

Abstract
Atherosclerosis (AS) is a chronic inflammatory disease characterized by accumulating deposition of lipids in the arterial intima. Notably, macrophages participate centrally in the pathogenesis of this deadly disease. In this study, we established AS mouse models in order to investigate the effect of microRNA-133b (miR-133b) on vulnerable plaque formation and vascular remodeling in AS and explore the potential functional mechanisms. The expression of miR-133b was altered or the Notch-signaling pathway was blocked in the AS mouse models in order to evaluate the proliferation, migration, and apoptosis of macrophages. It was observed that miR-133b was upregulated in AS, which might target MAML1 to regulate the Notch-signaling pathway. AS mice with downregulated miR-133b or inhibited Notch-signaling pathway presented with a reduced AS plaque area, a decreased positive rate of macrophages, and an increased positive rate of vascular smooth muscle cells. Moreover, Notch-signaling pathway blockade or miR-133b downregulation inhibited the macrophage viability and migration and accelerated the apoptosis. This study provides evidence that downregulated miR-133b expression may inhibit the immune responses of macrophages and attenuate the vulnerable plaque formation and vascular remodeling in AS mice through the MAML1-mediated Notch-signaling pathway, highlighting miR-133b as a novel therapeutic target for AS.
Keywords: microRNA-133b, atherosclerosis, Notch-signaling pathway, macrophage, plaque formation
Graphical Abstract
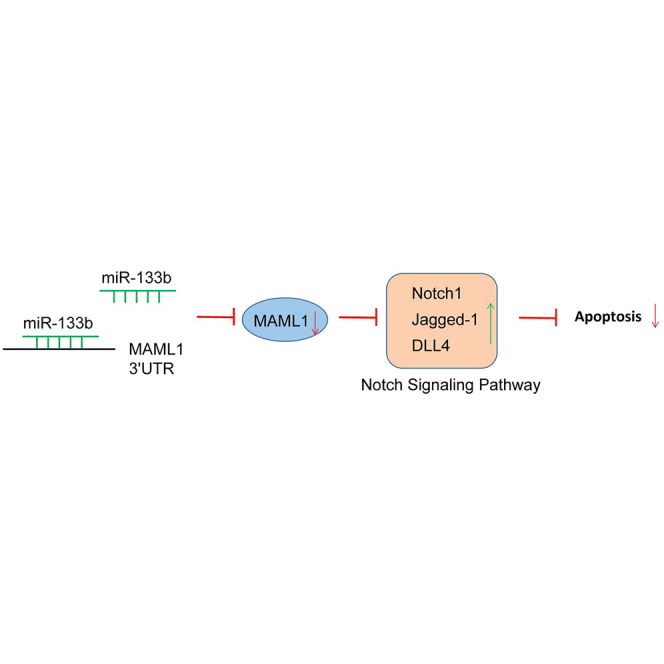
Introduction
Atherosclerosis (AS) is characterized as a chronic inflammatory disease caused by cardiovascular dysfunction, such as unstable angina, myocardial infarction, peripheral thromboses, stroke, and sudden cardiac death.1 It is typically stirred by the interplay between endothelial dysfunction and subendothelial lipoprotein retention, which occurs primarily at regions with blocked blood flow and in the inner membrane of medium-sized arteries, especially at the points of bifurcations and arterial branching.2, 3 The rupture of vulnerable atherosclerotic plaque accounts for the majority of clinically significant acute cardiovascular events in patients.4 The vascular lesions resemble early stages of AS, are regarded as acute atherosis, and are related to the impaired vascular remodeling of the spiral arteries.5 The present-day treatments for AS emphasize the limiting of its risk factors like hyperlipidemia or hypertension and modulating inflammation in the arterial wall.6 Therefore, it is necessary to provide potential molecular targets for the development of intervention strategies for AS.
MicroRNAs (miRNAs) have been introduced as biomarkers in the event of numerous cardiovascular diseases.7 Multiple studies have reported that miRNAs exert effects on cardiovascular cell functioning, and miR-133b is one of them that regulates cardiohypertrophy, embryonic heart development, and muscle cell proliferation.8 miR-133b is located at chromosome 6p12.2, and it modulates the expression of cardiac ion channels and cardiac myogenesis and development.9, 10 An increased level of miR-133a in patients with cardiovascular diseases primarily originates from injury to the myocardium, and it can serve as a marker for cardiomyocyte death in cardiovascular diseases.11
As bioinformatics websites and dual-luciferase reporter gene assay verified in the current study, mastermind-like 1 (MAML1) is a target gene of miR-133b. MAML1, as a member of the MAML family, is a transcriptional co-activator in the Notch-signaling pathway.12, 13 The Notch-signaling pathway is a key regulator of differentiation in a variety of organisms and tissue types, and it acts as a tumor suppressor in solid tumors.14 Studies have indicated that the Notch-signaling pathway is involved in the development and maintenance of the cardiovascular system as well as the modulation of inflammation, which plays a main role in cardiovascular diseases.15 For example, in 2012, Fukuda et al.16 reported that the Notch-signaling pathway is associated with the occurrence and progression of AS. In addition, Mao and Jiang17 demonstrated in a recent study of 2018 that the Notch-signaling pathway is activated in the vessels of AS animal models and inhibition of the Notch-signaling pathway can significantly improve AS. Moreover, Aquila et al.18 reported that Notch ligand Delta-like 4 (DLL4) and Jagged-1 participate in the progression of arterial disease.
From the above findings, we hypothesized that miR-133b, the MAML1 gene, and the Notch-signaling pathway are involved in AS. Therefore, this study was aimed at exploring the effects of miR-133b on the biological activities of macrophages in AS mouse models by binding to MAML1 via the Notch-signaling pathway.
Results
miR-133b Plays a Role in AS through the MAML1-Mediated Notch-Signaling Pathway
The R language package was employed in order to conduct differential analysis and screen the differential miRNAs from GEO: GSE26555. The expression heatmap of 10 differential miRNAs is shown in Figure 1A, indicating that miR-133b was significantly highly expressed in five AS samples (GEO: GSM931490, GSM931492, GSM931496, GSM931497, and GSM931498; red indicates high relative expression, and green indicates low relative expression). The expression patterns of the five AS samples were consistent, suggesting that the experimental conditions were stable and the data are reliable.
Figure 1.

miR-133b Affects AS by Targeting MAML1 and Regulating the Notch-Signaling Pathway
(A) Expression heatmap of 10 differential miRNAs in GEO: GSE26555. The abscissa represents sample number while the ordinate represents differential miRNAs. The upright histogram is color gradation, where each rectangle corresponds to one sample expression; red indicates high expression while green indicates low expression. (B) Comparison of target genes of miR-133b from TargetScan, miRDB, and DIANA. AS, atherosclerosis; miR-133b, microRNA-133b; MAML1, mastermind-like 1.
Next, the TargetScan, miRDB, and DIANA databases were used to predict the target genes of miR-133b. A total of 7 genes was selected from TargetScan based on the Cumulative weighted context2+ score < −0.5 and Aggregate probability of conserved targeting (PCT) > 0.9, while 29 predicted genes were obtained from miRDB according to Target Score > 95, and 132 genes were obtained from DIANA in accordance with miRNA target gene (miTG) score > 0.95. Subsequently, based on comparisons of target gene differences, a Venn map was plotted (Figure 1B), which demonstrated 2 intersection genes, namely, MAML1 and CELF4. miR-133b might regulate these 2 genes. The research of potential participant signaling pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database found that MAML1 is involved in the Notch-signaling pathway (map04330), yet research on CELF4 remained scarce. In addition, the Notch-signaling pathway has been previously reported to be vital for vascular diseases such as AS.16, 19, 20 Thus, it could be inferred that miR-133b targeted MAML1 to affect the Notch-signaling pathway in AS.
Successful Establishment of AS Mouse Models
The current study commenced experimentation with the establishment of AS mouse models. Four indexes of blood lipid profile were examined to verify the establishment of AS model, including low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), TC (total cholesterol), and triacylglycerol (TG) (Table 1). Significantly increased levels of LDL-C, TC, and TGs and a decreased level of HDL-C in modeled mice indicated toward successful establishment of AS models (p < 0.05).
Table 1.
Levels of Indexes of Blood Lipids (LDL-C, HDL-C, TC, and TGs) Reveal the Successful Establishment of AS Model in Mice
| Group | Case (n) | LDL-C (mmol/L, mean ± SD) | HDL-C (mmol/L, mean ± SD) | TC (mmol/L, mean ± SD) | TG (mmol/L, mean ± SD) |
|---|---|---|---|---|---|
| Healthy mice | 20 | 0.30 ± 0.03 | 0.63 ± 0.09 | 2.46 ± 0.15 | 0.61 ± 0.03 |
| AS mouse models | 20 | 10.11 ± 2.98∗ | 0.58 ± 0.93∗ | 18.38 ± 1.44∗ | 2.02 ± 0.57∗ |
LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; TC, total cholesterol; TG, triacylglycerol; AS, atherosclerosis.
p < 0.05, compared with the healthy mice.
