

Abstract
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disorder that is the leading genetic cause of infantile death. SMA is caused by homozygous deletion or mutation of the survival of motor neuron 1 gene (SMN1). The SMN2 gene is nearly identical to SMN1, however is alternatively spliced. The close relationship to SMN1 results in SMN2 being a very power genetic modifier of SMA disease severity and a target for therapies. We sought to identify the regulatory role individual HDAC proteins use to control expression of full length protein from the SMN2 genes. We used quantitative PCR to determine the effects shRNA silencing of individual HDACs on the steady state levels of a SMN2-luciferase reporter transcripts. We determined that reduction of individual HDAC proteins was sufficient to increase SMN protein levels in a transgenic reporter system. Knockdown of class I HDAC proteins preferentially activated the reporter by increased promoter transcription. Silencing of class II HDAC proteins maintained transcriptional activity; however silencing of HDAC 5 and 6 also appeared to enhance inclusion of an alternatively spliced exon. This work highlights HDAC proteins 2 and 6 as excellent investigative targets. These data are important to the basic understanding of SMN expression regulation and the refinements of current therapeutic compounds as well as the development of novel SMA therapeutics.
Keywords: Spinal muscular atrophy, RNAi, HDAC, Luciferase
1. Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disease that has a carrier frequency of 1 in 35 and affects approximately 1 in 6000 individuals. It is characterized by the progressive degeneration of alpha motor neurons and resulting weakness and deterioration of muscles. SMA is caused by the deletion or mutation of the survival motor neuron 1 (SMN1) gene [1]. Loss of SMN1 is partially compensated by SMN2, the result of a gene duplication event in humans. SMN2 pre-dominantly produces mRNA that is alternatively spliced, which expresses a truncated protein that lacks exon 7. This protein is unstable and is quickly degraded, however maintains the ability to rescue viability but cannot overcome neurological defects [2].
Alternative splicing of SMN2 is caused by a single C to T base change at the +6 position of SMN2 exon 7 [3,4]. Approximately 10% of transcripts produced from the SMN2 locus encode full length SMN protein, in contrast to SMN1 which produced greater than 95% exon 7 included transcripts. The C to T transition results in the abrogation of exonic splicing enhancers (ESE) and concurrently strengthen an exonic splicing silencer (ESS) [2,5–7]. These sites are populated by numerous protein complexes. Three proteins of note are hnRNP A1, hTra2b, and SF2/ASF. The latter two proteins have been shown to promote exon 7 inclusion while binding of the former inhibits efficient splicing [7].
SMA disease severity has an inverse correlation to SMN2 gene dosage [8,9]. As a result, the SMN2 gene is an excellent drug target for SMA therapy. Increases in full length protein expression or inclusion of exon 7 from the SMN2 gene can act to ameliorate disease symptoms. Compounds currently in clinical trials include a class of drugs that function as histone deacetylase (HDAC) inhibitors including phenyl butyrate [10–12], and valproic acid [12]. HDACs work by opposing histone acetyl-transferase proteins to determine the steady state levels of histone acetylation [13]. This post-translational modification of histone tails regulates the conformation of chromatin and partially controls the access of transcriptional machinery to DNA. HDAC proteins are categorized into four classes primarily based on homology to yeast deacetylase proteins. Class I HDACs (HDACs 1, 2, 3, and 8) are similar to the Saccharomyces cerevisiae transcriptional regulator RPD3. Class II HDACs (HDACs 4, 5, 6, 7, 9, and 10) are more closely related to yeast HDA1. Class III HDAC proteins are homologous to yeast SIR1 and are Nicotinamide adenine dinucleotide (NAD+) dependent. HDAC11 is a separate class as there is insufficient sequence or functional data to categorize it in any of the other three classes (reviewed in [14]).
Many HDAC inhibitors effective at increasing SMN expression target both class I and class II HDACs [15–17]. According to published data for M344 and suberoylanilide hydroxamic acid (SAHA/vorinostat), the most likely mechanism of action of these drugs is through improved access of transcription machinery to the SMN2 promoter and by promotion of exon 7 inclusion by the regulation splicing factors. M344 and SAHA are pan-HDAC inhibitors, targeting both class I and class II HDAC proteins.
