

Abstract
Antagonism of nicotinic acetylcholine receptors (nAChRs) in the medial habenula (MHb) or interpeduncular nucleus (IPN) triggers withdrawal-like behaviors in mice chronically exposed to nicotine, implying that nicotine dependence involves the sensitization of nicotinic signaling. Identification of receptor and/or neurophysiological mechanisms underlying this sensitization is important, as it could promote novel therapeutic strategies to reduce tobacco use. Using an approach involving photoactivatable nicotine, we previously demonstrated that chronic nicotine (cNIC) potently enhances nAChR function in dendrites of MHb neurons. However, whether cNIC modulates downstream components of the habenulo-interpeduncular (Hb-IP) circuit is unknown. In this study, cNIC-mediated changes to Hb-IP nAChR function were examined in mouse (male and female) brain slices using molecular, electrophysiological, and optical techniques. cNIC enhanced action potential firing and modified spike waveform characteristics in MHb neurons. Nicotine uncaging revealed nAChR functional enhancement by cNIC on proximal axonal membranes. Similarly, nAChR-driven glutamate release from MHb axons was enhanced by cNIC. In IPN, the target structure of MHb axons, neuronal morphology, and nAChR expression is complex, with stronger nAChR function in the rostral subnucleus [rostral IPN (IPR)]. As in MHb, cNIC induced strong upregulation of nAChR function in IPN neurons. This, coupled with cNIC-enhanced nicotine-stimulated glutamate release, was associated with stronger depolarization responses to brief (1 ms) nicotine uncaging adjacent to IPR neurons. Together, these results indicate that chronic exposure to nicotine dramatically alters nicotinic cholinergic signaling and cell excitability in Hb-IP circuits, a key pathway involved in nicotine dependence.
SIGNIFICANCE STATEMENT This study uncovers several neuropharmacological alterations following chronic exposure to nicotine in a key brain circuit involved in nicotine dependence. These results suggest that smokers or regular users of electronic nicotine delivery systems (i.e., “e-cigarettes”) likely undergo sensitization of cholinergic circuitry in the Hb-IP system. Reducing the activity of Hb-IP nAChRs, either volitionally during smoking cessation or inadvertently via receptor desensitization during nicotine intake, may be a key trigger of withdrawal in nicotine dependence. Escalation of nicotine intake in smokers, or tolerance, may involve stimulation of these sensitized cholinergic pathways. Smoking cessation therapeutics are only marginally effective, and by identifying cellular/receptor mechanisms of nicotine dependence, our results take a step toward improved therapeutic approaches for this disorder.
Keywords: 2-photon, acetylcholine, electrophysiology, glutamate, nicotine, nicotinic
Introduction
Despite widespread appreciation for their harmful actions, tobacco product use continues to exert an enormous toll on public health. Nicotine, a high-affinity agonist of nicotinic acetylcholine receptors (nAChRs) found naturally in tobacco, is the key psychoactive agent responsible for physical and psychological dependence on these products. Most smokers make multiple attempts at cessation, but withdrawal and subsequent relapse to nicotine use is the typical end result of a quit attempt. Despite being marketed as a cessation aid, electronic nicotine delivery systems (“e-cigarettes”) promote relapse to smoking (Young-Wolff et al., 2018). Withdrawal involves physical, affective, and cognitive disturbances in the hours and days following nicotine abstinence (Benowitz, 2009). Patients report symptoms such as perspiration, depression, anxiety, and a decline in attentional performance (Benowitz, 2008). Smoking cessation drugs, including varenicline and bupropion, are only marginally effective at fostering cessation (Caponnetto et al., 2012).
Aversive nicotine withdrawal symptoms may be required to produce escalated intake of nicotine (George et al., 2007; Gilpin et al., 2014), the latter being a cardinal feature of tobacco addiction. A significant effort among preclinical researchers has begun to identify the mechanistic basis for this aversive withdrawal response. β4 nAChR subunit knock-out mice exhibit decreased nicotine withdrawal behaviors (Salas et al., 2004). The conspicuous and selective expression of β4 in the habenulo-interpeduncular (Hb-IP) pathway (Marks et al., 1992) fueled speculation that this system played a role in nicotine dependence (Salas et al., 2004). This was later confirmed when blockade of nAChRs in the Hb-IP pathway precipitated nicotine withdrawal in mice chronically exposed to nicotine (Salas et al., 2009). Elegant subsequent work suggested that the Hb-IP pathway works to limit nicotine intake through α5-containing nAChRs (Fowler et al., 2011). Tapper and colleagues later demonstrated that optical activation of GABAergic Gad2+ neurons in the interpeduncular nucleus (IPN) is sufficient to reproduce withdrawal-like behavior (Zhao-Shea et al., 2013), but there is clear evidence that Gad2+ neurons are only a subset of all IPN neurons (Hsu et al., 2013; Shih et al., 2014; Ables et al., 2017; Morton et al., 2018).
Human brain-imaging studies have confirmed that smoking leads to the upregulation of high-affinity nAChRs, which is sustained for 3–4 weeks following the discontinuation of nicotine exposure (Mamede et al., 2007; Mukhin et al., 2008; Cosgrove et al., 2009). Blockade of nAChRs in the medial habenula (MHb) or IPN triggers withdrawal-like behavior in chronic nicotine (cNIC)-treated mice (Salas et al., 2009), implying that prolonged nicotine exposure sensitizes nAChRs directly and/or augments the downstream cellular effect of their activation. Despite the elegant circuitry work cited above, the nAChR and/or cellular sensitization mechanisms in MHb/IPN at play in nicotine dependence have not been identified. Moreover, how such sensitization maps onto the diverse cell types found in IPN (Zhao-Shea et al., 2015; Ables et al., 2017) is not at all clear. Identifying these mechanisms is a high priority, as this could promote discovery of improved tobacco cessation strategies. Using a combination of optical, physiological, and molecular approaches, we now show that chronic nicotine exposure enhances nAChR function and cellular excitability at multiple locations in the Hb-IP circuit.
Materials and Methods
Materials.
AAV5.hSyn.DIO.hM4D(Gi)-mCherry.WPRE.hGH was obtained from Addgene. (−)-Nicotine hydrogen tartrate salt (nicotine) was obtained from Glentham Life Sciences. Acetylcholine chloride (ACh), picrotoxin (PTX), and atropine sulfate (atropine) were obtained from Sigma-Aldrich. CGP 55845 (CGP), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX), d-AP5, donepezil hydrochloride (donepezil), tetrodotoxin (TTX), and QX314 chloride (QX314) were obtained from Tocris Bioscience. Photoactivatable nicotine (PA-Nic) was synthesized as previously described (Banala et al., 2018) and provided by the laboratory of Luke Lavis [Janelia Research Campus, Howard Hughes Medical Institute (HHMI)].
Mice.
All experimental protocols involving mice were approved by the Institutional Animal Care and Use Committee at Northwestern University (protocol #IS00003604). Procedures also followed the guidelines for the care and use of animals provided by the National Institutes of Health Office of Laboratory Animal Welfare. All efforts were made to minimize distress and suffering during experimental procedures, including during the use of anesthesia. Mice were housed at 22°C on a 12 h light/dark cycle with food and water available ad libitum. Mice were weaned on postnatal day 21 and housed with same-sex littermates. A tail sample was taken from each mouse for genotyping via PCR. The following mouse strains were obtained from The Jackson Laboratory: vGluT2-IRES-Cre (catalog #016963); Gad2-IRES-Cre (catalog #010802); Pvalb-tdTomato (tdT; catalog #027395); Sst-IRES-Cre (catalog #013044); Ai14 (catalog #007908); and C57BL/6J (catalog #000664). Mice expressing tdTomato in a Cre-dependent manner (vGluT2-IRES-Cre::Ai14, Gad2-IRES-Cre::Ai14) were obtained by crossing mice heterozygous for each mutation, which produced ∼25% double-heterozygous progeny. To fluorescently mark Sst+ neurons in IPN for targeted patch-clamp recordings, Sst-IRES-Cre mice were microinjected with AAV5.hSyn.DIO.hM4D(Gi)-mCherry in IPN. Male and female mice (age, 6–24 weeks) were used in approximately equal numbers.
Chronic nicotine treatment.
Mice were chronically exposed to nicotine either via their drinking water (Fig. 1; and see Figs. 3a–i, 4c–l) or an osmotic minipump (see Figs. 3j–m, 9). For data in Fig. 2f–h some mice were treated with nicotine via drinking water and some via minipumps. Previous work (Salas et al., 2009) suggests that these two methods are equivalent for the production of nicotine dependence in mice. Mice were treated with nicotine via drinking water as previously described (Banala et al., 2018), with minor modifications. Nicotine hydrogen tartrate or l-tartaric acid (control group) was dissolved in tap water, pH 7.0, supplemented with saccharin sodium (3 mg/ml) to mask the bitter taste of nicotine. The following treatment schedule was used for nicotine (reported as nicotine-free base) and tartaric acid, respectively (in μg/ml): days 1–2 (50, 75), days 3–4 (100, 150), day 5 and beyond (200, 300). The latter doses were maintained by replacing drinking water solutions every 2–3 d, and mice were treated for at least 28 d before experimentation. Mice were exposed to nicotine via minipumps as we described previously (Shih et al., 2014). Briefly, mice were implanted with primed osmotic minipumps (model 2004; Alzet) filled with sterile saline or (−)nicotine hydrogen-tartrate salt dissolved in sterile saline. Nicotine (free base) was delivered at a rate of 12 mg/kg/d. Mice were treated for 14–21 d before experimentation. We previously validated that osmotic minipump and nicotine drinking water result in identical nAChR upregulation in MHb neurons (Banala et al., 2018), supporting our approach using different treatment methods in the same study.
Figure 1.

Chronic nicotine exposure alters spontaneous action potential firing in MHb neurons. a, Representative cell-attached firing traces for MHb neurons from control and cNIC-treated mice. b, Summary data (control: n = 11 cells, n = 3 male mice; cNIC-treated: n = 10 cells, n = 4 male mice) of cell-attached firing in MHb neurons for control and cNIC-treated mice. c, Representative spontaneous action potentials for whole-cell patch-clamped MHb neurons for control and cNIC-treated mice, illustrating features quantified in subsequent panels. d, Representative spontaneous action potential phase plots for MHb neurons from control and cNIC-treated mice. e, Summary resting membrane potential data (control: n = 34 cells, n = 5 male mice; cNIC-treated: n = 31 cells, n = 7 male mice; the same mice were used for data in f–j) for MHb neurons from control and cNIC-treated mice. f, Summary action potential amplitude data (control: n = 32 cells; cNIC-treated: n = 30 cells) for MHb neurons from control and cNIC-treated mice. g, Summary action half-width data (control: n = 32 cells; cNIC-treated: n = 30 cells) for MHb neurons from control and cNIC-treated mice. h, Summary action potential threshold data (control: n = 32 cells; cNIC-treated: n = 29 cells) for MHb neurons from control and cNIC-treated mice. i, Summary action potential maximum rise slope data (control: n = 32 cells; cNIC-treated: n = 31 cells) for MHb neurons from control and cNIC-treated mice. j, Summary action potential maximum decay slope data (control: n = 32 cells; nicotine-treated: n = 31 cells) for MHb neurons from control and cNIC-treated mice.