Downregulated miR-133b Improves Pathological Changes in AS Mouse Models
To investigate whether miR-133b affected the pathological process of AS, the AS mice were injected with agomir or antagomir of miR-133b. Moreover, we also injected some control and AS mice with DAPT (N-[N-(3,5-difluorophenacetyl)-1-alanyl]-S-phenylglycine t-butyl ester), an inhibitor of the Notch-signaling pathway, to explore whether the Notch-signaling pathway participated in the influence of miR-133b on AS development. H&E staining was performed in order to measure the pathological changes in mice (Figure 2).
Figure 2.

Downregulated miR-133b Alleviates Pathological Changes of AS Mouse Models (400×)
Black arrow indicates toward typical atheromatous plaque. AS, atherosclerosis; miR-133b, microRNA-133b.
The vessel morphology in the control and control + DAPT groups was observed to be normal without AS plaque, and the endarterium was smooth without foam cells or inflammatory cells. The vessel wall in the AS and AS + miR-133b agomir + DAPT groups was thickened and the lumen was narrowed, with the appearance of vulnerable AS plaque characterized by multi-fibrous cap and enlarged lipid core. In the AS + miR-133b agomir group, when compared with the AS group, the AS plaque was observed to be further thickened and the lumen was narrower; more multi-fibrous cap, a larger lipid core, a bigger plaque area, and unstable plaque shape were observed. However, in the AS + DAPT and AS + miR-133b antagomir groups, the lumen was observed to be slightly narrowed, the tunica intima was mostly smooth, the multi-fibrous cap was thick, the lipid core was small, the plaque area was rather small, and plaque shape tended to be stable, which were suggestive of pathological improvement as compared with the AS group. Thus, these findings demonstrated that miR-133b depletion contributes to pathological improvement in AS mouse models.
Downregulated miR-133b and Inactivation of the Notch-Signaling Pathway Decrease the Positive Rate of Macrophages and Increase that of VSMCs in AS Mouse Models
Immunohistochemistry (Figure 3) and quantitative analysis (Table 2) revealed the positive rate of cells in different groups. There were no significant differences in the positive rates of macrophages and vascular smooth muscle cells (VSMCs) between the control group and the control + DAPT group (p > 0.05), while the positive rate of macrophages was found to be increased significantly and that of VSMCs was decreased remarkably in the rest of the groups (all p < 0.05). Compared with the AS group, the AS + miR-133b agomir group showed notably increased positive rates of macrophages and significantly decreased positive rates of VSMCs; the AS + DAPT group and the AS + miR-133b antagomir group exhibited significantly lower positive rates of macrophages and notably increased VSMCs (all p < 0.05); and the AS + miR-133b agomir + DAPT group presented no significantly different positive rates of macrophages and VSMCs (p > 0.05).
Figure 3.

Downregulated miR-133b Increases the Positive Rate of Macrophages and Decreases the Positive Rate of VSMCs in the AS Mouse Models
(A) The immunohistochemical staining of CD68 in each group (scale bar, 25 μM). (B) The immunohistochemical staining of α-SMA in each group (scale bar, 25 μM). AS, atherosclerosis; VSMC, vascular smooth muscle cell; miR-133b, microRNA-133b.
Table 2.
Quantitative Analysis for the Positive Rates of CD68 and α-SMA in Carotid Artery of Mice Detected by Immunohistochemistry
| Group | Case (n) | CD68 Positive Rate (%, mean ± SD) | α-SMA Positive Rate (%, mean ± SD) |
|---|---|---|---|
| Control | 20 | 20.23 ± 3.71 | 20.24 ± 1.37 |
| AS | 20 | 30.03 ± 3.48∗ | 13.56 ± 2.63∗ |
| Control + DAPT | 20 | 20.01 ± 2.99 | 20.48 ± 1.28 |
| AS + DAPT | 20 | 24.99 ± 1.07∗,# | 16.36 ± 2.94∗,# |
| AS + miR-133b agomir | 20 | 35.20 ± 2.83∗,# | 10.06 ± 1.38∗,# |
| AS + miR-133b antagomir | 20 | 23.18 ± 1.34∗,# | 16.27 ± 2.74∗,# |
| AS + miR-133b agomir + DAPT | 20 | 29.64 ± 1.26∗ | 12.58 ± 2.11∗ |
AS, atherosclerosis; VSMC, vascular smooth muscle cell; DAPT, N-[N-(3,5-difluorophenacetyl)-1-alanyl]-S-phenylglycine t-butyl ester; miR-133b, microRNA-133b.
p < 0.05, compared with the control group.
p < 0.05, compared with the AS group.
The aforementioned results showed that downregulation of miR-133b and inhibition of the Notch-signaling pathway might attenuate the immune reaction of macrophages, which could affect the formation of vulnerable AS plaque formation and vascular remodeling in AS mouse models.
MAML1 Is the Target Gene of miR-133b
The targeting relationship between miR-133b and MAML1 was predicted using bioinformatics websites (Figure 4A). As shown in Figure 4B, the dual-luciferase reporter gene assay revealed that cells co-transfected with miR-133b agomir and MAML1-wild-type (WT) plasmid exhibited significantly lowered luciferase activity compared to the cells co-transfected with miR-133b agomir-negative control (NC) and MAML1-WT plasmid (p < 0.05), while no obvious differences were observed in other groups (p > 0.05). The above results demonstrated that miR-133b targeted MAML1.
Figure 4.

MAML1 Is a Target Gene of miR-133b
(A) Binding sites of miR-133b to MAML1 based on bioinformatics prediction. (B) Luciferase activity of MAML1-WT and MAML1-Mut in the miR-133b agomir and miR-133b-NC groups. *p < 0.05, compared with the MAML1-WT + miR-133b agomir-NC group; the measurement data were presented as the mean ± SD; the experiment was repeated 3 times. miR-133b, microRNA-133b; NC, negative control; Mut, mutant; WT, wild-type; MAML1, mastermind-like 1.
miR-133b Downregulation Decreases Levels of MMP1 and MMP9 but Elevates TIMP1 Levels
Matrix metalloproteinases (MMPs) play a significant role in the stability of AS plaque and vascular remodeling. The current study employed ELISA to detect the levels of MMP1, MMP9, and tissue inhibitor of metalloproteinase 1 (TIMP1) (Table 3). There were no significant differences in the levels of MMP1, MMP9, and TIMP1 between the blank + DAPT group and blank group (p > 0.05); however, the levels of MMP1 and MMP9 were found to be increased significantly and those of TIMP1 decreased obviously in the rest of the groups as compared with the blank group (p < 0.05). In contrast to the AS group, the AS + miR-133b agomir group exhibited significantly increased levels of MMP1 and MMP9 and decreased TIMP1 levels; and the AS + DAPT and AS + miR-133b antagomir groups showed lowered levels of MMP1 and MMP9 and elevated TIMP1 levels (all p < 0.05). No significant differences were observed between the AS + miR-133b agomir + DAPT group and the AS group in the levels of MMP1, MMP9, and TIMP1 (p > 0.05). These results revealed that miR-133b antagomir could reduce AS plaque and vascular remodeling by regulating the levels of MMP1, MMP9, and TIMP1.
Table 3.
The Levels of MMP1, MMP9, and TIMP1 in Macrophages of Mice (n = 20)
| Group | MMP1 (μg/L, mean ± SD) | MMP9 (μg/L, mean ± SD) | TIMP1 (μg/L, mean ± SD) |
|---|---|---|---|
| Blank | 215.73 ± 10.12 | 279.63 ± 13.52 | 135.64 ± 10.37 |
| AS | 307.11 ± 15.63∗ | 593.77 ± 21.43∗ | 67.26 ± 6.33∗ |
| Blank + DAPT | 219.54 ± 11.23 | 283.56 ± 14.11 | 127.58 ± 10.14 |
| AS + DAPT | 267.14 ± 13.45∗,# | 510.33 ± 23.47∗,# | 89.69 ± 7.53∗,# |
| AS + miR-133b agomir | 347.56 ± 13.47∗,# | 669.63 ± 26.71∗,# | 35.47 ± 3.46∗,# |
| AS + miR-133b antagomir | 259.33 ± 13.41∗,# | 517.34 ± 24.12∗,# | 91.32 ± 7.44∗,# |
| AS + miR-133b agomir + DAPT | 306.75 ± 14.21∗ | 611.33 ± 21.69∗ | 66.19 ± 6.04∗ |
miR-133b, microRNA-133b; MMP, matrix metalloproteinase; TIMP1, tissue inhibitor of metalloproteinase 1; AS, atherosclerosis; DAPT, N-[N-(3,5-difluorophenacetyl)-1-alanyl]-S-phenylglycine t-butyl ester.
p < 0.05, compared with the blank group.
p < 0.05, compared with the AS group.
