biol 143:695–707. 10.1083/jcb.143.3.695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortosa E, Montenegro-Venegas C, Benoist M, Härtel S, González-Billault C, Esteban JA, Avila J (2011) Microtubule-associated protein 1B (MAP1B) is required for dendritic spine development and synaptic maturation. J Biol Chem 286:40638–40648. 10.1074/jbc.M111.271320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzoumaka E, Tischler AC, Sangameswaran L, Eglen RM, Hunter JC, Novakovic SD (2000) Differential distribution of the tetrodotoxin-sensitive rPN4/NaCh6/Scn8a sodium channel in the nervous system. J Neurosci Res 60:37–44. [DOI] [PubMed] [Google Scholar]
- Villarroel-Campos D, Gonzalez-Billault C (2014) The MAP1B case: an old MAP that is new again. Dev Neurobiol 74:953–971. 10.1002/dneu.22178 [DOI] [PubMed] [Google Scholar]
- Wagnon JL, Barker BS, Hounshell JA, Haaxma CA, Shealy A, Moss T, Parikh S, Messer RD, Patel MK, Meisler MH (2016) Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann Clin Transl Neurol 3:114–123. 10.1002/acn3.276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon JL, Barker BS, Ottolini M, Park Y, Volkheimer A, Valdez P, Swinkels ME, Patel MK, Meisler MH (2017) Loss-of-function variants of SCN8A in intellectual disability without seizures. Neurol Genet 3:e170. 10.1212/NXG.0000000000000170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weron A, Burnecki K, Akin EJ, Solé L, Balcerek M, Tamkun MM, Krapf D (2017) Ergodicity breaking on the neuronal surface emerges from random switching between diffusive states. Sci Rep 7:5404. 10.1038/s41598-017-05911-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker W, Faull R, Waldvogel H, Plumpton C, Burbidge S, Emson P, Clare J (1999) Localization of the type VI voltage-gated sodium channel protein in human CNS. Neuroreport 10:3703–3709. 10.1097/00001756-199911260-00044 [DOI] [PubMed] [Google Scholar]
- Yamada R, Kuba H (2016) Structural and functional plasticity at the axon initial segment. Front Cell Neurosci 10:250. 10.3389/fncel.2016.00250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Wu M, Xia P, Wang C, Yan P, Gao Q, Liu J, Wang H, Duan X, Yang X (2012) The role of microtubule-associated protein 1B in axonal growth and neuronal migration in the central nervous system. Neural Regen Res 7:842–848. 10.3969/j.issn.1673-5374.2012.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin K, Baillie GJ, Vetter I (2016) Neuronal cell lines as model dorsal root ganglion neurons: a transcriptomic comparison. Mol Pain 12:1–17. 10.1177/1744806916646111 [DOI] [PMC free article] [PubMed] [Google Scholar]