Figure 3.

Chronic nicotine enhances nicotine-stimulated glutamate release in IPN. a, Representative IPN neuron voltage-clamp recordings from mice treated with control or chronic nicotine. Recordings show Spontaneous excitatory postsynaptic current (sEPSCs) during superfusion of the slice with 0.06 μm nicotine. Insets show exploded view of example sEPSCs. b, sEPSC interevent interval cumulative distribution for a representative neuron from a control mouse before and after nicotine (0.06 μm) superfusion. c, sEPSC interevent interval cumulative distribution for a representative neuron from a cNIC-treated mouse before and after nicotine (0.06 μm) superfusion. d, sEPSC amplitude cumulative distribution for a representative neuron from a control mouse before and after nicotine (0.06 μm) superfusion. e, sEPSC amplitude cumulative distribution for a representative neuron from a cNIC-treated mouse before and after nicotine (0.06 μm) superfusion. f, Summary plots of sEPSC interevent interval for IPN neurons from control [0.03 μm, n = 6 cells; 0.06 μm, n = 5 cells; 0.12 μm, n = 5 cells; n = 7 mice (6 male/1 female)] and cNIC-treated [0.03 μm, n = 7 cells; 0.06 μm, n = 7 cells; 0.12 μm, n = 8 cells; n = 9 mice (5 male/4 female)] mice before and after superfusion of the slice with the indicated nicotine concentration. p Values (Wilcoxon matched-pairs signed rank tests) are shown for each group (blue, control mice; red, cNIC mice). The cells/mice used were also used to derive data in g, h, and i. g, Summary plots of sEPSC amplitude for IPN neurons from control and cNIC-treated mice before and after superfusion of the slice with the indicated nicotine concentration. p Values (Wilcoxon matched-pairs signed rank tests) are shown for each group (blue, control mice; red, cNIC mice). h, Summary baseline (no nicotine superfusion) sEPSC interevent interval data for all IPN recordings from control and cNIC-treated mice. p Value, Mann–Whitney test. i, Summary baseline (no nicotine superfusion) sEPSC amplitude data for all IPN recordings from control and cNIC-treated mice. p Value, Mann–Whitney test. j, Electrically eEPSCs in a voltage-clamped IPN neuron are sensitive to synaptic blockers (10 μm NBQX, 50 μm d-AP5). Inset, Recording configuration is shown for data in j through m. k, Superfusion of 0.12 μm nicotine enhances evoked glutamatergic transmission in IPN neurons. Inset, Summary plots of eEPSC amplitude before/after nicotine superfusion [n = 5 cells; n = 3 mice (2 male/1 female)]. p Value, Paired t test. l, Superfusion of 0.12 μm nicotine modifies paired-pulse facilitation in IPN neurons. Inset, Summary plots of paired-pulse eEPSC amplitude ratio (i.e., PPR) before/after nicotine superfusion [n = 5 cells; n = 4 mice (2 male/2 female)]. p Value, Paired t test. m, Selective modulation of paired-pulse facilitation by low nicotine concentrations in IPN neurons from cNIC-treated mice. Summary plots are shown for PPR measurements in IPN neurons of control [0.03 μm, n = 5 cells; 0.06 μm, n = 5 cells; 0.12 μm, n = 6 cells; n = 8 mice (5 male/3 female)] or cNIC-treated [0.03 μm, n = 7 cells; 0.06 μm, n = 6 cells; 0.12 μm, n = 7 cells; n = 9 mice (6 male/3 female)] mice before and after superfusion of the slice with the indicated nicotine concentration. *p < 0.05 (paired t test).
Figure 4.

Nicotine uncaging reveals nAChR functional upregulation in IPN neurons following chronic nicotine. a, PA-Nic was used to uncage nicotine with laser flash photolysis adjacent to IPN neurons in coronal brain slices of control and cNIC-treated mice. b, Variable IPN neuron morphology. Representative 2PLSM images of IPN neurons with complex (left image) and sparse (middle image) dendritic arbors are shown. Some neurons (middle image, boxed area exploded view in right image) have clear dendritic spines. c, Representative nicotine uncaging (50 ms, 2 mW, perisomatic stimulus) responses in an IPN neuron of a control and cNIC-treated mouse. Inset shows exploded view of the initial uncaging event. d, Summary data for all initial/first nicotine uncaging responses in IPN neurons of control [n = 11 cells; n = 3 mice (1 male/2 female)] and cNIC-treated [n = 11 cells; n = 4 mice (1 male/3 female)] mice. Cells/mice used were also used to derive data in c, e, f, and g. p Value, Unpaired t test. e, Summary time-series data for repeated (2 min interstimulus interval) nicotine uncaging responses in IPN neurons of control and cNIC-treated mice. Data at 0 min are the same data as in d but are replotted for clarity. Summary data from e for control (f) and cNIC-treated (g) mice are replotted on a normalized scale with individual cells shown in gray. h, Representative nicotine uncaging (50 ms, 2 mW; perisomatic stimulus) responses in IPN neurons from naive mice are shown for slices acutely treated with control ACSF or donepezil (1 μm; superfusion). Inset shows exploded view of the initial uncaging event. i, Summary data for all initial/first nicotine uncaging responses in control [n = 8 cells, 3 mice (2 male/1 female)] and donepezil-treated [n = 12 cells, 5 mice (3 male/2 female)] IPN neurons. p Value, Unpaired t test. j, Summary time-series data for repeated (2 min interstimulus interval) nicotine uncaging responses in control and donepezil-treated IPN neurons. Data at 0 min are the same data as in i but are replotted for clarity. k, l, Summary data from j for control (k) and donepezil-treated (l) slices are replotted on a normalized scale with individual cells shown in gray. m, Summary data for all initial/first nicotine uncaging responses in IPN neurons from cNIC-treated mice, where slices were either exposed to ACSF [control; n = 13 cells, 2 mice (1 male/1 female)] or donepezil [1 μm, superfusion; n = 11 cells, 2 mice (1 male/1 female)]. p Value, Unpaired t test.
Figure 9.
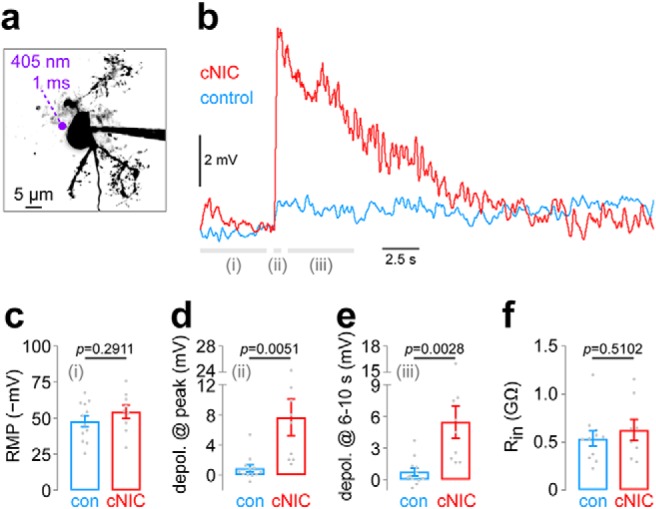
Chronic nicotine enhances IPR neuron excitability. a, Representative IPR neuron and perisomatic photolysis spot location. b, An averaged [control: n = 12 cells, n = 4 mice (2 male/2 female); cNIC: n = 9 cells, n = 6 mice (3 male/3 female)] current-clamp recording trace is shown in IPR neurons from control and cNIC-treated mice. PA-Nic (100 μm) was superfused, and photolysis (1 ms flash, 405 nm, 2 mW) was executed at a perisomatic location. Data from time periods i, ii, and iii are shown in c, d, and e, respectively. c, Summary plot of mean resting membrane potential during time period i (from −5 to −0.5 s before flash onset; see b) is shown for control and cNIC-treated neurons (see b for cell/mouse numbers). p Value, Unpaired t test. d, Summary plot showing the mean membrane potential change during time period ii (from 0.0 to +0.5 s after flash onset; see b) for control and cNIC-treated neurons. p Value, Unpaired t test. e, Summary plot showing the mean membrane potential change during time period iii (from +1.0 to +5.0 s after flash onset; see b) for control and cNIC-treated neurons. p Value, Unpaired t test. f, Summary plot showing input resistance for control and cNIC-treated IPR neurons. See b for cell/mouse numbers. p Value, Unpaired t test.
Figure 2.

nAChR functional upregulation in axons of MHb neurons. a, Representative 2PLSM image of a patch-clamped MHb neuron. b, 3D reconstruction of the neuron shown in a. Inset shows exploded view of reconstructed dendritic arbor. c, Sholl analysis for MHb neurons. For n = 24 MHb neurons (n = 17 mice [16 male/1 female]), morphology was reconstructed in 3D, and the number of Sholl intersections is plotted at each Sholl radius (1 μm step size). Shading indicates the 95% confidence interval. d, 3D reconstruction of a different MHb neuron with an intact axon. e, Nicotine uncaging along MHb neuron axons. A representative 2PLSM image of an MHb neuron with intact axon is shown, including approximate positions where PA-Nic (50 μm) laser flash photolysis was executed adjacent to the axonal membrane. f, Representative nAChR currents following nicotine uncaging along the axon of an MHb neuron from a control/cNIC-treated mouse. g, Summary nicotine uncaging-evoked current amplitudes for MHb neurons at the indicated distance from the soma along the axon (control: n = 7 cells, n = 4 mice [9 male/0 female]; cNIC-treated: n = 7 cells, n = 5 mice [5 male/0 female]). h, Input resistance in control and cNIC-treated MHb neurons (control: n = 11 cells, n = 4 mice [2 male/2 female]; cNIC-treated: n = 15 cells, n = 4 mice [4 male/0 female]).
Stereotaxic surgery.