Decreased miR-133b Inhibits Macrophage Proliferation
Macrophage viability of AS mouse models was measured using cell-counting kit-8 (CCK-8) (Figure 5). Compared with the blank group, the blank + DAPT group showed no significant differences in macrophage viability (p > 0.05), while the viability was accelerated in the other groups (p < 0.05). After 24 h of transfection, compared with the AS group, the AS + miR-133b agomir group exhibited notably increased macrophage viability; the AS + DAPT and AS + miR-133b antagomir groups presented with inhibited macrophage viability (p < 0.05); and the AS + miR-133b + DAPT group showed no significant changes in viability (p > 0.05). The CCK-8 results indicated that miR-133b inhibition decreased proliferation in macrophages.
Figure 5.

Downregulated miR-133b Inhibits Macrophage Growth and Proliferation by CCK-8 Assay
*p < 0.05, compared with the blank group; #p < 0.05, compared with the AS group;the measurement data were presented as the mean ± SD; the experiment was repeated 3 times; OD, optical density; AS, atherosclerosis; miR-133b, microRNA-133b; NC, negative control; CCK-8, cell counting kit-8.
Downregulation of miR-133b Suppresses Macrophage Migration
The migration rate of macrophages was assessed using scratch tests (Figure 6). Compared with the blank group, the blank + DAPT group showed no significant differences in macrophage migration (p > 0.05), while macrophage migration was significantly increased in the other groups (p < 0.05). Compared with the AS group, macrophage migration rate was significantly augmented in the AS + miR-133b agomir group but lowered in the AS + miR-133b antagomir and the AS + DAPT groups (p < 0.05), while no obvious changes were observed in the AS + miR-133b agomir + DAPT group (p > 0.05). The above results proved that miR-133b inhibition could attenuate the migration of macrophages.
Figure 6.

Downregulation of miR-133b Inhibits Macrophage Migration by Scratch Test
(A) Scratch test results of mouse macrophages in different groups. (B) Quantitative analysis for migration rate of mouse macrophages in different groups. *p < 0.05, compared with the blank group; #p < 0.05, compared with the AS group; the measurement data were presented as the mean ± SD; the experiment was repeated 3 times. AS, atherosclerosis; miR-133b, microRNA-133b.
Downregulated miR-133b Promotes Macrophage Apoptosis
FITC was then detected using flow cytometry at the wavelength of 480 and 530 nm and PI at the wavelength of over 575 nm (Figure 7; left lower quadrant, normal cells; left upper quadrant, dead cells; right lower quadrant, apoptotic cells at the early stage; and right upper quadrant, apoptotic cells at the advanced stage). The apoptosis of macrophages in the blank group was not significantly different from that in the blank + DAPT group (p > 0.05). However, as compared with the blank group, the AS, AS + miR-133b agomir, AS + DAPT, AS + miR-133b antagomir, and AS + miR-133 agomir + DAPT groups showed significantly increased macrophage apoptosis. Compared with the AS group, the AS + miR-133b agomir group showed decreased apoptosis; the AS + DAPT and AS + miR-133b antagomir groups presented with increased apoptosis rates (p < 0.05); while the AS + miR-133 agomir + DAPT group exhibited no significant changes in apoptosis (p > 0.05). These findings showed that miR-133b downregulation could promote apoptosis in macrophages.
Figure 7.

Downregulation of miR-133b Promotes Macrophage Apoptosis by Flow Cytometry
(A) Flow cytometric cell apoptosis of mouse macrophages in different groups. (B) Quantitative analysis for apoptosis rate of mouse macrophages in different groups. *p < 0.05, compared with the blank group; #p < 0.05, compared with the AS group; the measurement data were presented as the mean ± SD; the experiment was repeated 3 times. AS, atherosclerosis; miR-133b, microRNA-133b.
Downregulated miR-133b Elevates MAML1 and Inhibits the Activation of the Notch-Signaling Pathway
The mRNA and protein expressions of MAML1, the Notch-signaling pathway-related genes, and apoptosis-related genes in each group were detected using qRT-PCR (Figure 8A) and western blot analysis (Figures 8B and 8C). Compared with the blank group, the AS group exhibited upregulated expressions of miR-133b and downregulated mRNA and protein expressions of MAML1. Compared with the AS group, miR-133b expression was found to be elevated while the mRNA and protein expressions of MAML1 were decreased in the AS + miR-133b agomir and AS + miR-133b agomir + DAPT groups, while the opposite trends were observed in the AS + miR-133b antagomir group (all p < 0.05).
Figure 8.

Downregulation of miR-133b Induces Increased MAML1 Expression and Inactivated Notch-Signaling Pathway
(A) Relative expressions of miR-133b, Notch1, Jagged-1, DLL4, Bax, Bcl-2, and Caspase-3, as detected by qRT-PCR. (B) Gray values of MAML1, Notch1, Jagged-1, DLL4, Bax, Bcl-2, and Caspase-3, as determined by western blot analysis. (C) Statistical analysis of the gray values in (B). *p < 0.05, compared with the blank group; #p < 0.05, compared with the AS group; the measurement data were presented as the mean ± SD; the experiment was repeated 3 times. miR-133b, microRNA-133b; MAML1, mastermind-like 1; DLL4, Delta-like ligand 4; Bcl-2, B cell lymphoma-2; AS, atherosclerosis.
The mRNA and protein expressions of the Notch-signaling pathway-related genes (Notch1, Jagged-1, and DLL4) were downregulated in the blank + DAPT group, while they were upregulated in the remaining groups compared to the blank group (all p < 0.05). The mRNA and protein expressions of Notch1, Jagged-1, and DLL4 were not significantly different between the AS + miR-133b agomir + DAPT and AS groups (p > 0.05), but they were downregulated in the AS + DAPT and AS + miR-133b antagomir groups, while upregulated expressions were observed in the AS + miR-133b agomir group.
B cell lymphoma-2 (Bcl-2) is a negative regulator of cell apoptosis while Bax and Caspase-3 are positive regulatory genes of apoptosis, whose mRNA and protein expressions presented with no significant differences between the blank and the blank + DAPT groups (p > 0.05). The mRNA and protein expressions of Bcl-2 were upregulated, while those of Bax and Caspase-3 were downregulated in the remaining groups (p < 0.05) in comparison with the blank group. The mRNA and protein expressions of Bcl-2 were upregulated, while those of Bax and Caspase-3 were downregulated in the AS + miR-133b agomir group as compared with the AS group. The AS + DAPT and AS + miR-133b antagomir groups exhibited reduced Bcl-2 expression and increased expressions of Bax and Caspase-3 in comparison with the AS group (p < 0.05). The mRNA and protein expressions of Bcl-2, Bax, and Caspase-3 were not significantly different between the AS + miR-133b agomir + DAPT group and the AS group (p > 0.05). All these findings revealed that the downregulation of miR-133b inhibited the mRNA and protein expressions of Notch1, Jagged-1, DLL4, and Bcl-2 while promoting those of Bax and Caspase-3, thus inhibiting the Notch-signaling pathway and promoting apoptosis in macrophages.
Discussion
The accumulation of macrophages in the vascular wall responding to environmental stimuli, including modified lipids and cytokines, leads to AS lesions.21 Inflammatory pathway activation in macrophage lesions plays significant roles in promoting the proatherogenic process.3 In the current study, AS models were established in mice in order to explore the effect of miR-133b on the degradation, atrophy, and apoptosis of macrophages. Eventually, the study provided evidence revealing that miR-133b downregulation could inhibit the Notch-signaling pathway, thus promoting macrophage apoptosis and suppressing proliferation and migration in AS mouse models.