In this study we elucidate the role that individual HDAC proteins have on the regulation of SMN expression. We targeted HDACs 1–8 by the expression of short hairpin RNAs (shRNAs) and measured SMN protein levels using a SMN2 luciferase reporter cell line. We explored the effects of reduced HDAC protein levels on SMN2 transcription, exon 7 inclusion, and mRNA expression levels of SMN splicing modulators hTra2β, hnRNP A1, and SF2/ASF. Silencing of individual HDAC mRNA was sufficient to increase the amount of SMN reporter transcripts, and increase reporter activity. Changes in total and full length mRNA were differentially affected by the individual HDAC proteins silenced. While enhancing the basic understanding of SMA biology and SMN2 gene expression regulation, these data will also identify potential drug targets that may have less toxic properties than pan-HDACi while maintaining sufficient SMN protein induction.
2. Materials and methods
2.1. Tissue culture
Cells were incubated at 37 °C with 5% CO2. HEK-293 reporter SMN2-luciferase cells were grown in D-MEM (Gibco, #11995) with 10% fetal bovine serum (Sigma) and 1× Pen-Strep (Gibco, #15140) with 200 µg/mL hygromycin B (Invitrogen, #10687–010). Primary human SMA patient fibroblasts, 3813, were grown in D-MEM with 15% fetal bovine serum and 1× Pen-Strep.
2.2. shRNA vectors and transfection
All shRNA vectors were obtained from openbiosystems via the UMass Medical School RNAi Core Facility. All shRNAs were in the pGIPZ vector also expressing GFP under the CMV promoter. HDAC1 (V2LHS_61809, V3LHS_344564), HDAC2 (V2LHS_132136, V3LHS_382880), HDAC3 (V2LHS_53152, V3LHS_380875), HDAC4 (V2LHS_71327, V3LHS_340831), HDAC5 (V2LHS_68644, V3LHS_321382), HDAC6 (V2LHS_71188, V3LHS_330045), HDAC7 (V2LHS_96401, V3LHS_351666), and HDAC8 (V3LHS_355330, V3LHS_355335), pGIPZ non-silencing control. shRNAs (1 µg) were transfected into 200,000 SMN2-luciferase reporter cells in a 12 well tissue culture treated dish using Lipofectamine 2000 (Invitrogen) at a 1:4 DNA:Lipofectamine 2000 ratio.
2.3. Luciferase assays
Luciferase was measured 48 h post transfection by harvesting cells with 1 trypsin (Gibco, #15050–065) followed by trypsin inhibitor (Invitrogen, #17075–029) followed by PBS collection and centrifugation. Ten percent of cells by volume were equally distributed into 3 wells of a white tissue culture 96 well plate. The remaining 90% of cells were saved for RNA quantification. Promega Dual-glo luciferase assay system was used to determine SMN-luciferase activity. MS-275 was purchased through Selleck-Chem (#W12277).
2.4. Primary cell assays
Lentiviruses expressing HDAC shRNA clones were obtained from the UMass Medical School RNAi Core Facility. Lentivirus was added to 250,000 primary 3813 cells, plated in a 10 cm tissue culture dish, at a multiplicity of infection (MOI) of 10 in the presence of polybrene. Growth medium containing 10% FBS and 1% p/s was replaced 24 h following infection and allowed to grow an additional 48 h before harvesting. Reduced mRNA steady state levels were confirmed by semiquantitative endpoint PCR with the same primer sets and conditions as the reporter cell assays.