Male and female mice were used for surgery starting at 8 weeks of age. Mice were initially anesthetized with an intraperitoneal injection of a ketamine/xylazine mixture (120 mg/kg ketamine, 16 mg/kg xylazine). Mice were given additional “boost” injections of ketamine (100 mg/kg, i.p.) as needed. Alternatively, some mice were anesthetized with isoflurane, as follows: 3% (flow rate 500 ml/min) for induction and 1.5% (36 ml/min) for maintenance. Mice were secured into a stereotaxic frame and a small incision was made at the top of the head to expose the skull. Coordinates (unilateral) used for IPN injections were as follows (relative to bregma, in mm): mediolateral, +0.01 (or −0.01); anteroposterior (A/P), −3.5; and dorsoventral (D/V), −4.7. Exact coordinates were adjusted to account for slight differences in the head sizes of individual mice: the bregma/lambda distance measured for each mouse was divided by the reported bregma/lambda distance for C57 mice (4.21), then was multiplied by the A/P coordinate. The injection needle was slowly lowered through the drilled hole to the D/V coordinate. For adeno-associated viruses (AAVs), 500 nl of virus was infused at a rate of 50 nl/min. For all stereotaxic injections, the injection needle was left in place for 10 min after the infusion ended before slowly retracting the needle. Sutures were used to close the incision. At the conclusion of the surgery, mice were given ketoprofen (5 mg/kg, s.c.), and placed in a recovery cage, kept warm, and observed until they were ambulatory. Mice were single housed following virus injection surgery, and were given at least 14 d to recover and for the virus to express before beginning experimental procedures. For electrophysiology experiments, accurate targeting of IPN was determined via direct visualization of fluorescent neurons in brain slices during recordings.
mRNA in situ hybridization and expression analysis.
Mice were deeply anesthetized with Euthasol (sodium pentobarbital, 100 mg/kg; sodium phenytoin, 12.82 mg/kg) and decapitated. Brains were quickly removed on ice, snap frozen, and embedded in cryoembedding medium (O.C.T.). Brains were sectioned on a cryostat (model CM3050, Leica) into 20 μm sections, and sections were adhered to Superfrost Plus slides, kept at −20°C to dry for 60 min, and stored at −80°C until use. Sections were fixed with 4% paraformaldehyde and processed for RNAscope [Advanced Cell Diagnostics (ACD)] multichannel fluorescence in situ hybridization (FISH) according to the manufacturer manual for Multiplex assays. Sections were mounted with ProLong Gold Antifade Mountant with DAPI (Thermo Fisher Scientific). Probes for the detection of specific targets (Chrna2, Chrna5, Chrnb2, Chrnb4, Chrm3, Chrm5, Gad2, Slc17a6, Sst, Pvalb, Nacho/Tmem35a) were purchased from ACD (https://acdbio.com/). The probe targeting Nacho/Tmem35a (Gu et al., 2016; Kennedy et al., 2016; Matta et al., 2017) was designed de novo for this study in collaboration with ACD.
Sections were imaged on a Nikon A1 Confocal Microscope according to the following parameters: 1024 × 1024 pixels, ∼200 nm/pixel, 20× 0.75 numerical aperture (NA) objective. Nikon system images were processed with custom scripts in ImageJ (NIH). All images to be used for FISH quantification were acquired and processed in the same manner. FISH quantification used the “fluorescence coverage (%)” method (Wallace et al., 2017; Yan et al., 2018), which reports the fraction of fluorescent pixels to total pixels in a cellular region of interest (ROI). An ImageJ script used DAPI staining to locate nuclei for automated and unbiased creation of cellular ROIs. The DAPI image was filtered with a Gaussian blur filter (σ = 3), thresholded (ImageJ “default” threshold), and the thresholded nuclei were dilated slightly [MorphoLibJ (ImageJ) dilation filter; disc, radius = 2] to allow the capture of the RNA fluorescence signal just outside the nucleus but still presumably within the cell. A watershed algorithm was then applied to the filtered, binary DAPI image to isolate/separate adjacent nuclei. Finally, ROIs were detected in ImageJ (Analyze Particles algorithm; size = 20 to infinity; circularity = 0.5–1.0) and saved for application to fluorescence channel images. FISH channel images were each processed as follows: Gaussian blur filter (σ = 1); Mexican Hat filter (radius = 2); and threshold (Otsu algorithm). ROIs from the DAPI image routine were then applied to the filtered/thresholded FISH channel and a raw “% coverage” value was derived for each ROI. For each channel, these raw % coverage values for each ROI were then scaled to the single ROI in the dataset with the highest % coverage. This normalization step accounted for differences in probe performance and target gene expression levels. Using these transformed/normalized datasets, a cutoff value of 10% “normalized percentage coverage” was uniformly assigned to each distribution. Scatter plots in Figures 6 and 7 show this cutoff for each channel as a horizontal and vertical line at 10% “normalized percentage coverage.” Exactly n = 3 mice were sampled for each condition, and 2 images of the rostral IPN (IPR) subnucleus were analyzed per mouse.
Figure 6.

Cholinergic components in rostral IPN subnucleus. a, A Dodt contrast image of the IPN is shown with patch-clamp electrode tip in the IPR. b, Representative traces and summary data for ACh-evoked currents in IPR and IPC neurons [IPR, n = 12 cells; IPC, n = 5 cells; n = 4 mice (3 male/1 female)]. c, Representative triple-label FISH images in IPN probing for Chrna5, Chrnb4, and Gad2. d, Example image of Chrna5, Chrnb4, and Gad2 FISH signals in individual IPR neurons. Exploded view of numbered/boxed cells are shown at bottom right. e, Scatterplot of Chrna5 (abscissa) vs Chrnb4 (ordinate) “% coverage” for all nuclei in IPR FISH images (n = 3 male mice). Gad2 % coverage for each nucleus is represented via the indicated dot color. f, Left, Pie graph of Chrna5+ nuclei showing fraction of Chrnb4+ and Chrnb4− nuclei. Right, Pie graph of Chrnb4+ nuclei showing fraction of Chrna5+ and Chrna5− nuclei. g, Scatterplot of Chrna5 (abscissa) vs Chrnb2 (ordinate) % coverage for all nuclei in IPR FISH images [n = 3 mice (2 male/1 female)]. Inset, Pie graph of Chrna5+ nuclei showing fraction of Chrnb2+ and Chrnb2− nuclei. h, Scatterplot of Chrna5 (abscissa) vs Chrna2 (ordinate) % coverage for all nuclei in IPR FISH images [n = 3 mice (2 male/1 female)]. Inset, Pie graph of Chrna5+ nuclei showing fraction of Chrnb2+ and Chrna2− nuclei. i, Scatterplot of Chrna5 (abscissa) vs Nacho (ordinate) % coverage for all nuclei in IPR FISH images [n = 3 mice (2 male/1 female)]. Inset, Pie graph of Chrna5+ nuclei showing fraction of Nacho+ and Nacho− nuclei. j, Scatterplot of Chrna5 (abscissa) vs Chrm3 (ordinate) % coverage for all nuclei in IPR FISH images [n = 3 mice (2 male/1 female)]. Inset, Pie graph of Chrna5+ nuclei showing fraction of Chrm3+ and Chrm3− nuclei. k, Scatterplot of Chrna5 (abscissa) vs Chrm5 (ordinate) % coverage for all nuclei in IPR FISH images [n = 3 mice (2 male/1 female)]. Inset, Pie graph of Chrna5+ nuclei showing fraction of Chrm5+ and Chrm5− nuclei. See Table 1 for full summary of FISH results.
Figure 7.

nAChR expression in rostral IPN neurons. a, Representative FISH image of Chrna5 and Gad2 FISH signals in IPR. b, Scatterplot of Gad2 (abscissa) vs Chrna5 (ordinate) % coverage for all nuclei in IPR FISH images (these data are from the same experiment shown in Fig. 6e). c, Pie graph of Gad2+ and Gad2− nuclei showing fraction of Chrna5+ and Chrna5− nuclei. d, Representative FISH image of Chrna5 and SstFISH signals in IPR. e, Scatterplot of Sst (abscissa) vs Chrna5 (ordinate) % coverage for all nuclei in IPR FISH images (n = 3 male mice). f, Pie graph of Sst+ and Sst− nuclei showing fraction of Chrna5+ and Chrna5− nuclei. g, Representative FISH image of Chrna5 and Pvalb FISH signals in IPR. h, Scatterplot of Pvalb (abscissa) vs Chrna5 (ordinate) % coverage for all nuclei in IPR FISH images (n = 3 male mice). i, Pie graph of Pvalb+ and Pvalb− nuclei showing fraction of Chrna5+ and Chrna5− nuclei. j, Representative FISH image of Chrna5 and Slc17a6 FISH signals in IPR. k, Scatterplot of Slc17a6 (abscissa) vs Chrna5 (ordinate) % coverage for all nuclei in IPR FISH images (n = 3 male mice). l, Pie graph of Slc17a6+ and Slc17a6− nuclei showing fraction of Chrna5+ and Chrna5− nuclei.
Brain slice preparation and recording solutions.
Brain slices were prepared as described previously (Engle et al., 2012). Mice were anesthetized with Euthasol (sodium pentobarbital, 100 mg/kg; sodium phenytoin, 12.82 mg/kg) before transcardiac perfusion with oxygenated (95% O2/5% CO2), 4°C N-methyl-d-glucamine (NMDG)-based recovery solution that contains the following (in mm): 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 10 MgSO4·7H2O, and 0.5 CaCl2·2H2O, at 300–310 mOsm and pH 7.3–7.4. Brains were immediately dissected after the perfusion and held in oxygenated 4°C recovery solution for 1 min before cutting a brain block containing the MHb or IPN and sectioning the brain with a vibratome (model VT1200S; Leica). Coronal slices (200–250 μm) were sectioned through the brain area of interest and transferred to an oxygenated 33°C recovery solution for 12 min. Slices were then kept in holding solution (containing the following, in mm: 92 NaCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 2 MgSO4·7H2O, and 2 CaCl2·2H2O, at 300–310 mOsm and pH 7.3–7.4) at 23°C for ≥60 min before recordings.
Brain slices were transferred to a recording chamber being continuously superfused at a rate of 1.5–2.0 ml/min with oxygenated 32°C recording solution. The recording solution [artificial cerebrospinal fluid (ACSF)] contained the following (in mm): 124 NaCl, 2.5 KCl, 1.2 NaH2PO4, 24 NaHCO3, 12.5 glucose, 2 MgSO4·7H2O, 2 CaCl2·2H2O, and 0.001 atropine, at 300–310 mOsm and pH 7.3–7.4. For the following datasets/figure panels, this recording solution was supplemented with the following: Figure 3: PTX (100 μm); Figures 4 and 5: TTX (0.5 μm); CNQX (10 μm); d-AP5 (30 μm); PTX (75 μm); and CGP (10 μm). Patch pipettes were pulled from borosilicate glass capillary tubes (1B150F-4, World Precision Instruments) using a programmable microelectrode puller (P-97, Sutter Instrument). Tip resistance ranged from 5.0 to 10.0 MΩ when filled with internal solution. The following internal solution was used (in mm): 135 potassium gluconate, 5 EGTA, 0.5 CaCl2, 2 MgCl2, 10 HEPES, 2 MgATP, and 0.1 GTP, pH adjusted to 7.25 with Tris base, and osmolarity adjusted to 290 mOsm with sucrose. For data in Figures 2, f and g, and 4c–l, this internal solution also contained QX-314 (2 mm) for improved voltage control.