Initially, our findings revealed that, in the macrophages transfected with DAPT or miR-133b antagomir, the lumen was slightly narrowed, the tunica intima was mostly smooth, the multi-fibrous cap was thick, the lipid core was small, the plaque area was rather small, and the plaque shape tended to be stable, all of which were suggestive of pathological improvement of AS conditions. These findings demonstrated that downregulation of miR-133b or inhibition of the Notch-signaling pathway decreased the area of vulnerable plaque and vessel thickness. Vulnerable plaques are defined as precursors to lesions that rupture, while plaque rupture results in about 75% of all fatal coronary thrombi.22, 23
There are numerous causes of plaque development, including the development of smooth muscle cells and endothelial cells, collagens, MMPs, fibronectin, elastin, and excessive foam cells.1 A previous study reported the existence of alterations in the expressions of specific miRNAs in human atherosclerotic plaques, and it suggested that miRNAs may regulate the evolution of atherosclerotic plaque toward instability and rupture.24 In addition, our data suggested that the downregulation of miR-133b and inhibition of the Notch-signaling pathway led to higher positive rates of macrophages and notably decreased VSMCs; moreover, they lowered the levels of MMP1 and MMP9 and elevated that of TIMP1.
VSMCs are engaged in arterial wall remodeling, which would maintain blood flow in affected vessels caused by atherosclerotic alteration and function flexibly responding to vascular injury.25 The transformation of VSMCs into foam cells leads to increased plaque size and decreased stability, and interleukin-19-induced miR133a expression stimulated VSMCs, representing a link between vascular lipid metabolism and inflammation.26 Additionally, miR-133b has been reported to directly target MMP9, the silencing of which inhibited viability and migration in human non-small-cell lung cancer.27 Similarly, MMPs play critical roles in the pathogenesis of coronary artery disease, a stage of AS involving atheromas inside the arterial walls, thus resulting in narrowing of the arteries and culminating in decreased blood flow (p: 23105899). MMPs and TIMPs are associated with AS and plaque rupture, and increased levels of MMP1 and MMP9 were observed in atheromatous, vulnerable plaques.28 Supportively, a previous study indicated that miR-133b could inhibit the expression of MMP9.29 Therefore, downregulation of miR-133b or inhibition of the Notch-signaling pathway could attenuate the vulnerable plaque in AS.
Also, concluding from the qRT-PCR and western blot analyses, downregulation of miR-133b decreased the expressions of MAML, Notch1, Jagged-1, DLL4, and Bcl-2 while promoting those of Bax and Caspase-3, thus inhibiting the Notch-signaling pathway and promoting apoptosis in macrophages. Similarly, a previous study indicated that miR-133b regulated death receptor-induced apoptosis by targeting anti-apoptotic genes.30 Strikingly, one study reported that the activation of macrophages leads to the pathogenesis of inflammatory diseases such as AS, while Notch3 can increase during macrophage differentiation, and Notch3 knockdown lowered the transcription levels of genes that promoted inflammation.31 Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses.32 MAML genes are the primary components of the Notch-signaling pathway, regulating cellular events involved in both normal development and oncogenesis.33 Bcl-2 is a protein that is a critical component of the apoptotic pathway and functions chiefly as an apoptosis inhibitor.34 Previous studies have indicated that transfection of miR-133b could inhibit the activation of Caspase-3 and reverse the decrease in the Bcl-2:Bax ratio.35, 36 Jagged-1 and DLL4 are two members of the Notch family in mammals, and DLL4 is a basic factor during the formation process of mature vasculature.37 Downregulation of miR-19a inhibited metastatic renal cell carcinoma by inactivating the Notch-signaling pathway in vitro, as reported by another research.38
Our findings also indicated that the downregulation of miR-133b could inhibit the proliferation and migration while promoting apoptosis in macrophages in AS. miR-133a can serve as a biomarker to predict cardiac hypertrophy in patients with chronic hemodialysis.39 The aberrant expression of miR-133 is implicated in the nicotine-induced cardiomyocyte apoptosis via the mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK)-signaling pathway.40
In conclusion, the current study provides evidence suggesting that the downregulation of miR-133b could suppress proliferation and migration while promoting apoptosis in macrophages, to attenuate the vulnerable plaque formation and vascular remodeling by inactivating the Notch-signaling pathway via binding to MAML1 in AS mouse models. Therefore, our study further elucidates the molecular mechanism of miR-133b in AS, and it can pave the path for better treatment regimens of AS. However, further studies are warranted in order to adequately define the detailed mechanisms by which miR-133b and the Notch-signaling pathway interact and influence AS.
Materials and Methods
Ethics Statement
All animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the NIH,41 and the ethical approval was issued by the Animal Ethics Committee of Zhejiang Provincial People’s Hospital, People’s Hospital of Hangzhou Medical College. Additionally, all efforts were made to minimize the suffering of the included animals.
Microarray-Based Analysis
AS-related miRNA microarray dataset GEO: GSE26555 was obtained using the GEO database (GEO: GSE26555). Next, the R language package was applied for differential analysis, and the affy package (http://www.bioconductor.org/packages/release/bioc/html/affy.html) was utilized for standardized pretreatment of expression data. Next, the limma package (http://master.bioconductor.org/packages/release/bioc/html/limma.html) was used to conduct differential screening, and pheatmap package (https://cran.r-project.org/web/packages/pheatmap/index.html) was employed to plot the differential expression heatmap. After miRNA was selected, TargetScan (http://www.targetscan.org/vert_71/), miRDB (http://www.mirdb.org/), and DIANA (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=microT_CDS/index) databases were employed to predict the target genes of miRNA. Subsequently, the Venn online analysis tool Calculate and draw custom Venn diagrams (http://bioinformatics.psb.ugent.be/webtools/Venn/) was utilized to screen the results of the target gene. Kyoto Encyclopedia of Genes and Genomes (KEGG) PATHWAY Database (https://www.genome.jp/kegg/pathway.html) was used to research the potentially related signaling pathway.
Animal Grouping and Model Establishment of AS Model in Mice
A total of 140 healthy male ApoE−/− mice, aged 8 weeks (SCXK-Su-2011-0003, Cavens Lab Animal, Jiangsu, China), was included in the current study. Among them, 20 mice were randomly regarded as the control group, while another 20 mice served as the control + DAPT group, which were all fed basic diets for a duration of 12 weeks. The remaining 100 mice were fed high-fat feedstuff (2% cholesterol, 0.5% bile acid, 3% lard, 0.2% propylthiouracil, and 94.3% basic diet) for 12 weeks in order to establish AS models. After 12 weeks, 20 healthy mice and AS-modeled mice were selected, and venous blood samples were drawn to test the levels of LDL-C, HDL-C, TC, and TG using an automatic blood biochemical analyzer (AU5800, Beckman Coulter, CA, USA). Increased levels of LDL-C, TC, and TGs and decreased levels of HDL-C in modeled mice in comparison with the healthy mice indicated the successful establishment of AS models.
Subsequently, the modeled mice were randomly assigned into the 5 following groups with 20 mice in each group: the AS group, the AS + DAPT group (injected with DAPT, an inhibitor of the Notch-signaling pathway), the AS + miR-133b agomir group (injected with miR-133b agomir), the AS + miR-133b antagomir (injected with miR-133b antagomir), and the AS + miR-133b agomir + DAPT group (injected with miR-133b agomir and DAPT). From the second week after the successful establishment of AS models, mice in the AS + miR-133b agomir, AS + miR-133b antagomir, and AS + miR-133b agomir + DAPT groups were injected with the same volumes of miR-agomir or antagomir (dissolved in 0.2 mL normal saline, purchased from RiboBio, Guangdong, China) through the caudal vein every 2 weeks at a dose of 20 mg/kg, while the AS and the AS + DAPT groups were injected with the same dosages of normal saline. From the sixth week after modeling, mice in the control + DAPT, AS + DAPT, and AS + miR-133b agomir + DAPT groups were hypodermically injected with DAPT (dissolved in DMSO, 160 μL/mouse) every alternate day, while mice in the remaining groups were treated with hypodermic injection of DMSO (140 μL/mouse) every alternate day. Both injections were administered every other day and lasted for a total of 5 weeks.
H&E Staining
The aorta was collected from mice and fixed in 4% paraformaldehyde, followed by dehydration and permeabilization. The paraffin-embedded aorta was sliced into 0.6-mm-thick sections. A total of 3 sections were selected from each mouse, stained with H&E (Beijing Solarbio Science & Technology, Beijing, China) for 2–3 min, and dissimilated in 1% hydrochloric acid alcohol for 2–3 s. After being soaked in warm water for 10 min until a blue coloration was observed, the sections were stained with eosin for 2 min, dehydrated with gradient alcohol, and cleared in xylene. Subsequently, histopathologic changes were observed using an optical microscope after the sections were sealed with neutral balsam (the nucleus was stained blue and the cytoplasm was stained red).