2.5. PCR and qRT-PCR
RNA was isolated from cells using Trizol Reagent (Invitrogen, #15596–026). cDNA was generated using the Improm-II Reverse Transcription System (Promega, #A3801). Primer sequences: Exon5/Xho-forward: catttccttctggaccactcgag, Exon6-forward: gcccaaatctgctccatggaac, Luciferase-reverse: atagcttctgccaac cgaacgg, Exon7-reverse: taaggaatgtgagcaccttccttc, Exon8-reverse: gatctgtctgatcgtttctttagtgg, GAPDH-reverse (G3A): tccaccaccctgttgctgta, GAPDH-forward (G3S): accacagtccatgccatcac. The primer pairs to determine splicing factor mRNA changes were as follows (forward, reverse), hTra2β (cacatcgaccggcgacagca, ccccgatccgtgagcacttcc), SF2/ASF (ttagatctcatgagggagaaactgcc, ggcttctgctacg actacggc), hnRNP A1 (agggcgaaggtaggctggca, gcttcctcagctgttcgggct).qPCR was performed as described in the protocol for iQ SybrGreen Supermix (BioRad, #170–8882) in an Eppendorf Mastercycler ep Realplex 4 real-time PCR machine. Reactions were incubated for a 10 min 94 °C hot start followed by 45 cycles of the following: 94 °C for 45 s, 60 °C for 15 s, 72 °C for 45 s. The Pfaffl method was used to determine the change in transcript levels relative to the DMSO or non-silencing shRNA controls and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
2.6. Chromosome immunoprecipitation (ChIP)
ChIP protocol was performed as described (Upstate Cell Signaling) using anti-acetyl histone H3 (Upstate, #06–599) and RNA Polymerase II CTD (abcam, #ab5095). Antibodies were bound and washed using Protein A Dynabeads (Invitrogen, #10002D). DNA was purified using Qiagen Quick PCR Clean-Up Kit. qPCR was performed as above. Percent input bound was calculated based on a 5 point standard dilution curve of each relative input. Primer sequences: Forward (promoter): ggccaccgtactgttccgc, reverse (exon 2): ccaaatgtcagaatcatcgctctgg.
2.7. Statistics
Two tailed t-test was used to calculate statistical significance relative to DMSO or non-silencing shRNA controls. P-values – (*** – <0.0001, *** – 0.0001–0.001, ** – 0.0011–0.01, * – 0.011– 0.05). Error bars are SEM.
3. Results
3.1. SMN2-luciferase reporter responds to HDAC inhibitors
We theorized that individual HDAC proteins have distinct contributions to the regulation of SMN expression. Increases in SMN protein levels by a pan-HDAC inhibitor may be the result of the collective and potentially additive effects by suppression of multiple HDAC proteins. We wished to characterize how a class I specific HDAC inhibitor would respond in our reporter cell system. We compared the selective HDAC inhibitor MS-275 (Entinostat) with two pan-HDAC inhibitors, SAHA and M344. Both have been reported to increase SMN levels. The reporter used in this study is expressed from the 3.4 kB SMN2 promoter followed by exons 1–5 cDNA linked to a SMN2-luciferase splicing cassette [18]. The luciferase protein is only properly translated when SMN exon 7 is included in the processed mRNA. When exon 7 is excluded, the exon 6–8 junction shifts the reading frame and translation is prematurely terminated early in the luciferase gene. Each of these drugs were tested at four concentrations, 370 nM, 1.1, 3.3 and 10 µM. Both SAHA and M344 increased SMN luciferase levels up to 10-fold. MS-275 activity was less active, only achieving 4-fold increase of the fusion protein (data not shown). Activation of the reporter by the class I specific drug MS-275 implies that inhibition of a subset of HDAC proteins is sufficient to increase SMN protein levels.
To further elucidate the mechanism that the individual HDAC inhibitors use to increase the SMN expression, we quantified changes in the reporter mRNA. Primers used to measure the total levels of reporter mRNA which included both exon 7 included and excluded messages. Separate reactions were used to independently measure the increase or decrease of exon 7 included or full length mRNA. An increase of exon 7 included reporter mRNA greater than that observed for total reporter mRNA infers a shift in the splicing of exon 7 because changes in splicing of the reporter mRNA should not affect the amount of mRNA present, only the ratio of total reporter mRNA to exon 7 excluded transcripts.
SAHA and M344 increased both total and exon 7 included mRNA in the reporter cells. As the drug concentration increased there was a greater difference between exon 7 included transcripts and total mRNA (Fig. 1). This suggests that at lower concentrations the pan-HDAC inhibitors may directly affect the acetylation state of the histones populating the SMN2 reporter promoter. With the increase of drug, there is also an increased effect on relevant targets potentially including the splicing factors that modify SMN2 splicing. Reporter mRNA analysis agrees with previously published reporter reporting these drugs not only increase the transcription activity at the SMN2 promoter but also increase exon 7 inclusion.
Fig. 1.