Figure 5.

Prolonged inward currents in IPN neurons are specific to nicotine. a, Nicotine (100 μm) or ACh (300 μm) was applied to naive IPN neurons in slices via pressure ejection application. Repeated application at 2 or 10 min interstimulus intervals was used. b, Representative nicotine-evoked currents [2 min interstimulus interval; time points shown (min), 0, 6, 12). c, Summary time-series data for nicotine pressure ejection (2 min interstimulus interval). Individual cell responses [n = 10 cells; n = 4 mice (2 male/2 female)] are shown in gray. d, Summary time-series data for nicotine pressure ejection (10 min interstimulus interval). Individual cell responses [n = 8 cells; n = 3 mice (1 male/2 female)] are shown in gray. e, Representative ACh-evoked currents [2 min interstimulus interval; time points shown (min), 0, 6, 12]. f, Summary time-series data for ACh pressure ejection (2 min interstimulus interval). Individual cell responses [n = 12 cells; n = 4 mice (3 male/1 female)] are shown in gray. g, Representative ACh- and nicotine-evoked inward currents in IPN neurons are plotted on the same time scale. h, Summary rise time data comparing ACh [n = 12 cells; n = 4 mice (3 male/1 female)] and nicotine [n = 12 cells; n = 4 mice (2 male/2 female)] pressure ejection application. p Value, Unpaired t test. ACh data are replotted at right on a different scale. i, Summary decay time data comparing ACh [n = 12 cells; n = 4 mice (3 male/1 female)] and nicotine [n = 11 cells; n = 4 mice (2 male/2 female)] pressure ejection application. p Value, Unpaired t test.
Standard patch-clamp electrophysiology.
Neurons within brain slices were first visualized with infrared or visible differential interference contrast, followed in some cases by fluorescence microscopy to identify neurons expressing fluorescent proteins. Electrophysiology experiments were conducted using a Nikon Eclipse FN-1 Microscope or a Scientifica SliceScope. A computer running pCLAMP 10 software was used for recordings along with a Multiclamp 700B or Axopatch 200B amplifier and an analog-to-digital (A/D) converter (Digidata 1440A or Digidata 1550A). pClamp software, Multiclamp/Axopatch amplifiers, and Digitata A/D converters were from Molecular Devices. Data were sampled at 10 kHz and low-pass filtered at 1 kHz. Immediately before gigaseal formation, the junction potential between the patch pipette and the superfusion medium was nulled. Series resistance was uncompensated. A light-emitting diode (LED) light source (X-Cite 110 LED, Excelitas Technologies) coupled to excitation filters (400/40 nm, 470/40 nm, and 560/40 nm bandpass) was used to search for fluorescent neurons. To record physiological events following the local application of drugs, a drug-filled pipette was moved to within 20–40 μm of the recorded neuron using a second micromanipulator. A Picospritzer (General Valve) dispensed drug (dissolved in recording solution) onto the recorded neuron via a pressure ejection. Ejection volume, duration, and ejection pressure varied depending on the goal of the experiment.
Two-photon laser-scanning microscopy, electrophysiology, and nicotine uncaging.
PA-Nic photolysis was performed as described previously (Banala et al., 2018; Yan et al., 2018; Arvin et al., 2019). An Olympus BX51 upright microscope and a 60× (1.0 NA) water-dipping (2 mm working distance) objective was used to visualize cells. Prairie View 5.4 (Bruker Nano) software was used for image acquisition, photostimulation, and electrophysiology acquisition via a Multiclamp 700B patch-clamp amplifier. Analog signals were sampled at 5 kHz and low-pass filtered at 1 kHz, and an A/D converter (PCI-NI6052e, National Instruments) was used for digitization. Patch-clamp recordings were carried out using the internal solution mentioned above, except that Alexa Fluor 488, 568, or 594 (hydrazide salt; 50–200 μm) was also included in the recording pipette to visualize cells using two-photon laser-scanning microscopy (2PLSM). After break-in, the internal solution with the Alexa Fluor dye was allowed to equilibrate for 15–20 min before imaging was initiated. A Mai Tai HP1040 (Spectra-Physics) was used to excite Alexa Fluor 488, 568, or 594. These dyes were used during uncaging experiments for cell imaging at 965, 790, and 800 nm, respectively. The laser was pulsed at 90 MHz (pulse duration, ∼250 fs), and a M350–80-02-BK Pockels cell (ConOptics) was used for power attenuation. The dual-channel, two-photon fluorescence was detected by two non-descanned detectors; green and red channels (dual emission filters, 525/70 and 595/50 nm) were detected by the following Hamamatsu photomultiplier tubes, respectively: end-on GaAsP (7422PA-40) and side-on multi-alkali (R3896). A 405 nm Continuous Wave laser (100 mW OBIS LX, Coherent), incorporated into a laser launch system, was used for photostimulation/uncaging via a second set of x–y galvanometers incorporated into the scanhead (Cambridge Technologies). The 405 nm laser power was measured below the sample but above the condenser using a FieldMaster GS (with LM10 HTD sensor head, Coherent). PA-Nic [Fig. 2f,g: 50 μm; Fig. 4c–l (and also see Fig. 9a–d: 100 μm[scap])] was applied to the slice via superfusion. The Markpoints module of Prairie View 5.4α software was used to select spots in the field of view (∼1 μm diameter) for focal uncaging of nicotine via 405 nm laser light flashes (voltage clamp: 50 ms, 2 mW; current clamp: 1 ms, 2 mW). For some data [Figs. 2a,e, 4b (and see Figs. 8b,d,f,h, 9a)], a Z-series 2PLSM image of the cellular morphology was acquired after the completion of electrophysiological recordings. For Figure 2, these z-stacks were used to reconstruct neurons in 3D using either Simple Neurite Tracer (ImageJ plugin; Fig. 2c) or Neutube (Janelia Farm, HHMI; Fig. 2b,d). Sholl analysis was performed with Simple Neurite Tracer.
Figure 8.

Functional nAChRs in IPR neurons. a, Representative ACh (300 μm)-evoked currents in Gad2+ and adjacent Gad2− IPR neurons. Inset, Plot of ACh-evoked current amplitude in all tested Gad2+ and Gad2− IPR neurons (Gad2+, n = 9 cells; Gad2−, n = 10 cells; n = 2 male mice). b, Example 2PLSM images of Gad2+ IPR neurons. c, Representative ACh (300 μm)-evoked currents in Sst+ and adjacent Sst− IPR neurons. Inset, Plot of ACh-evoked current amplitude in all tested Sst+ and Sst− IPR neurons [Sst+, n = 10 cells; Sst−, n = 9 cells; n = 3 mice (1 male/2 female)]. d, Example 2PLSM images of Sst+ IPR neurons. e, Representative ACh (300 μm)-evoked currents in Pvalb+ and adjacent Pvalb− IPR neurons. Inset, Plot of ACh-evoked current amplitude in all tested Pvalb+ and Pvalb− IPR neurons (Pvalb+, n = 9 cells; Pvalb−, n = 11 cells; n = 2 male mice). f, Example 2PLSM images of Pvalb+ IPR neurons. g, Representative ACh (300 μm)-evoked currents in vGluT2+ and adjacent vGluT2− IPR neurons. Inset, Plot of ACh-evoked current amplitude in all tested vGluT2+ and vGluT2− IPR neurons (vGluT2+, n = 7 cells; vGluT2−, n = 8 cells; n = 3 male mice). h, Example 2PLSM images of vGluT2+ IPR neurons.
Experimental design and statistical analysis.
The α level was set to 0.05 for all statistical tests, which were conducted with GraphPad Prism 7 software. Experimenters were blinded to the treatment condition for the data described in Figure 1 (and see Figure 9). Statistical tests included two-sided unpaired Student's t test [Figs. 1b,e–j, 2g,h, 4d,i,m, 5h,i, Fig. 6b (and see Fig. 9c–f)], two-sided paired t test (Fig. 3k–m), nonparametric two-tailed Wilcoxon matched-pairs signed rank test (Fig. 3f,g), and nonparametric two-tailed Mann–Whitney test (Fig. 3h,i). Error bars denote SEM. Individual data points in scatter plots represent independent replicates/cells. Image analysis was performed with ImageJ (NIH). Analysis of electrophysiology data was performed with Clampfit (Molecular Devices) and custom scripts written in MATLAB (MathWorks). Resting membrane potential (Fig. 1e) was determined by simply deriving the mean membrane potential for each cell from a continuous 20 s recording. Input resistance was calculated from the change in steady-state current evoked by a voltage step from the holding voltage (Ables et al., 2017). Action potential characteristics were calculated/derived by first detecting spikes in recording traces using the threshold search feature of Clampfit. All automatically detected spikes were checked and manually accepted or rejected. Spike amplitude was derived by finding the difference between the peak spike voltage and the baseline, prespike voltage. The action potential threshold was defined as the voltage at which dV/dt (voltage changes over time) exceeded 20 mV/ms (Yu et al., 2008). Spontaneous excitatory postsynaptic current (sEPSCs) were detected via automated detection using Mini Analysis (Synaptosoft) followed by manual verification of detected events. Paired-pulse experiments were conducted using a 50 ms interpulse interval. Rise time and decay time (Fig. 5) was the time from 10% to 90% of peak response, or 90% of peak to 10% of peak response, respectively. Throughout the figure legends, the number of individual neurons tested is stated immediately before the number of animals from which those neurons were derived.
Results
Chronic nicotine enhances nAChR function on axons of MHb neurons
To examine Hb-IP changes brought about by cNIC, we recorded from ventral inferior MHb (MHbVI) neurons exhibiting spontaneous action potential firing. The MHb principally targets the IPN, which removes the requirement for any retrograde labeling of IPN-projecting MHb neurons; all or nearly all MHb neurons send projections to IPN. We previously reported that cNIC increased the MHbVI neuron firing rate using the cell-attached recording configuration (Shih et al., 2015). We found the same effect in this study (Fig. 1a,b; t(19) = 4.036, p = 0.0007), validating this prior result. In whole-cell configuration, action potential waveforms differed in control and cNIC-treated MHbVI neurons. Figure 1c shows representative spike traces from a control and cNIC neuron, and Figure 1d shows representative phase plots for spikes recorded from control and cNIC neurons. Compared with control neurons, cNIC neurons exhibited a depolarized resting membrane potential (Fig. 1e; t(63) = 2.395, p = 0.0196). Compared with spikes recorded in control neurons, cNIC spikes have reduced amplitude (Fig. 1f; t(60) = 2.166, p = 0.0343), increased half-width (Fig. 1g; t(60) = 3.033, p = 0.0036), a depolarized membrane potential at which spikes reached firing threshold (Fig. 1h; t(59) = 2.413, p = 0.0189), a decreased rise slope (Fig. 1i; t(61) = 3.328, p = 0.0015), and a decreased decay slope (Fig. 1j; t(61) = 2.725, p = 0.0084).