Immunohistochemistry
A total of 5 aorta sections was collected from each group, treated with xylene, dehydrated with gradient ethanol, and incubated with 3% H2O2 at room temperature for 10 min. Next, the sections were immersed in a citrate buffer (0.01 nmol/L [pH 6.0]) and heated to 96°C–98°C in a microwave oven. Then the sections were blocked with 10% normal goat serum for 20 min at room temperature, and they were incubated with primary antibodies rabbit polyclonal antibody to α-SMA (dilution ratio of 1:100–300, A03744, Boster Biological Technology, Wuhan, China) and CD68 (dilution ratio of 1:2,000, ab125212, Abcam, Cambridge, MA, USA) overnight at 4°C, with PBS serving as the NC. Subsequently, the sections were added with biotin-labeled goat anti-rabbit immunoglobulin G (IgG) secondary antibody (dilution ratio of 1:2,000, ab205718, Abcam, Cambridge, MA, USA) at room temperature for 30 min.
After that, the sections were incubated with streptavidin-biotin complex (SABC) and developed with diaminobenzidine (DAB), followed by being counterstained with hematoxylin. Then, the sections were dehydrated with gradient ethanol, cleared with xylene, sealed with neutral balsam, and observed under a light microscope. The cytoplasm of smooth muscle cells (SMCs) and macrophage-positive cells was stained with pale yellow, brownish yellow, or brown coloration. Ten fields of view were selected from each slice in order to calculate the percentage of cells with positive expression to the total cells. The CIMA multiscale true color pathological image analysis system software (Image Processing Center, School of Astronautics, Beihang University, Beijing, China) was employed for image analysis.
Macrophage Separation and Culture
Mice were collected from both the control and AS groups (n = 5 in each group), and intraperitoneally injected with 3% gelatinous broth prepared with double distilled water (1.5–2 mL per mouse) for a duration of 4 days. Next, the mice were euthanized by spinal cord dislocation, and then a tiny incision was made on the lower abdomen using surgical scissors to expose the peritoneum. After that, 5 mL RPMI-1640 medium was injected into the abdominal cavity of mice. Then, the culture medium was extracted into the centrifuge tube after filtration with a 70-μMb filter. The above operations were repeated 3 times until the medium turned totally clear. Next, the extracted medium was centrifuged and added with serum-free medium, which was observed and counted under the microscope. Then, the resuspended solution was inoculated into the culture plate with the original culture medium changed after 3 h. The cells were further incubated with the addition of basic medium containing 10% fetal bovine serum (FBS).
Next, the cells were inoculated in a 6-well plate and identified until cell confluency reached over 90%. In brief, the cells were trypsinized, centrifuged, and blocked with goat serum. Next, the cells were incubated with the F4/80 antibody prepared with 200 μL PBS (dilution ratio of 1:100) for 30 min, avoiding light exposure. Finally, the sample was added with 500 μL PBS and tested with flow cytometry. After verification, the macrophages were incubated in DMEM containing 10% FBS (Gibco, Grand Island, NY, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin in a 5% CO2 incubator at 37°C.
Then, the incubated cells were assigned into the 7 following groups: the blank group (macrophages from the control group), the blank + DAPT group (macrophages from the control group treated with DAPT), the AS group (macrophages from the AS group), the AS + DAPT group (macrophages from the AS group treated with DAPT), the AS + miR-133b agomir group (macrophages from the AS group transfected with miR-133b agomir), the AS + miR-133b antagomir group (macrophages from the AS group transfected with miR-133b antagomir), and the AS + miR-133b agomir + DAPT group (macrophages from the AS group transfected with miR-133b agomir and treated with DAPT). Then the cells were treated with DAPT as follows: DAPT was primarily added into the cell culture medium for incubation (Selleck Chemicals, Houston, TX, USA), which was mixed with 1.2 mL DMSO to obtain a 10 mmol/L solution, and then diluted to 10 μmol/L. Transfection of miR-133b agomir and antagomir was performed using Lipofectamine 2000 (Invitrogen, Gaithersburg, MD, USA), according to the manufacturer’s instructions. At last, the cells were collected for further experimentation after incubation.
Dual-Luciferase Reporter Gene Assay
The target relationship between miR-133b and MAML1 was predicted using the bioinformatics prediction website, http://www.microrna.org/microrna/home.do. Total RNA content from the macrophages was extracted from each group, the potential target genes were amplified using PCR, and Hind II and Pme I cleavage sites were added to both ends of the above amplification products. Next, the pMIR-Report Luciferase vector (Biovector107902, Biovector Science Lab, Beijing, China) was treated with restriction enzymes Hind III and Pme I, and the large fragments were recycled after electrophoresis. The target gene was connected to the vector via Ligase 4 to obtain MAML1-WT and MAML1 mutant (Mut) plasmids, which were transfected into the competent cell of Escherichia col. After Escherichia coli was identified by PCR, the bacterial colony containing target gene fragments was examined by an extraction kit to get the plasmids that were later sent to be validated by gene sequencing.
Subsequently, the mouse macrophages were assigned into the following groups: the MAML1-WT + miR-133b agomir-NC group, the MAML1-Mut + miR-133b agomir-NC group, the MAML1-WT + miR-133b agomir group, and the MAML1-Mut + miR-133b agomir group. Cell transfection was performed using Lipofectamine 2000 (Invitrogen, Gaithersburg, MD, USA), according to the manufacturer’s instructions. The original solution was changed 6 h after transfection, and the cells were collected after 24 h. After collection, the fluorescence intensity was measured using a Firefly Luciferase Reporter Gene Assay Kit (RG005, Beyotime Institute of Biotechnology, Shanghai, China) and a microplate reader (MK3, Thermo, Pittsburgh, PA, USA) at an excitation wavelength of 560 nm.
ELISA
Levels of MMP1, MMP9, and TIMP1 in the supernatant of cell samples from each group were measured using MMP1 ELISA (R&D Systems, Minneapolis, MN, USA), MMP9 ELISA (ZY-MMP9-Mu, ImmunoClone, USA), and TIMP1 ELISA kits (R&D Systems, Minneapolis, MN, USA), respectively, according to the manufacturer’s instructions. The optical density (OD) value of each well was measured at an excitation wavelength of 450 nm using a microplate reader (MK3, Thermo, Pittsburgh, PA, USA), and the sample concentration was calculated according to the relationship between sample concentration and OD value.
CCK-8
The cell suspension (100 μL) was inoculated into a 96-well plate at a density of 2 × 105 cells/mL, and 3 duplicate wells were set for each group. The cells were detected at various time intervals (0, 12, 24, and 48 h) after transfection. During detection, 100 μL culture medium containing 10 μL CCK-8 solution (7sea Biotech, Shanghai, China) was added to each well and incubated for 1 h at 37°C. The OD value (A490) of each well was detected using an ELISA kit (DG5031, Kehuai Instruments, Shanghai, China). The experiment was repeated three times.
Scratch Test
Cells (n = 100 μL) were seeded in a 6-well plate at a density of 2 × 105 cells/mL. After transfection, the cells were incubated for 24 h in a serum-free medium, and then a pipette tip was used to scratch a line according to the division of the selected colony. Next, the cells were photographed and incubated in the serum-free medium for 24 h. The experiment was repeated three times.
Flow Cytometry
The apoptosis rate of macrophages in each group was detected using an Annexin V-FITC/propidium iodide (PI) (Annexin V-FITC/PI) double staining kit (556547, Shanghai Solja Technology, Shanghai, China), according to the manufacturer’s instructions. The cells were collected, centrifuged, and resuspended with pre-cooled 1× PBS, followed by the addition of 300 μL 1× binding buffer. After that, the cells were stained with 5 μL Annexin V-FITC and 1% PI (40710ES03, Shanghai Qianchen Biotechnical, Shanghai, China), respectively, avoiding light exposure. Then, FITC was detected using flow cytometry at the wavelength of 480 and 530 nm and PI at the wavelength over 575 nm (Figure 7; left lower quadrant, normal cells; left upper quadrant, dead cells; right lower quadrant, apoptotic cells at the early stage; and right upper quadrant, apoptotic cells at the advanced stage). All apoptotic cells at the right upper and lower quadrants were collected to analyze the cell apoptotic rate.
RNA Extraction and qPCR Analysis
Total RNA content was extracted from macrophages with a Trizol Reagent kit (Takara, Tokyo, Japan). The extracted RNA was reverse transcribed into cDNA using a reverse transcription kit (Takara Biotechnology, Liaoning, Dalian, China). The miR-133b expression was detected using Quanti-Tect SYBR Green PCR kit with U6 serving as the internal reference, while the mRNA expression of other genes was detected using the SYBR Premix Ex TaqII kit (Takara, Japan), with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) serving as the internal reference, on an ABI 7500 qPCR system (ABI, Oyster Bay, NY, USA). The primers used are listed in Table 4. The 2−ΔΔCt method was employed to determine the relative expression of each gene as follows: ΔΔCt = ΔCt experiment group − Ct control group; ΔCt = Ct target gene − Ct internal gene.