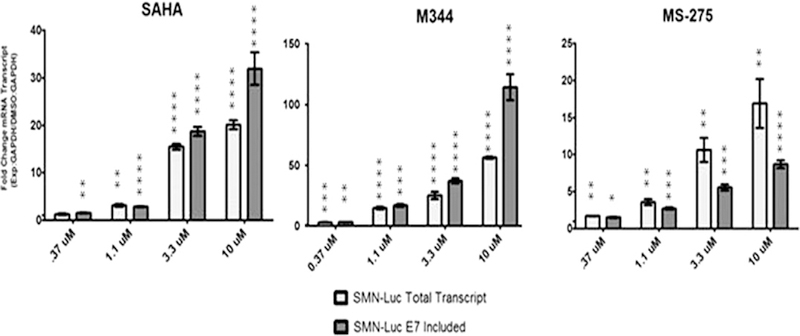
HDAC inhibition increases SMN-luciferase reporter mRNA levels. qRT-PCR was used to measure increases of SMN-luciferase mRNA following treatment with HDAC inhibitors. Fold increase of mRNA was normalized to GAPDH.
Analysis of mRNA changes following MS-275 treatment revealed a different profile than what was observed with the paninhibitors. Class I specific HDAC inhibition increased both total and exon 7 included mRNA. However, the fold increases of the total transcript exceeded that of exon 7 included (Fig. 1). Therefore, we propose that the increase in full-length exon 7 included message corresponds to the MS-275 dependent increase of total SMN2 reporter transcription and not an increase in exon 7 splicing efficiency.
3.2. Silencing of HDAC proteins increases SMN reporter protein and mRNA levels
Analysis of mRNA increases with MS-275 and pan-HDAC inhibitors SAHA and M334 suggested key mechanistic differences between the class I and II HDACs. Therefore, we targeted HDACs 1–8 using shRNA to explore the possible role that each plays in regulating SMN levels. We confirmed knockdown using semi-quantitative endpoint-PCR (data not shown). Reduced levels of the class I HDAC1, 2, 3, and 8, all increased SMN-luciferase reporter by at least 1.5-fold (Fig. 2A). Knockdown of the individual class II HDAC proteins, HDAC4, 6, and 7 resulted in an increase of reporter activty, with HDAC6 showing the highest gain. HDAC5 shRNA did not result in activation of the reporter.
Fig. 2.
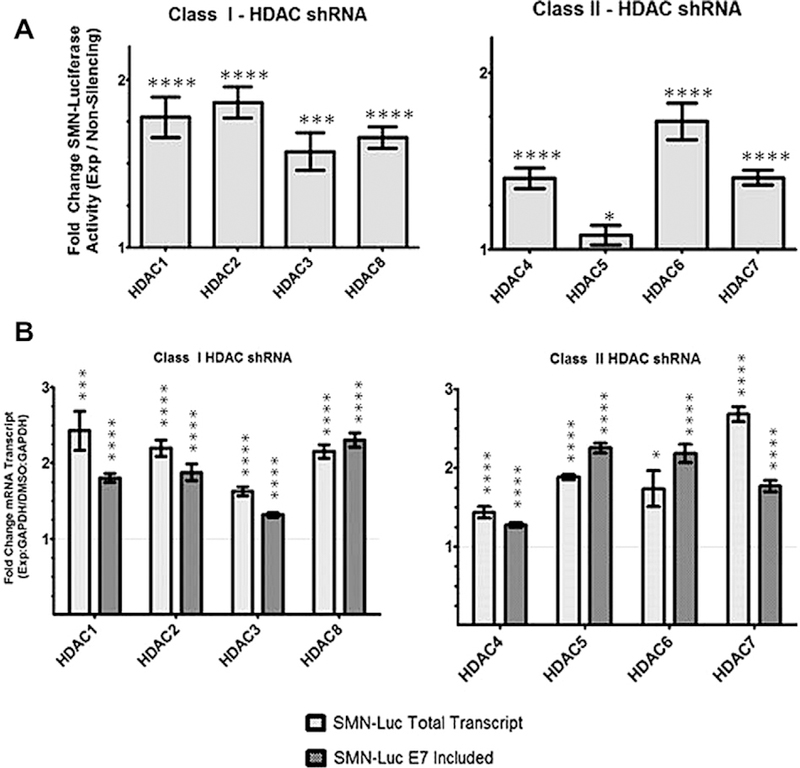
shRNA silencing of individual HDACs increase SMN-luciferase activity and mRNA. (A) HDAC shRNAs increase SMN-luciferase activity levels. SMN2 reporter cells were treated for 48 h with shRNA. HDAC2 silencing increase luciferase activity the greatest at nearly 2-fold. HDAC5 knockdown appears to have no effect on luciferase activity (B) qRT-PCR was used to measure increases of SMN-luciferase mRNA following treatment with HDAC shRNA. Fold increase of mRNA was normalized to GAPDH.