We recently showed that nAChRs are functionally upregulated in proximal dendrites of MHbVI cells following cNIC (Banala et al., 2018), and our prior work revealed strong nAChR subunit expression in MHb proximal axons as they enter the fasciculus retroflexus (Shih et al., 2014). To further study axonal nAChRs in MHb neurons, we reconstructed MHb neuronal morphology following 2PLSM during whole-cell recordings (Fig. 2a,b). Sholl analysis revealed that MHb neurons have approximately two primary dendrites and a compact dendritic arbor (Fig. 2c), which is consistent with previous work (Kim and Chang, 2005). Although dendrites are often found dorsal and medial to the soma, a narrow and unbranching process (presumably the axon) was found in n = 11 of 24 reconstructed neurons that invariably projected ventrally (Fig. 2d). Using laser flash photolysis of PA-Nic during voltage-clamp recordings and 2PLSM (Banala et al., 2018; Arvin et al., 2019), we rapidly (50 ms flash duration; 2 mW) evoked nAChR activity by uncaging nicotine adjacent to the axon (Fig. 2e) of MHb neurons from control and cNIC-treated animals (Fig. 2f; 45 μm from soma along the axon). MHb axonal nAChR function was strongly enhanced in cNIC neurons compared with control neurons when the uncaging spot was placed at 15 μm (t(12) = 2.206, p = 0.047), 30 μm (t(12) = 3.235, p = 0.0072), 45 μm (t(12) = 3.285, p = 0.0065), and 60 μm (t(11) = 3.238, p = 0.0079) from the soma (Fig. 2g). Mean input resistance was not different in MHb control neurons compared with cNIC-treated neurons (t(24) = 0.5985, p = 0.5551; Fig. 2h).
Chronic nicotine boosts glutamate transmission in IPN through nAChRs
cNIC-enhanced nAChR function in the proximal axons of MHb neurons (Fig. 2) suggests that nAChRs may also be upregulated in more distal axonal or presynaptic compartments of these cells. We tested this by recording EPSCs in IPN neurons, which are modulated by presynaptic nAChRs on MHb fibers (McGehee et al., 1995; Grady et al., 2009; Frahm et al., 2015). Using PTX (100 μm) to suppress fast GABAergic transmission, we examined sEPSCs before and after application of 0.03 μm, 0.06 μm (Fig. 3a, representative traces after 0.06 μm nicotine superfusion), or 0.12 μm nicotine. Cumulative distribution plots of sEPSC interevent interval (IEI; Fig. 3b,c) and amplitude (Fig. 3d,e) are shown for representative control and cNIC IPN neurons. Mean sEPSC IEI reduction (increased sEPSC frequency) occurred with 0.03 and 0.06 μm nicotine in IPN neurons from cNIC-treated animals (Fig. 3f, red symbols; 0.03 μm: W = −28, p = 0.0156; 0.06 μm: W = −26, p = 0.0312), but IEI reduction was not observed, in a statistical sense, in IPN neurons from control animals at these concentrations (Fig. 3f, blue symbols; 0.03 μm: W = 3, p = 0.8438; 0.06 μm: W = −9, p = 0.3125). A higher nicotine concentration (0.12 μm) was associated with a statistical trend toward IEI reduction in both treatment groups (control 0.12 μm: W = −15, p = 0.0625; cNIC 0.12 μm: W = −26, p = 0.0781). Nicotine bath application did not alter sEPSC amplitude in control (0.03 μm: W = 7, p = 0.5625; 0.06 μm: W = 1, p > 0.9999; 0.12 μm: W = −1, p > 0.9999) or cNIC (0.03 μm: W = 16, p = 0.2188; 0.06 μm: W = −10, p = 0.4688; 0.12 μm: W = 16, p = 0.3125) groups (Fig. 3g), and there was no significant difference (though a trend appeared) in baseline (prenicotine) sEPSC IEI (Fig. 3h; control: median = 1206 ms, n = 16; cNIC: median = 794 ms, n = 22; U = 115, p = 0.0734) or amplitude (Fig. 3i; control: median = −11.1 pA, n = 16; cNIC: median = −10.8 pA, n = 22; U = 164.5, p = 0.7424) in control versus cNIC animals. These results are consistent with the upregulation of presynaptic nAChRs in glutamatergic MHb axons, so we tested this more directly by recording electrically evoked excitatory postsynaptic current (eEPSCs) in IPN neurons. Local electrical stimulation produced inward currents that were sensitive to excitatory synaptic blockers and that could be partially recovered after drug washout (Fig. 3j). These glutamatergic, single-pulse eEPSCs could be enhanced by bath application of 0.12 μm nicotine (Fig. 3k; t(4) = 3.566, p = 0.0235), suggesting a role for nAChRs in presynaptic facilitation. This is corroborated by results showing that bath-applied nicotine (0.12 μm) also reduced the eEPSC paired-pulse ratio (PPR; Fig. 3l; t(4) = 3.673, p = 0.0213). Finally, we recorded eEPSCs before/after bath nicotine and derived PPRs (50 ms interval) in control and cNIC-treated animals. Only 0.12 μm nicotine could lower the PPR in control IPN neurons (0.03 μm: t(4) = 0.9462, p = 0.3976; 0.06 μm: t(4) = 1.662, p = 0.1719; 0.12 μm: t(5) = 2.762, p = 0.0398), whereas all tested nicotine concentrations lowered the PPR in IPN neurons from cNIC-treated animals (0.03 μm: t(6) = 2.532, p = 0.0446; 0.06 μm: t(5) = 6.552, p = 0.0012; 0.12 μm: t(6) = 3.669, p = 0.0105; Fig. 3m). Together with data on proximal axon nAChR upregulation (Fig. 2), these results indicate that chronic exposure to nicotine enhances nAChR functional activity in MHb axons and presynaptic terminals.
Enhanced postsynaptic nAChR function in IPN neurons
Given nicotine-mediated enhancement of presynaptic nAChR function in MHb axons (Figs. 2,3), we next asked whether cNIC modified nAChR function in the IPN, the main projection target for MHb neurons. We used PA-Nic laser flash photolysis in IPN at perisomatic locations during patch-clamp recording and 2PLSM (Fig. 4a). We found IPN neurons to be morphologically diverse; some neurons had extensive dendritic arbors, while others had simpler dendrites that were decorated with dendritic spines (Fig. 4b). The following results were not taken from any specific IPN subnucleus or morphological type. A cocktail of pharmacological blockers, as described in Materials and Methods, was used to isolate nAChR-activated currents. PA-Nic laser flash photolysis evoked inward currents of modest amplitude and slow recovery kinetics (Fig. 4c, control trace) relative to nicotine uncaging responses in MHb (Banala et al., 2018) and ventral tegmental area (VTA; Yan et al., 2018) neurons. cNIC treatment substantially increased the inward current amplitude following nicotine uncaging (t(20) = 4.638, p = 0.0002; Fig. 4c,d). Interestingly, repeated (2 min interval) perisomatic PA-Nic photolysis resulted in attenuation of the evoked inward current amplitude, which was not differentially affected in control versus cNIC-treated neurons (Fig. 4e–g). This attenuation precluded us from analyzing nAChR currents in dendritic compartments. We speculated that if IPN nAChRs are enhanced by chronic nicotine treatment, we might be able to pharmacologically mimic this effect in naive tissue. To block acetylcholinesterase (AChE) and increase local ACh levels, naive brain slices were continuously (≥30 min) superfused with donepezil (1 μm) during recordings. In the presence of donepezil, PA-Nic photolysis evoked much larger inward current amplitudes compared with untreated IPN neurons (t(18) = 2.246, p = 0.0375; Fig. 4h,i). Donepezil did not alter the current amplitude attenuation seen with repeated nicotine uncaging (Fig. 4j–l). Last, we asked whether the effect of cNIC treatment and donepezil were additive on nAChR current amplitudes. We measured uncaging-evoked currents in IPN neurons from control and donepezil-treated slices derived from cNIC-treated mice. Donepezil did not further enhance nAChR currents (t(22) = 1.000, p = 0.3280; Fig. 4m). Together, these data indicate that cNIC treatment enhances activity of perisomatic nAChRs in IPN neurons, and acute AChE treatment recapitulates this effect of cNIC.
To determine whether the attenuation of inward current amplitude was due to photodamage (from repeated laser flashes), we conducted control experiments using pressure ejection application of ACh or nicotine to IPN neurons (Fig. 5a). Nicotine (100 μm) pressure ejection to IPN neurons at an interevent interval of 2 min or even 10 min was also associated with inward current amplitude attenuation (Fig. 5b–d), ruling out photodamage as the cause of the attenuation. To determine whether this nicotine-associated attenuation is a generalized artifact of our recording procedures, ACh was applied via pressure ejection with a 2 min interevent interval. Strikingly, inward current amplitude did not attenuate with repeated ACh (300 μm) pressure ejection (Fig. 5e,f). We noted that equivalent pressure ejection times for ACh and nicotine, using concentrations of similar efficacy (Banala et al., 2018), resulted in dramatically different activation and decay kinetics in IPN neurons (Fig. 5g). Nicotine application was associated with slower rise time (t(22) = 2.492, p = 0.0207) and slower decay time (t(21) = 6.641, p = 0.0000014205) compared with ACh (Fig. 5h,i), perhaps because nicotine is not metabolized as ACh is by AChE. These control experiments suggest that the actions of nicotine at IPN neurons are markedly prolonged compared with those of ACh.
Potent increase in nicotine sensitivity in rostral IPN neurons following chronic nicotine
Although we restricted our recordings to the medial aspects of IPN, EPSC (Fig. 3), and PA-Nic, photolysis recordings (Fig. 4) were not made in a specific subnucleus within this medial aspect. Recent work suggests a remarkable diversity of neuronal types in various IPN subnuclei (Hsu et al., 2013; Morton et al., 2018), some of which have differential responses to nicotine (Ables et al., 2017). Before further cNIC studies, we explored the regional differences in the expression of various components of the cholinergic system. IPR neurons displayed substantially higher nAChR function compared with neurons of the caudal subnucleus [i.e., caudal IPN (IPC); t(15) = 2.968, p = 0.0096; Fig. 6a,b]. This relatively high nAChR function in IPR neurons, combined with their dense projections to areas involved in motivated behavior (Hsu et al., 2013; Morton et al., 2018) as well as their role in modulating nicotine reward (Ables et al., 2017), caused us to focus subsequent efforts on this subnucleus. α5 nAChR subunits are strongly expressed in local and projection neurons of the IPR (Hsu et al., 2013; Ables et al., 2017), so we used this as a convenient molecular marker for this part of the IPN. Using fluorescent in situ hybridization, we found strong expression of Chrna5 and Chrnb4 mRNA specifically in IPR, with weak but measurable Gad2 mRNA expression therein (Fig. 6c–e). Chrnb4 mRNA is expressed in a majority, but not all, IPR neurons that express Chrna5 mRNA (Fig. 6e,f). The vast majority of Chrna5+ IPR neurons coexpress β2 and α2 nAChR subunit mRNA, as well as the recently identified (Gu et al., 2016) nAChR chaperone Nacho/Tmem35a (Fig. 6g–i). Muscarinic ACh receptors, if present in IPN neurons, would also respond to ACh released from MHb cholinergic axons (Grady et al., 2009; Ren et al., 2011). Most Chrna5+ IPR neurons coexpress mRNA for muscarinic M3 AChRs, but only a minority expressed the M5 AChR mRNA (Fig. 6j,k). Full FISH results are summarized in Table 1.