Table 4.
Primer Sequences for qRT-PCR
| Gene | Sequence | |
|---|---|---|
| miR-133b | forward | 5′-TTGGTCCCCTTCAACCAGCTA-3′ |
| reverse | 5′-CAGTGCGTGTCGTGGAGT-3′ | |
| U6 | forward | 5′-CTCGCTTCGGCAGCACA-3′ |
| reverse | 5′-ACGCTTCACGAATTTGCGT-3′ | |
| Notch1 | forward | 5′-GATGGCCTCAATGGGTACAAG-3′ |
| reverse | 5′-TCGTTGTTGTTGATGTCACAGT-3′ | |
| Jagged-1 | forward | 5′-ATGCAGAACGTGAATGGAGAG-3′ |
| reverse | 5′-GCGGGACTGATACTCCTTGAG-3′ | |
| DLL4 | forward | 5′-TTCCAGGCAACCTTCTCCGA-3′ |
| reverse | 5′-ACTGCCGCTATTCTTGTCCC-3′ | |
| Bax | forward | 5′-GTTTCATCCAGGATCGAGCAG-3′ |
| reverse | 5′-CCCCAGTTGAAGTTGCCATC-3′ | |
| Bcl-2 | forward | 5′-GGTACCGGAGAGCGTTCAGT-3′ |
| reverse | 5′-CTGCTGCATTGTTCCCGTAG-3′ | |
| Caspase-3 | forward | 5′-AGCTTCTTCAGAGGCGACTA-3′ |
| reverse | 5′-GGACACAATACACGGGATCT-3′ | |
| MAML1 | forward | 5′-CTGCCTCCCCTCAGAATGG-3′ |
| reverse | 5′-CTCCCGGCGTGTCTTCTTAT-3′ | |
| GAPDH | forward | 5′-GGGAAATTCAACGGCACAGT-3′ |
| reverse | 5′-AGATGGTGATGGGCTTCCC-3′ | |
miR-133b, microRNA-133b; DLL4, Delta-like ligand 4; Bcl-2, B cell lymphoma-2; Bax, Bcl2-associated X protein; MAML1, mastermind-like 1, GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Western Blot Analysis
Total protein content was extracted using mammalian protein extraction reagent (M-PER) containing protease inhibitors (Pierce, Rockford, IL, USA), and the concentration of the extracted protein was detected using a bicinchoninic acid (BCA) kit (20201ES76, Yeasen Biotech, Shanghai, China). After that, the proteins were separated by SDS-PAGE (10%) and transferred to a nitrocellulose membrane. After being blocked with 5% skimmed milk overnight at 4°C, the membrane was incubated with the following diluted primary antibodies overnight at 4°C: MAML1 (1:1,000, ab155786), Notch1 (1:500, ab8925), Jagged-1 (1:500, ab7771), DLL4 (1:2,000, ab7280), Bax (1:2,000, ab32503), Bcl-2 (1:1,000, ab59348), and Caspase-3 (1:500, ab13847). Next, the membrane was further incubated with the horseradish peroxidase (HRP)-labeled goat anti-rabbit IgG secondary antibody (1:5,000, ab6789) at room temperature for 1 h. Finally, the enhanced chemiluminescence (ECL) reaction reagent (Pierce, Waltham, MA, USA) was used to visualize the results, and GAPDH (1:2,500, ab9485) was regarded as the internal control. All aforementioned antibodies were purchased from Abcam (Cambridge, MA, USA). The gray value was analyzed by ImageJ.
Statistical Analysis
Statistical analyses were performed using the SPSS 21.0 software (IBM, Armonk, NY, USA). Measurement data were presented as the mean ± SD. Comparisons between two groups were analyzed by t test and among multiple groups by one-way ANOVA, followed by variance homogeneity test. When the results presented with significant differences, the q test was employed for pair comparison. Otherwise, a non-parametric rank test was adopted. The detection standard was set as α = 0.05. A p value < 0.05 was considered to be statistically significant.
Author Contributions
C.-G.Z., B.-Y.C., and R.-H.S. designed the study. X.-Z.M., F.H., and Q.L. collated the data, carried out data analyses, and produced the initial draft of the manuscript. H.-J.H., J.-Q.L., and Y.-X.T. contributed to drafting and polishing the manuscript. All authors read and approved the final submitted manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We would like to thank all participants enrolled in the present study.
References
- 1.Husain K., Hernandez W., Ansari R.A., Ferder L. Inflammation, oxidative stress and renin angiotensin system in atherosclerosis. World J. Biol. Chem. 2015;6:209–217. doi: 10.4331/wjbc.v6.i3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]; Husain, K., Hernandez, W., Ansari, R.A., and Ferder, L. (2015). Inflammation, oxidative stress and renin angiotensin system in atherosclerosis. World J. Biol. Chem. 6, 209-217. [DOI] [PMC free article] [PubMed]
- 2.Moore K.J., Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; Moore, K.J., and Tabas, I. (2011). Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341-355. [DOI] [PMC free article] [PubMed]
- 3.Tabas I., García-Cardeña G., Owens G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015;209:13–22. doi: 10.1083/jcb.201412052. [DOI] [PMC free article] [PubMed] [Google Scholar]; Tabas, I., Garcia-Cardeña, G., and Owens, G.K. (2015). Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 209, 13-22. [DOI] [PMC free article] [PubMed]
- 4.Motz J.T., Fitzmaurice M., Miller A., Gandhi S.J., Haka A.S., Galindo L.H., Dasari R.R., Kramer J.R., Feld M.S. In vivo Raman spectral pathology of human atherosclerosis and vulnerable plaque. J. Biomed. Opt. 2006;11:021003. doi: 10.1117/1.2190967. [DOI] [PubMed] [Google Scholar]; Motz, J.T., Fitzmaurice, M., Miller, A., Gandhi, S.J., Haka, A.S., Galindo, L.H., Dasari, R.R., Kramer, J.R., and Feld, M.S. (2006). In vivo Raman spectral pathology of human atherosclerosis and vulnerable plaque. J. Biomed. Opt. 11, 021003. [DOI] [PubMed]
- 5.Staff A.C., Dechend R., Pijnenborg R. Learning from the placenta: acute atherosis and vascular remodeling in preeclampsia-novel aspects for atherosclerosis and future cardiovascular health. Hypertension. 2010;56:1026–1034. doi: 10.1161/HYPERTENSIONAHA.110.157743. [DOI] [PubMed] [Google Scholar]; Staff, A.C., Dechend, R., and Pijnenborg, R. (2010). Learning from the placenta: acute atherosis and vascular remodeling in preeclampsia-novel aspects for atherosclerosis and future cardiovascular health. Hypertension 56, 1026-1034. [DOI] [PubMed]
- 6.Zarzycka B., Nicolaes G.A., Lutgens E. Targeting the adaptive immune system: new strategies in the treatment of atherosclerosis. Expert Rev. Clin. Pharmacol. 2015;8:297–313. doi: 10.1586/17512433.2015.1025052. [DOI] [PubMed] [Google Scholar]; Zarzycka, B., Nicolaes, G.A., and Lutgens, E. (2015). Targeting the adaptive immune system: new strategies in the treatment of atherosclerosis. Expert Rev. Clin. Pharmacol. 8, 297-313. [DOI] [PubMed]
- 7.Repetto E., Lichtenstein L., Hizir Z., Tekaya N., Benahmed M., Ruidavets J.B., Zaragosi L.E., Perret B., Bouchareychas L., Genoux A. RNY-derived small RNAs as a signature of coronary artery disease. BMC Med. 2015;13:259. doi: 10.1186/s12916-015-0489-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; Repetto, E., Lichtenstein, L., Hizir, Z., Tekaya, N., Benahmed, M., Ruidavets, J.B., Zaragosi, L.E., Perret, B., Bouchareychas, L., Genoux, A., et al. (2015). RNY-derived small RNAs as a signature of coronary artery disease. BMC Med. 13, 259. [DOI] [PMC free article] [PubMed]
- 8.Gao S., Wassler M., Zhang L., Li Y., Wang J., Zhang Y., Shelat H., Williams J., Geng Y.J. MicroRNA-133a regulates insulin-like growth factor-1 receptor expression and vascular smooth muscle cell proliferation in murine atherosclerosis. Atherosclerosis. 2014;232:171–179. doi: 10.1016/j.atherosclerosis.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gao, S., Wassler, M., Zhang, L., Li, Y., Wang, J., Zhang, Y., Shelat, H., Williams, J., and Geng, Y.J. (2014). MicroRNA-133a regulates insulin-like growth factor-1 receptor expression and vascular smooth muscle cell proliferation in murine atherosclerosis. Atherosclerosis 232, 171-179. [DOI] [PMC free article] [PubMed]