Increased mRNA expression profiles for each class of protein mirrored the results from the drug assays. Knockdown of the class I proteins HDAC1, 2, 3, and 8 increased total mRNA greater than exon 7 included transcript (Fig. 2B). This is consistent with results from MS-275 treatment and supports our hypothesis that the class I HDACs plays a role in transcriptional regulation. Analysis of mRNA increases following shRNA transfection of the class II HDAC protein revealed a more diverse set of profiles. Silencing of HDACs 4 and 7 resulted in a pattern consistent with transcriptional regulation, while HDAC5 and 6 shRNA treatment displayed a more subdued response that suggests that these proteins play a role in recognition and inclusion of exon 7 (Fig. 2B). HDAC5 shRNA treatment resulted in an increase of SMN-luciferase mRNA transcripts without any noticeable impact on SMN-luciferase activity (Fig. 2A). The reported function of HDAC5 does not indicate an explanation for this observation. It is possible that silencing of HDAC5 increased reporter mRNA and promoted inclusion of exon 7, but concurrently inhibited translation, caused a premature termination, or translational frame shift.
3.3. Analysis of changes of endogenous splicing factor mRNAs
Increases in mRNA and protein levels of the known SMN splicing factors Tra2b, hnRNAP A1, and SF2/ASF have been reported to promote inclusion of SMN exon 7 in SMN2 transcripts. To investigate whether these same mechanisms are operative in the reporter cells; we measured the mRNA levels of these splicing factors following drug or shRNA treatment. hnRNP A1, an inhibitory splicing factor, was decreased following treatment with each of the three chemical HDAC inhibitors tested. At 10 µM M344, hnRNP A1 transcripts were elevated relative to the lower levels observed at lower drug concentrations. hTra2b transcript levels were increased in response to both M344 and SAHA, with no effect in response to MS- 275 treatment. SF2/ASF was only significantly increased when reporter cells were treated with M344 (Fig. 3A). This is in agreement with data in Fig. 1 showing a more potent response in fold increase of exon 7 included mRNA in response to M344 over that of SAHA at the same concentration. Analysis of these same transcripts following shRNA treatments did not reveal any direct pattern associated with the mechanisms determined by SMN transcripts analysis. shRNAs specific to HDAC1 or HDAC2, both suspected transcriptional regulators, show nearly identical effects on total and exon 7 included SMN reporter mRNAs in Fig. 2; however, in this experiment low levels of HDAC1 significantly increased hTra2b and SF2/ ASF, while HDAC2 shRNA treatment was inconsequential (Fig. 3B). Additionally, silencing of HDAC5 and 6 similarly increase the levels of endogenous splicing factors as HDAC1; yet analysis of the SMN reporter transcripts argues the mechanism of SMN increase differs between the two HDACs. Therefore, there is no direct correlation of these splicing factor mRNA levels to the affect on SMN reporter mRNA. Modulation of SMN splicing is not exclusive to these three proteins measured. The entirety of proteins required to promote exon 7 inclusion may not be sufficiently modified by single HDAC knockdown. Hence, the reason why more potent pan-HDAC inhibitors may be required to directly modify SMN exon 7 splicing.
Fig. 3.