Table 1.
Rostral IPN (IPR) fluorescence in situ hybridization results. The number of IPR nuclei that are positive and negative for expression of the indicated probes (criteria for positive and negative is described in Materials and Methods) is shown for each triple-probe combination FISH experiment. Data from n = 3 mice were pooled for each experiment
| Probe 1 | Probe 2 | Probe 3 | Total | Probe 1(+) | Probe 1(-) | Probe 1(+) |
Probe 1(-) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Probe 2(+) | Probe 2(-) | Probe 2(+) | Probe 2(-) | Probe 2(+) | Probe 2(-) | Probe 2(+) | Probe 2(-) | ||||||
| Probe 3(+) | Probe 3(+) | Probe 3(-) | Probe 3(-) | Probe 3(+) | Probe 3(+) | Probe 3(-) | Probe 3(-) | ||||||
| Gad2 | Chrna5 | Chrna2 | 1260 | 831/1260 | 429/1260 | 608/831 | 50/831 | 113/831 | 60/831 | 104/429 | 84/429 | 24/429 | 217/429 |
| Gad2 | Chrna5 | Chrnb2 | 1128 | 741/1128 | 387/1128 | 515/741 | 74/741 | 84/741 | 68/741 | 73/387 | 55/387 | 45/387 | 214/387 |
| Gad2 | Chrna5 | Chrnb4 | 868 | 602/868 | 266/868 | 321/602 | 7/602 | 227/602 | 47/602 | 38/266 | 13/266 | 45/266 | 170/266 |
| Chrm3 | Chrna5 | Chrnb4 | 944 | 487/944 | 457/944 | 310/487 | 5/487 | 137/487 | 35/487 | 96/457 | 14/457 | 112/457 | 235/457 |
| Chrm5 | Chrna5 | Chrnb4 | 1024 | 328/1024 | 696/1024 | 138/328 | 2/328 | 148/328 | 40/328 | 280/696 | 16/696 | 179/696 | 221/696 |
| Pvalb | Chrna5 | Chrnb4 | 911 | 100/911 | 811/911 | 42/100 | 7/100 | 14/100 | 37/100 | 372/811 | 18/811 | 208/811 | 213/811 |
| Sst | Chrna5 | Chrnb4 | 976 | 208/976 | 768/976 | 85/208 | 4/208 | 85/208 | 34/208 | 252/768 | 14/768 | 218/768 | 284/768 |
| Slc17a6 | Chrna5 | Chrnb4 | 1039 | 98/1039 | 941/1039 | 33/98 | 3/98 | 16/98 | 46/98 | 450/941 | 18/941 | 247/941 | 226/941 |
| Gad2 | Chrna5 | Nacho | 1164 | 871/1164 | 293/1164 | 728/871 | 45/871 | 80/871 | 18/871 | 82/293 | 104/293 | 17/293 | 90/293 |
The IPR is composed of multiple cell types. Gad2+ cells are abundant throughout the IPR (Fig. 7a,b), Sst+ cells are less common and found only in the dorsal half of IPR (Fig. 7d,e), and Pvalb+ (Fig. 7g,h) and Slc17a6+ (Fig. 7j,k) cells are sparse and found only in ventral IPR. Each of these cell types coexpress α5 nAChR subunits to differing degrees. Gad2+ cells nearly all express Chrna5 mRNA (Fig. 7c), and >80% of Sst+ cells also coexpress Chrna5 (Fig. 7f). In contrast, only ∼50% of Pvalb+ (Fig. 7i) and Slc17a6+ (Fig. 7l) cells in IPR coexpress Chrna5. Full FISH results are summarized in Table 1.
To corroborate and extend these mRNA expression results, we made targeted patch-clamp recordings in these four IPR cell types, where we examined nAChR currents and neuronal morphology via simultaneous 2PLSM imaging. Gad2+ and vGluT2+ IPR neurons were identified via Cre-dependent expression of tdT in Ai14 reporter mice that were crossed to Gad2-Cre or vGluT2-Cre mice (Yan et al., 2018), respectively. Pvalb+ IPR neurons were identified via tdT expression that was driven directly by the Pvalb promoter in bacterial artificial transgenic Pvalb-tdT mice. To mark Sst+ IPR neurons, Sst-Cre mice were microinjected in IPR with an AAV expressing mCherry-tagged DREADDs (designer receptor exclusively activated by designer drugs), which provide a fluorescent signal that is enriched in infected neuronal somata (Yan et al., 2018). In each of the above four slice preparations, we measured ACh-activated currents in fluorescent and nearby nonfluorescent IPR neurons. Gad2+ neurons, along with neighboring Gad2− neurons, exhibited robust functional nAChR expression (Fig. 8a). Morphologically, Gad2+ neurons have complex dendritic arbors that are commonly studded with dendritic spines (Fig. 8b). Sst+ neurons have low nAChR functional expression compared with nearby Sst− neurons (Fig. 8c). Sst+ neurons are morphologically simpler than Gad2+ neurons, having few dendritic processes and little evidence of dendritic spines (Fig. 8d). The nAChR functional expression profile for Pvalb+ neurons was similar to that of Sst+ cells (Fig. 8e), but Pvalb+ neuronal morphology was more variable (Fig. 8f). Finally, vGluT2+ IPR neurons also exhibited low functional expression of nAChRs compared with their neighboring vGluT2− cells (Fig. 8g). vGluT2+ neuron morphology was most similar to Sst+ cells, with low dendritic complexity commonly seen (Fig. 8h). Together, these results indicate that IPR Gad2+ neurons are plentiful and exhibit relatively high nAChR expression compared with three other minor cell types in IPN. We cannot rule out that these cell types may overlap in the expression of these markers, as we did not examine coexpression of mRNAs for Gad2, Sst, Pvalb, or Slc17a6.
Finally, we asked whether cNIC-mediated nAChR upregulation in IPR neurons has an impact on cellular excitability. During 2PLSM imaging, current-clamp recordings were made without ectopic current injection and with only atropine present, permitting presynaptic and postsynaptic mechanisms to influence the membrane potential of the recorded cell while we recorded EPSPs following perisomatic nicotine uncaging. These experiments were conducted in neurons that were not spontaneously firing, which was the case for the majority of IPR neurons we encountered. A representative example of such neurons is shown (Fig. 9a), including a typical perisomatic uncaging location. Using a very brief (1 ms) flash duration for nicotine uncaging, we noted much stronger depolarization in IPR neurons from cNIC-treated animals compared with control animals (t(19) = 3.164, p = 0.0051; Fig. 9b,d; averaged traces). Even with a brief pulse duration, depolarizations were prolonged in cNIC neurons, lasting several seconds (t(19) = 3.424, p = 0.0028; Fig. 9e). These results were not influenced by a differential resting membrane potential between the two treatment groups, as this parameter was not different between the two (t(19) = 1.086, p = 0.2911; Fig. 9c). Likewise, input resistance did not differ between control and cNIC-treated neurons (t(17) = 0.6727, p = 0.5102; Fig. 9f). Together, these results indicate that IPR neurons exposed chronically to nicotine are profoundly sensitized and poised to potently modulate local or long-range projection targets following a subsequent exposure to nicotine.
Discussion
In this study, we demonstrate that cNIC sensitizes the Hb-IP pathway at multiple locations. MHb neuron action potential firing is enhanced and spike waveform is substantially altered by cNIC. Axonal nAChRs are upregulated proximal to MHb neuronal somata, and in more distal presynaptic compartments in IPN. As in MHb neurons, nAChRs are functionally upregulated in IPN neurons. Finally, nicotine-elicited depolarization in IPN neurons is augmented by cNIC, likely via a combined action of presynaptic and postsynaptic nAChRs.
Results from multiple laboratories suggest that neuronal/nAChR activity in MHb and/or IPN is sensitized by exposure to cNIC, since blocking such activity is sufficient to induce withdrawal-like behaviors. This effect is durable, having been shown for blockade of (1) MHb/IPN nAChRs (Salas et al., 2009), (2) habenular neurokinin signaling (Dao et al., 2014), and (3) pacemaker firing in MHb neurons (Görlich et al., 2013). We did not examine withdrawal, but our past results (Shih et al., 2014, 2015; Banala et al., 2018) and present results are consistent with this sensitization hypothesis. cNIC increases pacemaker firing (Fig. 1a,b) and depolarizes the membrane potential of MHb neurons (Fig. 1c–e). We speculate that a depolarized membrane potential reduces the number of voltage-gated sodium channels available for activation, leading to changes in spike waveform such as reduced spike amplitude (Fig. 1f), spike widening (Fig. 1g), reduced spike rise and decay slope (Fig. 1c,d,i,j), and a shift in spike threshold to a more depolarized membrane potential (Fig. 1d,h). Conversely, it is possible that cNIC initiates a signaling cascade that downregulates sodium channels. Future work will be required to distinguish these various scenarios.
cNIC sensitizes axons from MHb neurons at multiple locations. Our demonstration of functional nAChRs on axonal membranes is corroborated by recent work (Passlick et al., 2018), but extends those data and our own results (Banala et al., 2018) by showing axonal upregulation following chronic nicotine administration (Fig. 2). Axonal nAChR upregulation extends to dual cholinergic/glutamatergic (Ren et al., 2011) presynaptic terminals in IPN, where chronic nicotine sensitizes their responsiveness to smoking-relevant concentrations (∼30 nm) of nicotine (Fig. 3). Paired-pulse experiments show that acute nicotine administration reduces PPR by increasing the amplitude of the first pulse relative to the second (Fig. 3l), suggesting that nAChR activation may enhance Ca2+ entry into presynaptic terminals to facilitate glutamate release. cNIC enables lower nicotine concentrations to be effective (Fig. 3m), perhaps via increased trafficking of functional nAChRs from somatodendritic compartments (Fig. 2e–g) to the presynaptic terminal. Cholinergic mechanisms may also play a role in presynaptic facilitation by nAChRs. Enhanced MHb neuron firing rate (Fig. 1a,b) and spike widening (Fig. 1c,g) could enhance ACh release from MHb axons in IPN (Grady et al., 2009; Ren et al., 2011), priming presynaptic terminals via heightened nAChR activity.