- 9.Crawford M., Batte K., Yu L., Wu X., Nuovo G.J., Marsh C.B., Otterson G.A., Nana-Sinkam S.P. MicroRNA 133B targets pro-survival molecules MCL-1 and BCL2L2 in lung cancer. Biochem. Biophys. Res. Commun. 2009;388:483–489. doi: 10.1016/j.bbrc.2009.07.143. [DOI] [PMC free article] [PubMed] [Google Scholar]; Crawford, M., Batte, K., Yu, L., Wu, X., Nuovo, G.J., Marsh, C.B., Otterson, G.A., and Nana-Sinkam, S.P. (2009). MicroRNA 133B targets pro-survival molecules MCL-1 and BCL2L2 in lung cancer. Biochem. Biophys. Res. Commun. 388, 483-489. [DOI] [PMC free article] [PubMed]
- 10.Li X., Wan X., Chen H., Yang S., Liu Y., Mo W., Meng D., Du W., Huang Y., Wu H. Identification of miR-133b and RB1CC1 as independent predictors for biochemical recurrence and potential therapeutic targets for prostate cancer. Clin. Cancer Res. 2014;20:2312–2325. doi: 10.1158/1078-0432.CCR-13-1588. [DOI] [PubMed] [Google Scholar]; Li, X., Wan, X., Chen, H., Yang, S., Liu, Y., Mo, W., Meng, D., Du, W., Huang, Y., Wu, H., et al. (2014). Identification of miR-133b and RB1CC1 as independent predictors for biochemical recurrence and potential therapeutic targets for prostate cancer. Clin. Cancer Res. 20, 2312-2325. [DOI] [PubMed]
- 11.Kuwabara Y., Ono K., Horie T., Nishi H., Nagao K., Kinoshita M., Watanabe S., Baba O., Kojima Y., Shizuta S. Increased microRNA-1 and microRNA-133a levels in serum of patients with cardiovascular disease indicate myocardial damage. Circ Cardiovasc Genet. 2011;4:446–454. doi: 10.1161/CIRCGENETICS.110.958975. [DOI] [PubMed] [Google Scholar]; Kuwabara, Y., Ono, K., Horie, T., Nishi, H., Nagao, K., Kinoshita, M., Watanabe, S., Baba, O., Kojima, Y., Shizuta, S., et al. (2011). Increased microRNA-1 and microRNA-133a levels in serum of patients with cardiovascular disease indicate myocardial damage. Circ Cardiovasc Genet 4, 446-454. [DOI] [PubMed]
- 12.Quaranta R., Pelullo M., Zema S., Nardozza F., Checquolo S., Lauer D.M., Bufalieri F., Palermo R., Felli M.P., Vacca A. Maml1 acts cooperatively with Gli proteins to regulate sonic hedgehog signaling pathway. Cell Death Dis. 2017;8:e2942. doi: 10.1038/cddis.2017.326. [DOI] [PMC free article] [PubMed] [Google Scholar]; Quaranta, R., Pelullo, M., Zema, S., Nardozza, F., Checquolo, S., Lauer, D.M., Bufalieri, F., Palermo, R., Felli, M.P., Vacca, A., et al. (2017). Maml1 acts cooperatively with Gli proteins to regulate sonic hedgehog signaling pathway. Cell Death Dis. 8, e2942. [DOI] [PMC free article] [PubMed]
- 13.Kuncharin Y., Sangphech N., Kueanjinda P., Bhattarakosol P., Palaga T. MAML1 regulates cell viability via the NF-κB pathway in cervical cancer cell lines. Exp. Cell Res. 2011;317:1830–1840. doi: 10.1016/j.yexcr.2011.05.005. [DOI] [PubMed] [Google Scholar]; Kuncharin, Y., Sangphech, N., Kueanjinda, P., Bhattarakosol, P., and Palaga, T. (2011). MAML1 regulates cell viability via the NF-κB pathway in cervical cancer cell lines. Exp. Cell Res. 317, 1830-1840. [DOI] [PubMed]
- 14.Klinakis A., Lobry C., Abdel-Wahab O., Oh P., Haeno H., Buonamici S., van De Walle I., Cathelin S., Trimarchi T., Araldi E. A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature. 2011;473:230–233. doi: 10.1038/nature09999. [DOI] [PMC free article] [PubMed] [Google Scholar]; Klinakis, A., Lobry, C., Abdel-Wahab, O., Oh, P., Haeno, H., Buonamici, S., van De Walle, I., Cathelin, S., Trimarchi, T., Araldi, E., et al. (2011). A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature 473, 230-233. [DOI] [PMC free article] [PubMed]
- 15.Aquila G., Pannella M., Morelli M.B., Caliceti C., Fortini C., Rizzo P., Ferrari R. The role of Notch pathway in cardiovascular diseases. Glob. Cardiol. Sci. Pract. 2013;2013:364–371. doi: 10.5339/gscp.2013.44. [DOI] [PMC free article] [PubMed] [Google Scholar]; Aquila, G., Pannella, M., Morelli, M.B., Caliceti, C., Fortini, C., Rizzo, P., and Ferrari, R. (2013). The role of Notch pathway in cardiovascular diseases. Glob. Cardiol. Sci. Pract. 2013, 364-371. [DOI] [PMC free article] [PubMed]
- 16.Fukuda D., Aikawa E., Swirski F.K., Novobrantseva T.I., Kotelianski V., Gorgun C.Z., Chudnovskiy A., Yamazaki H., Croce K., Weissleder R. Notch ligand delta-like 4 blockade attenuates atherosclerosis and metabolic disorders. Proc. Natl. Acad. Sci. USA. 2012;109:E1868–E1877. doi: 10.1073/pnas.1116889109. [DOI] [PMC free article] [PubMed] [Google Scholar]; Fukuda, D., Aikawa, E., Swirski, F.K., Novobrantseva, T.I., Kotelianski, V., Gorgun, C.Z., Chudnovskiy, A., Yamazaki, H., Croce, K., Weissleder, R., et al. (2012). Notch ligand delta-like 4 blockade attenuates atherosclerosis and metabolic disorders. Proc. Natl. Acad. Sci. USA 109, E1868-E1877. [DOI] [PMC free article] [PubMed]
- 17.Mao Y.Z., Jiang L. Effects of Notch signalling pathway on the relationship between vascular endothelial dysfunction and endothelial stromal transformation in atherosclerosis. Artif. Cells Nanomed. Biotechnol. 2018;46:764–772. doi: 10.1080/21691401.2017.1337030. [DOI] [PubMed] [Google Scholar]; Mao, Y.Z., and Jiang, L. (2018). Effects of Notch signalling pathway on the relationship between vascular endothelial dysfunction and endothelial stromal transformation in atherosclerosis. Artif. Cells Nanomed. Biotechnol. 46, 764-772. [DOI] [PubMed]
- 18.Aquila G., Fortini C., Pannuti A., Delbue S., Pannella M., Morelli M.B., Caliceti C., Castriota F., de Mattei M., Ongaro A. Distinct gene expression profiles associated with Notch ligands Delta-like 4 and Jagged1 in plaque material from peripheral artery disease patients: a pilot study. J. Transl. Med. 2017;15:98. doi: 10.1186/s12967-017-1199-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; Aquila, G., Fortini, C., Pannuti, A., Delbue, S., Pannella, M., Morelli, M.B., Caliceti, C., Castriota, F., de Mattei, M., Ongaro, A., et al. (2017). Distinct gene expression profiles associated with Notch ligands Delta-like 4 and Jagged1 in plaque material from peripheral artery disease patients: a pilot study. J. Transl. Med. 15, 98. [DOI] [PMC free article] [PubMed]
- 19.Liu Z.J., Tan Y., Beecham G.W., Seo D.M., Tian R., Li Y., Vazquez-Padron R.I., Pericak-Vance M., Vance J.M., Goldschmidt-Clermont P.J. Notch activation induces endothelial cell senescence and pro-inflammatory response: implication of Notch signaling in atherosclerosis. Atherosclerosis. 2012;225:296–303. doi: 10.1016/j.atherosclerosis.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu, Z.J., Tan, Y., Beecham, G.W., Seo, D.M., Tian, R., Li, Y., Vazquez-Padron, R.I., Pericak-Vance, M., Vance, J.M., Goldschmidt-Clermont, P.J., et al. (2012). Notch activation induces endothelial cell senescence and pro-inflammatory response: implication of Notch signaling in atherosclerosis. Atherosclerosis 225, 296-303. [DOI] [PMC free article] [PubMed]