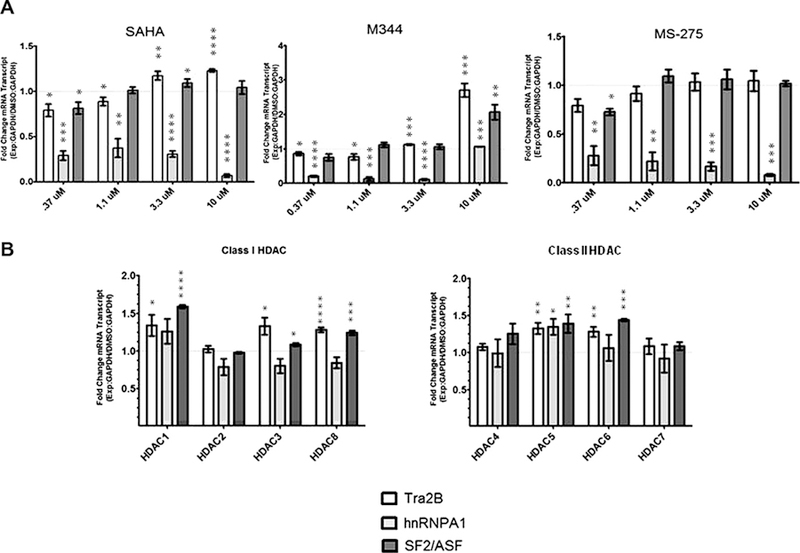
Quantitative PCR of endogenous splicing factors in response to HDAC inhibitor treatment in SMN2-luciferase reporter cells. SMN2-luciferase reporter cells were treated with 4 concentrations of HDAC inhibitors SAHA, M344, and MS-275. mRNA levels of splicing factors hTra2b (white bars), hnRNPA1 (gray bars), and SF2/ASF (dark gray bars). qRT-PCR was used to measure fold changes in transcript levels relative to DMSO carrier treated cells. GAPDH was used for normalization. (B) SMN2-luciferase reporter cells were treated with HDAC specific shRNA. mRNA levels of splicing factors hTra2b (white bars), hnRNPA1 (gray bars), and SF2/ASF (dark gray bars). qRT-PCR was used to measure fold changes in transcript levels relative to a non-silencing shRNA control. GAPDH was used for normalization.
3.4. Quantification of acetyl-histone H3 and RNA Polymerase II at the reporter promoter
We measured the amount of acetyl-histone H3 and RNA Polymerase II (RNAP) present at the reporter promoter to determine the effects shRNA or HDAC inhibitor treatment imparts on transcriptional modulators. We treated reporter cells with a single dosage of each HDACi as well as each shRNA. We performed chromosome immunoprecipitation (ChIP) using an acetyl-histone H3 and RNAPII antibodies and subsequent qPCR to measure the percent of input DNA bound by acetyl-H3 and RNAP. Treatment of the cells with each of the three HDAC inhibitors resulted in increases of approximately 1.5-fold in RNAP ChIP at the reporter promoter. Increased ChIP of acetyl-H3 was moderate with SAHA and MS-275 treatment (Fig. 3). Increases of acetyl-H3 with HDACi in the reporter cells were consistent with published reports. HDAC shRNA treatment did not yield any statistically significant increases of acetyl-H3 or RNAP ChIP with the exception of HDAC7 (Fig. 4). HDAC7 silencing increased the amount of RNAP Polymerase ChIP at the SMN2 reporter promoter 1.4-fold. This result is comparable to drug treatment and supports the data that HDAC7 primarily regulates SMN transcription (Fig. 2B).
Fig. 4.
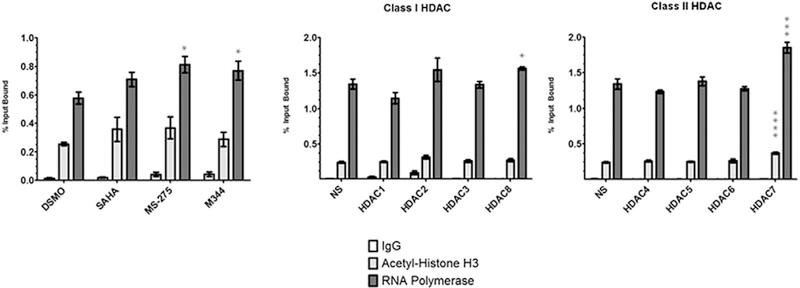
Quantitative chromosome immunoprecipitation of the SMN2-luciferase promoter. The SMN promoter was isolated from HDAC inhibitor and HDAC shRNA treated HEK293T SMN2-luciferase reporter cells using chromosome immunoprecipitation. Irrelevant rabbit IgG was used for a negative control. qPCR was used to accurate determine the percent of input DNA bound by anti-acetyl-H3 and anti-RNA Polymerase CTD antibodies.