At the end of this Hb-IP pathway, IPN neurons exhibit somatodendritic nAChR functional upregulation (Fig. 4), resulting in IPR neurons that have a dramatically enhanced responsiveness to brief nicotine pulses (Fig. 9). This is consistent with chronic nicotine lowering the threshold for nicotine-mediated excitation (Nashmi et al., 2007). cNIC may also enhance cholinergic tone in the Hb-IP pathway, as facilitation of ACh signaling with donepezil treatment (Fig. 4h–l) was sufficient to reproduce increased IPN nAChR function. To the extent these mouse studies model the human response to nicotine in tobacco products, this state of enhanced nicotine responsiveness reflects the condition of “current smokers.” The first exposure to nicotine after nightly cessation (i.e., sleep) is expected to potently activate the pathway, with the smoker subsequently titrating their nicotine intake to optimally stimulate the pathway to maximize its benefits and reduce its aversive effects. Nicotine-mediated activation of cNIC-sensitized IPR neurons is expected to potently modulate serotonin and glutamatergic neurons of the dorsal raphe as well as glutamatergic and cholinergic neurons of the lateral dorsal tegmental nucleus (LDTg; Hsu et al., 2013; Ables et al., 2017; Ren et al., 2018). The IPN-to-LDTg circuit, which impinges on the VTA, was recently shown to play an important role in nicotine aversion (Wolfman et al., 2018).
Our results mapping the expression and functional activity of IPN nAChRs help to resolve a recent discrepancy. One group reported that optical activation of α5+ IPN neurons was insufficient to induce withdrawal-like behaviors (Morton et al., 2018), whereas another reported that optical activation of Gad2+ IPN neurons was sufficient to induce withdrawal (Zhao-Shea et al., 2013). Our results, and other studies (Hsu et al., 2013; Ables et al., 2017; Morton et al., 2018), show that whereas α5+ neurons are found predominantly in IPR, Gad2 expression is only weak/modest in IPR and strongest in IPC (Fig. 6c). Our data indicate that these groups were likely examining different types of IPN neurons, highlighting the importance of considering various IPN subnuclei and neurochemical cell types when dissecting behavioral phenomena. Relatedly, Zhao-Shea et al. (2013) speculated that Sst+ IPR neurons and their resident nAChRs play a specialized role in triggering nicotine withdrawal. However, our demonstration that Sst+ IPR neurons have much lower levels of functional nAChRs compared with Gad2+ cells (Fig. 7a,c) suggests that the latter cell type may play a more dominant role in the response of the IPR to nicotine. Gad2− cells also show substantial nAChR activity (Fig. 8a), suggesting the existence of an additional, unidentified neurochemical cell type that could be explored in future studies. Regardless, our results illustrate a key point: the IPN is a diverse and complex structure with numerous neurochemical cell types that have varying levels of nAChR functional activity. Chronic nicotine may therefore act nonuniformly on the Hb-IP system, potentially shifting the balance of activity from one group of cells/circuits to another.
IPN components, especially α5-containing nAChRs, are emerging as key mediators of nicotine dependence-associated behaviors. α5 subunits are nearly 10-fold more abundant in IPN than in any other brain area (Forget et al., 2018), and α5 knock-out mice do not exhibit nicotine withdrawal (Salas et al., 2009) or attenuated nicotine self-administration of aversive doses of nicotine (Fowler et al., 2011). Rats expressing an α5 sequence variant associated with human nicotine dependence and lung cancer show more facile relapse to nicotine-seeking behavior as well as an inverse correlation between IPN neuronal activity and relapse behavior (Forget et al., 2018). These results support our data demonstrating sensitized neuronal and nAChR activity in the IPN following exposure to nicotine, and point to α5-containing nAChRs as a possible therapeutic target for smoking cessation (Picciotto and Kenny, 2013). Unfortunately, there has been no reliable way to pharmacologically dissect α5-containing nAChRs because this subunit is an “accessory” subunit that does not participate in forming the orthosteric ligand binding site (Groot-Kormelink et al., 2001). Targeting α5-containing nAChRs may therefore involve manipulation of other subunits in α5-containing pentamers, such as α2. α2 subunits, which are implicated in human nicotine dependence (Sullivan et al., 2004), are required for precipitated withdrawal following cNIC treatment (Salas et al., 2009) and may negatively regulate nicotine intake (Lotfipour et al., 2013). We found that α5 and α2 are strongly coexpressed in IPR neurons (Fig. 6h). Given that α2 mRNA expression is enhanced by cNIC in a subpopulation of IPN GABAergic neurons that are found in IPR (Ables et al., 2017), the nAChR functional enhancement we identified in IPR neurons (Figs. 4, 9) could involve α2α5β4 nAChRs.
The importance of the Hb-IP pathway extends beyond nicotine dependence. For example, altered cholinergic activity in this circuit is implicated in withdrawal from morphine (Neugebauer et al., 2013), blockade of MHb or IPN nAChRs is sufficient to precipitate withdrawal from chronic alcohol exposure (Perez et al., 2015), and psychostimulants likely influence cholinergic signaling in IPN by altering ACh release (Hussain et al., 2008). To advance the field, future studies examining the mechanisms involved in nicotine withdrawal should include optical recording (e.g., fiber photometry, microendoscopy) from distinct IPN cells during behavior. Accurate targeting of IPN neurons with viruses or cannulae, while simultaneously avoiding activation or destruction of the overlying VTA, will be a critical challenge. Understanding the role of synaptically released acetylcholine in normal and pathological IPN transmission will also be important. To date, most examinations of Hb-IP ACh release have relied on ChAT (choline acetyltransferase)-ChR2 (channelrhodopsin 2) bacterial artificial chromosome transgenic mice that have abnormal cholinergic transmission (Nagy and Aubert, 2012; Crittenden et al., 2014). Examination of the mechanism of upregulation should also be performed. MHb nAChR upregulation appears to occur via increased receptor number on the cell surface and not via a change in receptor sensitivity (Banala et al., 2018), and a similar investigation in IPN would be useful to conduct. Perhaps the nAChR trafficking protein NACHO/TMEM35a plays a role in upregulation following cNIC exposure. Finally, our IPN morphology results clearly show a variety of novel cell types in IPN subnuclei, and it will be fascinating to connect these morphological types with functional roles in health and disease. These studies reveal a collection of striking adaptations to chronic nicotine in Hb-IP circuitry, while also highlighting the importance of accounting for neurochemical cell types and anatomical subnuclei in these circuits. Although the multiplicity of nicotine-elicited changes suggests that uncovering a single unifying mechanism for nicotine dependence will be difficult, having a variety of mechanisms to target therapeutically could ultimately be advantageous.
Footnotes
This work was supported by National Institutes of Health Grants DA-040626 and DA-044460 to R.M.D. M.C.A. was supported by a PhRMA Foundation Fellowship.
The authors declare no competing financial interests.
References
- Ables JL, Görlich A, Antolin-Fontes B, Wang C, Lipford SM, Riad MH, Ren J, Hu F, Luo M, Kenny PJ, Heintz N, Ibañez-Tallon I (2017) Retrograde inhibition by a specific subset of interpeduncular α5 nicotinic neurons regulates nicotine preference. Proc Natl Acad Sci U S A 114:13012–13017. 10.1073/pnas.1717506114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvin MC, Wokosin DL, Banala S, Lavis LD, Drenan RM (2019) Probing nicotinic acetylcholine receptor function in mouse brain slices via laser flash photolysis of photoactivatable nicotine. J Vis Exp. Advance online publication. Retrieved January 25 2019. doi: 10.3791/58873. 10.3791/58873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banala S, Arvin MC, Bannon NM, Jin XT, Macklin JJ, Wang Y, Peng C, Zhao G, Marshall JJ, Gee KR, Wokosin DL, Kim VJ, McIntosh JM, Contractor A, Lester HA, Kozorovitskiy Y, Drenan RM, Lavis LD (2018) Photoactivatable drugs for nicotinic optopharmacology. Nat Methods 15:347–350. 10.1038/nmeth.4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz NL. (2008) Neurobiology of nicotine addiction: implications for smoking cessation treatment. Am J Med 121 [4 Suppl 1]:S3–S10. 10.1016/j.amjmed.2008.01.015 [DOI] [PubMed] [Google Scholar]
- Benowitz NL. (2009) Pharmacology of nicotine: addiction, smoking-induced disease, and therapeutics. Annu Rev Pharmacol Toxicol 49:57–71. 10.1146/annurev.pharmtox.48.113006.094742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caponnetto P, Russo C, Polosa R (2012) Smoking cessation: present status and future perspectives. Curr Opin Pharmacol 12:229–237. 10.1016/j.coph.2012.02.005 [DOI] [PubMed] [Google Scholar]
- Cosgrove KP, Batis J, Bois F, Maciejewski PK, Esterlis I, Kloczynski T, Stiklus S, Krishnan-Sarin S, O'Malley S, Perry E, Tamagnan G, Seibyl JP, Staley JK (2009) beta2-nicotinic acetylcholine receptor availability during acute and prolonged abstinence from tobacco smoking. Arch Gen Psychiatry 66:666–676. 10.1001/archgenpsychiatry.2009.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden JR, Lacey CJ, Lee T, Bowden HA, Graybiel AM (2014) Severe drug-induced repetitive behaviors and striatal overexpression of VAChT in ChAT-ChR2-EYFP BAC transgenic mice. Front Neural Circuits 8:57. 10.3389/fncir.2014.00057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao DQ, Perez EE, Teng Y, Dani JA, De Biasi M (2014) Nicotine enhances excitability of medial habenular neurons via facilitation of neurokinin signaling. J Neurosci 34:4273–4284. 10.1523/JNEUROSCI.2736-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle SE, Broderick HJ, Drenan RM (2012) Local application of drugs to study nicotinic acetylcholine receptor function in mouse brain slices. J Vis Exp 68:e50034. 10.3791/50034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forget B, Scholze P, Langa F, Morel C, Pons S, Mondoloni S, Besson M, Durand-de Cuttoli R, Hay A, Tricoire L, Lambolez B, Mourot A, Faure P, Maskos U (2018) A human polymorphism in CHRNA5 is linked to relapse to nicotine seeking in transgenic rats. Curr Biol 28:3244–3253.e7. 10.1016/j.cub.2018.08.044 [DOI] [PubMed] [Google Scholar]
- Fowler CD, Lu Q, Johnson PM, Marks MJ, Kenny PJ (2011) Habenular α5 nicotinic receptor subunit signalling controls nicotine intake. Nature 471:597–601. 10.1038/nature09797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frahm S, Antolin-Fontes B, Görlich A, Zander JF, Ahnert-Hilger G, Ibañez-Tallon I (2015) An essential role of acetylcholine-glutamate synergy at habenular synapses in nicotine dependence. Elife 4:e11396. 10.7554/eLife.11396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George O, Ghozland S, Azar MR, Cottone P, Zorrilla EP, Parsons LH, O'Dell LE, Richardson HN, Koob GF (2007) CRF-CRF1 system activation mediates withdrawal-induced increases in nicotine self-administration in nicotine-dependent rats. Proc Natl Acad Sci U S A 104:17198–17203. 10.1073/pnas.0707585104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Whitaker AM, Baynes B, Abdel AY, Weil MT, George O (2014) Nicotine vapor inhalation escalates nicotine self-administration. Addict Biol 19:587–592. 10.1111/adb.12021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görlich A, Antolin-Fontes B, Ables JL, Frahm S, Slimak MA, Dougherty JD, Ibañez-Tallon I (2013) Reexposure to nicotine during withdrawal increases the pacemaking activity of cholinergic habenular neurons. Proc Natl Acad Sci U S A 110:17077–17082. 10.1073/pnas.1313103110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Moretti M, Zoli M, Marks MJ, Zanardi A, Pucci L, Clementi F, Gotti C (2009) Rodent habenulo-interpeduncular pathway expresses a large variety of uncommon nAChR subtypes, but only the α3β4* and α3β3β4* subtypes mediate acetylcholine release. J Neurosci 29:2272–2282. 10.1523/JNEUROSCI.5121-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groot-Kormelink PJ, Boorman JP, Sivilotti LG (2001) Formation of functional α3β4α5 human neuronal nicotinic receptors in Xenopus oocytes: a reporter mutation approach. Br J Pharmacol 134:789–796. 10.1038/sj.bjp.0704313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S, Matta JA, Lord B, Harrington AW, Sutton SW, Davini WB, Bredt DS (2016) Brain alpha7 nicotinic acetylcholine receptor assembly requires NACHO. Neuron 89:948–955. 10.1016/j.neuron.2016.01.018 [DOI] [PubMed] [Google Scholar]
- Hsu YW, Tempest L, Quina LA, Wei AD, Zeng H, Turner EE (2013) Medial habenula output circuit mediated by α5 nicotinic receptor-expressing GABAergic neurons in the interpeduncular nucleus. J Neurosci 33:18022–18035. 10.1523/JNEUROSCI.2927-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain RJ, Taraschenko OD, Glick SD (2008) Effects of nicotine, methamphetamine and cocaine on extracellular levels of acetylcholine in the interpeduncular nucleus of rats. Neurosci Lett 440:270–274. 10.1016/j.neulet.2008.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BC, Dimova JG, Dakoji S, Yuan LL, Gewirtz JC, Tran PV (2016) Deletion of novel protein TMEM35 alters stress-related functions and impairs long-term memory in mice. Am J Physiol Regul Integr Comp Physiol 311:R166–R178. 10.1152/ajpregu.00066.2016 [DOI] [PubMed] [Google Scholar]
- Kim U, Chang SY (2005) Dendritic morphology, local circuitry, and intrinsic electrophysiology of neurons in the rat medial and lateral habenular nuclei of the epithalamus. J Comp Neurol 483:236–250. 10.1002/cne.20410 [DOI] [PubMed] [Google Scholar]
- Lotfipour S, Byun JS, Leach P, Fowler CD, Murphy NP, Kenny PJ, Gould TJ, Boulter J (2013) Targeted deletion of the mouse α2 nicotinic acetylcholine receptor subunit gene (Chrna2) potentiates nicotine-modulated behaviors. J Neurosci 33:7728–7741. 10.1523/JNEUROSCI.4731-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamede M, Ishizu K, Ueda M, Mukai T, Iida Y, Kawashima H, Fukuyama H, Togashi K, Saji H (2007) Temporal change in human nicotinic acetylcholine receptor after smoking cessation: 5IA SPECT study. J Nucl Med 48:1829–1835. 10.2967/jnumed.107.043471 [DOI] [PubMed] [Google Scholar]
- Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, Collins AC (1992) Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci 12:2765–2784. 10.1523/JNEUROSCI.12-07-02765.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta JA, Gu S, Davini WB, Lord B, Siuda ER, Harrington AW, Bredt DS (2017) NACHO mediates nicotinic acetylcholine receptor function throughout the brain. Cell Rep 19:688–696. 10.1016/j.celrep.2017.04.008 [DOI] [PubMed] [Google Scholar]
- McGehee DS, Heath MJ, Gelber S, Devay P, Role LW (1995) Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science 269:1692–1696. 10.1126/science.7569895 [DOI] [PubMed] [Google Scholar]
- Morton G, Nasirova N, Sparks DW, Brodsky M, Sivakumaran S, Lambe EK, Turner EE (2018) Chrna5-expressing neurons in the interpeduncular nucleus mediate aversion primed by prior stimulation or nicotine exposure. J Neurosci 38:6900–6920. 10.1523/JNEUROSCI.0023-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhin AG, Kimes AS, Chefer SI, Matochik JA, Contoreggi CS, Horti AG, Vaupel DB, Pavlova O, Stein EA (2008) Greater nicotinic acetylcholine receptor density in smokers than in nonsmokers: a PET study with 2–18F-FA-85380. J Nucl Med 49:1628–1635. 10.2967/jnumed.108.050716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy PM, Aubert I (2012) Overexpression of the vesicular acetylcholine transporter increased acetylcholine release in the hippocampus. Neuroscience 218:1–11. 10.1016/j.neuroscience.2012.05.047 [DOI] [PubMed] [Google Scholar]
- Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, Huang Q, McClure-Begley T, Lindstrom JM, Labarca C, Collins AC, Marks MJ, Lester HA (2007) Chronic nicotine cell specifically upregulates functional α4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci 27:8202–8218. 10.1523/JNEUROSCI.2199-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer NM, Einstein EB, Lopez MB, McClure-Begley TD, Mineur YS, Picciotto MR (2013) Morphine dependence and withdrawal induced changes in cholinergic signaling. Pharmacol Biochem Behav 109:77–83. 10.1016/j.pbb.2013.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passlick S, Thapaliya ER, Chen Z, Richers MT, Ellis-Davies GCR (2018) Optical probing of acetylcholine receptors on neurons in the medial habenula with a novel caged nicotine drug analogue. J Physiol 596:5307–5318. 10.1113/JP276615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez E, Quijano-Cardé N, De Biasi M (2015) Nicotinic mechanisms modulate ethanol withdrawal and modify time course and symptoms severity of simultaneous withdrawal from alcohol and nicotine. Neuropsychopharmacology 40:2327–2336. 10.1038/npp.2015.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Kenny PJ (2013) Molecular mechanisms underlying behaviors related to nicotine addiction. Cold Spring Harb Perspect Med 3:a012112. 10.1101/cshperspect.a012112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Qin C, Hu F, Tan J, Qiu L, Zhao S, Feng G, Luo M (2011) Habenula “cholinergic” neurons co-release glutamate and acetylcholine and activate postsynaptic neurons via distinct transmission modes. Neuron 69:445–452. 10.1016/j.neuron.2010.12.038 [DOI] [PubMed] [Google Scholar]
- Ren J, Friedmann D, Xiong J, Liu CD, Ferguson BR, Weerakkody T, DeLoach KE, Ran C, Pun A, Sun Y, Weissbourd B, Neve RL, Huguenard J, Horowitz MA, Luo L (2018) Anatomically defined and functionally distinct dorsal raphe serotonin sub-systems. Cell 175:472–487.e20. 10.1016/j.cell.2018.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas R, Pieri F, De Biasi M (2004) Decreased signs of nicotine withdrawal in mice null for the β4 nicotinic acetylcholine receptor subunit. J Neurosci 24:10035–10039. 10.1523/JNEUROSCI.1939-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas R, Sturm R, Boulter J, De Biasi M (2009) Nicotinic receptors in the habenulo-interpeduncular system are necessary for nicotine withdrawal in mice. J Neurosci 29:3014–3018. 10.1523/JNEUROSCI.4934-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih PY, Engle SE, Oh G, Deshpande P, Puskar NL, Lester HA, Drenan RM (2014) Differential expression and function of nicotinic acetylcholine receptors in subdivisions of medial habenula. J Neurosci 34:9789–9802. 10.1523/JNEUROSCI.0476-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih PY, McIntosh JM, Drenan RM (2015) Nicotine dependence reveals distinct responses from neurons and their resident nicotinic receptors in medial habenula. Mol Pharmacol 88:1035–1044. 10.1124/mol.115.101444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Neale BM, van den Oord E, Miles MF, Neale MC, Bulik CM, Joyce PR, Straub RE, Kendler KS (2004) Candidate genes for nicotine dependence via linkage, epistasis, and bioinformatics. Am J Med Genet B Neuropsychiatr Genet 126B:23–36. 10.1002/ajmg.b.20138 [DOI] [PubMed] [Google Scholar]
- Wallace ML, Saunders A, Huang KW, Philson AC, Goldman M, Macosko EZ, McCarroll SA, Sabatini BL (2017) Genetically distinct parallel pathways in the entopeduncular nucleus for limbic and sensorimotor output of the basal ganglia. Neuron 94:138–152.e5. 10.1016/j.neuron.2017.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfman SL, Gill DF, Bogdanic F, Long K, Al-Hasani R, McCall JG, Bruchas MR, McGehee DS (2018) Nicotine aversion is mediated by GABAergic interpeduncular nucleus inputs to laterodorsal tegmentum. Nat Commun 9:2710. 10.1038/s41467-018-04654-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Peng C, Arvin MC, Jin XT, Kim VJ, Ramsey MD, Wang Y, Banala S, Wokosin DL, McIntosh JM, Lavis LD, Drenan RM (2018) Nicotinic cholinergic receptors in VTA glutamate neurons modulate excitatory transmission. Cell Rep 23:2236–2244. 10.1016/j.celrep.2018.04.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young-Wolff KC, Klebaner D, Folck B, Tan ASL, Fogelberg R, Sarovar V, Prochaska JJ (2018) Documentation of e-cigarette use and associations with smoking from 2012 to 2015 in an integrated healthcare delivery system. Prev Med 109:113–118. 10.1016/j.ypmed.2018.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Shu Y, McCormick DA (2008) Cortical action potential backpropagation explains spike threshold variability and rapid-onset kinetics. J Neurosci 28:7260–7272. 10.1523/JNEUROSCI.1613-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao-Shea R, Liu L, Pang X, Gardner PD, Tapper AR (2013) Activation of GABAergic neurons in the interpeduncular nucleus triggers physical nicotine withdrawal symptoms. Curr Biol 23:2327–2335. 10.1016/j.cub.2013.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao-Shea R, DeGroot SR, Liu L, Vallaster M, Pang X, Su Q, Gao G, Rando OJ, Martin GE, George O, Gardner PD, Tapper AR (2015) Increased CRF signalling in a ventral tegmental area-interpeduncular nucleus-medial habenula circuit induces anxiety during nicotine withdrawal. Nat Commun 6:6770. 10.1038/ncomms7770 [DOI] [PMC free article] [PubMed] [Google Scholar]