4. Discussion
HDAC inhibitors most effective at increasing SMN protein levels do not discriminate between HDAC classes. These drugs act globally, and as a result increase expression of the SMN2 gene and the levels of other modulating proteins such as splicing factors. However, the pitfalls of off-target effects need to be addressed. To examine this and gain a better understanding of the contributions of individual HDAC proteins, we targeted individual HDAC mRNAs and measured the effects of shRNA treatment on SMN2 transcription and splicing.
In a HEK293 SMN2-luciferase reporter cell line we were able to detect changes in SMN2 mRNA and protein in response to individual HDAC knockdown. We were able to demonstrate that targeted silencing of individual HDAC proteins increased full length SMN protein. Our experiments support that each HDAC protein, with the exception of HDAC5, contributes to increasing SMN protein with potential of overlapping of mechanisms. Inhibition of class I HDACs or class II HDACs 4 and 7, appeared to modulate SMN2 reporter transcription, while exon 7 inclusion was regulated by the class II HDAC proteins 5 and 6.
However, HDAC2 and HDAC 6 may be the most compelling targets for SMA therapy. Recent works have identified these two HDACs as having neuroprotective properties. Specifically, HDAC2 deficiency resulted in increased synapse number as well as improved memory formation [19]. Interestingly, HDAC2, but not HDAC1, populates promoters of genes directly associated with synaptic plasticity. Further highlighting the role of HDAC2 in synaptic plasticity, SAHA was unable to change synapse numbers in HDAC2 deficient mice in contrast to treated wild-type animals [19]. This suggests that the transcriptional effect of SAHA on SMN2 expression may be dependent on HDAC2.
HDAC6 is an interesting drug target because a neuron-specific function has been extensively established. Regulation of HDAC6 levels or microtubule related activity could drastically alter a motor neurons ability to respond to external stimuli and either promote or inhibit survival. HDAC6’s role in preventing autophagy is linked to association with poly-ubiquitinated proteins and promoting their degradation reducing aggregation related neurondegredation [20,21]. It is possible that inhibition of HDAC6 alters the stability of the SMN protein by countering any mechanism that may promote the proteosome based degradation of SMN. In combination with proposed neuroprotective properties, as well as unique mechanisms to increase full length SMN protein, inhibition of one or both of these two HDAC proteins may have profound implications in SMA therapy.
The advantage of pan-inhibitor compounds is that transcriptional and splicing pathways are being targeted simultaneously. This is most likely why the most potent HDACi drugs currently under investigation for SMA fall into this class of compounds. However, many of these same drugs mentioned, including SAHA, MS-275, phenyl butyrate, valproic acid, and sodium butyrate are currently used or are in clinical trials for cancer therapy. It is this class of drugs propensity to induce toxic effects on cell cycle and also cell death that make them valuable in this context. Compounds that are more specific in could be more effective in SMA therapy and lack potential undesirable cytotoxic side effects.
Development of more effective and targeted therapies depends of the complete understanding of HDACs are involved in SMN regulation. This study has shown that inhibition of an individual HDAC is sufficient to increase SMN2 transcription as well as promote the inclusion of exon 7. Based on our observations, it should be possible to identify compounds that increase SMN expression while avoiding the broad genomic effects of the pan-HDAC inhibitors it and preventing any associated toxicity. A compound that is capable of achieving this, while maintaining potency would be extremely beneficial to SMA patients and families.
References
- [1].Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. , Identification and characterization of a spinal muscular atrophy-determining gene, Cell 80 (1995) 155–165. [DOI] [PubMed] [Google Scholar]
- [2].Lorson CL, Androphy EJ, An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN, Hum. Mol. Genet 9 (2000) 259–265. [DOI] [PubMed] [Google Scholar]
- [3].Lorson CL, Hahnen E, Androphy EJ, Wirth B, A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy, Proc. Natl. Acad. Sci. USA 96 (1999) 6307–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, McPherson JD, A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2, Hum. Mol. Genet 8 (1999) 1177–1183. [DOI] [PubMed] [Google Scholar]
- [5].Kashima T, Manley JL, A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy, Nat. Genet 34 (2003) 460–463. [DOI] [PubMed] [Google Scholar]
- [6].Singh NN, Androphy EJ, Singh RN, An extended inhibitory context causes skipping of exon 7 of SMN2 in spinal muscular atrophy, Biochem. Biophys. Res. Commun 315 (2004) 381–388. [DOI] [PubMed] [Google Scholar]
- [7].Cartegni L, Krainer AR, Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1, Nat. Genet 30 (2002) 377–384. [DOI] [PubMed] [Google Scholar]
- [8].Taylor JE, Thomas NH, Lewis CM, Abbs SJ, Rodrigues NR, Davies KE, Mathew CG, Correlation of SMNt and SMNc gene copy number with age of onset and survival in spinal muscular atrophy, Eur. J. Hum. Genet 6 (1998) 467–474. [DOI] [PubMed] [Google Scholar]
- [9].Feldkotter M, Schwarzer V, Wirth R, Wienker TF, Wirth B, Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy, Am. J. Hum. Genet 70 (2002) 358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mercuri E, Bertini E, Messina S, Solari A, D’Amico A, Angelozzi C, Battini R, Berardinelli A, Boffi P, Bruno C, Cini C, Colitto F, Kinali M, Minetti C, Mongini T, Morandi L, Neri G, Orcesi S, Pane M, Pelliccioni M, Pini A, Tiziano FD, Villanova M, Vita G, Brahe C, Randomized, double-blind, placebocontrolled trial of phenylbutyrate in spinal muscular atrophy, Neurology 68 (2007) 51–55. [DOI] [PubMed] [Google Scholar]
- [11].Brahe C, Vitali T, Tiziano FD, Angelozzi C, Pinto AM, Borgo F, Moscato U, Bertini E, Mercuri E, Neri G, Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients, Eur. J. Hum. Genet 13 (2005) 256–259. [DOI] [PubMed] [Google Scholar]
- [12].Swoboda KJ, Scott CB, Reyna SP, Prior TW, LaSalle B, Sorenson SL, Wood J, Acsadi G, Crawford TO, Kissel JT, Krosschell KJ, D’Anjou G, Bromberg MB, Schroth MK, Chan GM, Elsheikh B, Simard LR, Phase II open label study of valproic acid in spinal muscular atrophy, PLoS One 4 (2009) e5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wade PA, Transcriptional control at regulatory checkpoints by histone deacetylases: molecular connections between cancer and chromatin, Hum. Mol. Genet 10 (2001) 693–698. [DOI] [PubMed] [Google Scholar]
- [14].de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB, Histone deacetylases (HDACs): characterization of the classical HDAC family, Biochem. J 370 (2003) 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hahnen E, Eyupoglu IY, Brichta L, Haastert K, Trankle C, Siebzehnrubl FA, Riessland M, Holker I, Claus P, Romstock J, Buslei R, Wirth B, Blumcke I, In vitro and ex vivo evaluation of second-generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy, J. Neurochem 98 (2006) 193–202. [DOI] [PubMed] [Google Scholar]
- [16].Riessland M, Ackermann B, Forster A, Jakubik M, Hauke J, Garbes L, Fritzsche I, Mende Y, Blumcke I, Hahnen E, Wirth B, SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy, Hum. Mol. Genet 19 (2010) 1492–1506. [DOI] [PubMed] [Google Scholar]
- [17].Riessland M, Brichta L, Hahnen E, Wirth B, The benzamide M344, a novel histone deacetylase inhibitor, significantly increases SMN2 RNA/protein levels in spinal muscular atrophy cells, Hum. Genet 120 (2006) 101–110. [DOI] [PubMed] [Google Scholar]
- [18].Zhang ML, Lorson CL, Androphy EJ, Zhou J, An in vivo reporter system for measuring increased inclusion of exon 7 in SMN2 mRNA: potential therapy of SMA, Gene Ther 8 (2001) 1532–1538. [DOI] [PubMed] [Google Scholar]
- [19].Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH, HDAC2 negatively regulates memory formation and synaptic plasticity, Nature 459 (2009) 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pandey UB, Batlevi Y, Baehrecke EH, Taylor JP, HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration, Autophagy 3 (2007) 643–645. [DOI] [PubMed] [Google Scholar]
- [21].Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP, HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS, Nature 447 (2007) 859–863. [DOI] [PubMed] [Google Scholar]