- 20.Gamrekelashvili J., Limbourg F.P. Rules of attraction: endothelial Notch signalling controls leucocyte homing in atherosclerosis via VCAM1. Cardiovasc. Res. 2016;112:527–529. doi: 10.1093/cvr/cvw207. [DOI] [PubMed] [Google Scholar]; Gamrekelashvili, J., and Limbourg, F.P. (2016). Rules of attraction: endothelial Notch signalling controls leucocyte homing in atherosclerosis via VCAM1. Cardiovasc. Res. 112, 527-529. [DOI] [PubMed]
- 21.Chinetti-Gbaguidi G., Colin S., Staels B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 2015;12:10–17. doi: 10.1038/nrcardio.2014.173. [DOI] [PubMed] [Google Scholar]; Chinetti-Gbaguidi, G., Colin, S., and Staels, B. (2015). Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 12, 10-17. [DOI] [PubMed]
- 22.Thim T., Hagensen M.K., Bentzon J.F., Falk E. From vulnerable plaque to atherothrombosis. J. Intern. Med. 2008;263:506–516. doi: 10.1111/j.1365-2796.2008.01947.x. [DOI] [PubMed] [Google Scholar]; Thim, T., Hagensen, M.K., Bentzon, J.F., and Falk, E. (2008). From vulnerable plaque to atherothrombosis. J. Intern. Med. 263, 506-516. [DOI] [PubMed]
- 23.Virmani R., Burke A.P., Kolodgie F.D., Farb A. Vulnerable plaque: the pathology of unstable coronary lesions. J. Interv. Cardiol. 2002;15:439–446. doi: 10.1111/j.1540-8183.2002.tb01087.x. [DOI] [PubMed] [Google Scholar]; Virmani, R., Burke, A.P., Kolodgie, F.D., and Farb, A. (2002). Vulnerable plaque: the pathology of unstable coronary lesions. J. Interv. Cardiol. 15, 439-446. [DOI] [PubMed]
- 24.Cipollone F., Felicioni L., Sarzani R., Ucchino S., Spigonardo F., Mandolini C., Malatesta S., Bucci M., Mammarella C., Santovito D. A unique microRNA signature associated with plaque instability in humans. Stroke. 2011;42:2556–2563. doi: 10.1161/STROKEAHA.110.597575. [DOI] [PubMed] [Google Scholar]; Cipollone, F., Felicioni, L., Sarzani, R., Ucchino, S., Spigonardo, F., Mandolini, C., Malatesta, S., Bucci, M., Mammarella, C., Santovito, D., et al. (2011). A unique microRNA signature associated with plaque instability in humans. Stroke 42, 2556-2563. [DOI] [PubMed]
- 25.Chistiakov D.A., Orekhov A.N., Bobryshev Y.V. Vascular smooth muscle cell in atherosclerosis. Acta Physiol. (Oxf.) 2015;214:33–50. doi: 10.1111/apha.12466. [DOI] [PubMed] [Google Scholar]; Chistiakov, D.A., Orekhov, A.N., and Bobryshev, Y.V. (2015). Vascular smooth muscle cell in atherosclerosis. Acta Physiol. (Oxf.) 214, 33-50. [DOI] [PubMed]
- 26.Gabunia K., Herman A.B., Ray M., Kelemen S.E., England R.N., DeLa Cadena R., Foster W.J., Elliott K.J., Eguchi S., Autieri M.V. Induction of MiR133a expression by IL-19 targets LDLRAP1 and reduces oxLDL uptake in VSMC. J. Mol. Cell. Cardiol. 2017;105:38–48. doi: 10.1016/j.yjmcc.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gabunia, K., Herman, A.B., Ray, M., Kelemen, S.E., England, R.N., DeLa Cadena, R., Foster, W.J., Elliott, K.J., Eguchi, S., and Autieri, M.V. (2017). Induction of MiR133a expression by IL-19 targets LDLRAP1 and reduces oxLDL uptake in VSMC. J. Mol. Cell. Cardiol. 105, 38-48. [DOI] [PMC free article] [PubMed]
- 27.Zhen Y., Liu J., Huang Y., Wang Y., Li W., Wu J. miR-133b Inhibits Cell Growth, Migration, and Invasion by Targeting MMP9 in Non-Small Cell Lung Cancer. Oncol. Res. 2017;25:1109–1116. doi: 10.3727/096504016X14800889609439. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhen, Y., Liu, J., Huang, Y., Wang, Y., Li, W., and Wu, J. (2017). miR-133b Inhibits Cell Growth, Migration, and Invasion by Targeting MMP9 in Non-Small Cell Lung Cancer. Oncol. Res. 25, 1109-1116. [DOI] [PMC free article] [PubMed]
- 28.Tanindi A., Sahinarslan A., Elbeg S., Cemri M. Relationship Between MMP-1, MMP-9, TIMP-1, IL-6 and Risk Factors, Clinical Presentation, Extent and Severity of Atherosclerotic Coronary Artery Disease. Open Cardiovasc. Med. J. 2011;5:110–116. doi: 10.2174/1874192401105010110. [DOI] [PMC free article] [PubMed] [Google Scholar]; Tanindi, A., Sahinarslan, A., Elbeg, S., and Cemri, M. (2011). Relationship Between MMP-1, MMP-9, TIMP-1, IL-6 and Risk Factors, Clinical Presentation, Extent and Severity of Atherosclerotic Coronary Artery Disease. Open Cardiovasc. Med. J. 5, 110-116. [DOI] [PMC free article] [PubMed]
- 29.Qiu T., Zhou X., Wang J., Du Y., Xu J., Huang Z., Zhu W., Shu Y., Liu P. MiR-145, miR-133a and miR-133b inhibit proliferation, migration, invasion and cell cycle progression via targeting transcription factor Sp1 in gastric cancer. FEBS Lett. 2014;588:1168–1177. doi: 10.1016/j.febslet.2014.02.054. [DOI] [PubMed] [Google Scholar]; Qiu, T., Zhou, X., Wang, J., Du, Y., Xu, J., Huang, Z., Zhu, W., Shu, Y., and Liu, P. (2014). MiR-145, miR-133a and miR-133b inhibit proliferation, migration, invasion and cell cycle progression via targeting transcription factor Sp1 in gastric cancer. FEBS Lett. 588, 1168-1177. [DOI] [PubMed]
- 30.Bhattacharjya S., Roy K.S., Ganguly A., Sarkar S., Panda C.K., Bhattacharyya D., Bhattacharyya N.P., Roychoudhury S. Inhibition of nucleoporin member Nup214 expression by miR-133b perturbs mitotic timing and leads to cell death. Mol. Cancer. 2015;14:42. doi: 10.1186/s12943-015-0299-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bhattacharjya, S., Roy, K.S., Ganguly, A., Sarkar, S., Panda, C.K., Bhattacharyya, D., Bhattacharyya, N.P., and Roychoudhury, S. (2015). Inhibition of nucleoporin member Nup214 expression by miR-133b perturbs mitotic timing and leads to cell death. Mol. Cancer 14, 42. [DOI] [PMC free article] [PubMed]
- 31.Fung E., Tang S.M., Canner J.P., Morishige K., Arboleda-Velasquez J.F., Cardoso A.A., Carlesso N., Aster J.C., Aikawa M. Delta-like 4 induces notch signaling in macrophages: implications for inflammation. Circulation. 2007;115:2948–2956. doi: 10.1161/CIRCULATIONAHA.106.675462. [DOI] [PubMed] [Google Scholar]; Fung, E., Tang, S.M., Canner, J.P., Morishige, K., Arboleda-Velasquez, J.F., Cardoso, A.A., Carlesso, N., Aster, J.C., and Aikawa, M. (2007). Delta-like 4 induces notch signaling in macrophages: implications for inflammation. Circulation 115, 2948-2956. [DOI] [PubMed]
- 32.Wang Y.C., He F., Feng F., Liu X.W., Dong G.Y., Qin H.Y., Hu X.B., Zheng M.H., Liang L., Feng L. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res. 2010;70:4840–4849. doi: 10.1158/0008-5472.CAN-10-0269. [DOI] [PubMed] [Google Scholar]; Wang, Y.C., He, F., Feng, F., Liu, X.W., Dong, G.Y., Qin, H.Y., Hu, X.B., Zheng, M.H., Liang, L., Feng, L., et al. (2010). Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res. 70, 4840-4849. [DOI] [PubMed]
- 33.Wu L., Griffin J.D. Modulation of Notch signaling by mastermind-like (MAML) transcriptional co-activators and their involvement in tumorigenesis. Semin. Cancer Biol. 2004;14:348–356. doi: 10.1016/j.semcancer.2004.04.014. [DOI] [PubMed] [Google Scholar]; Wu, L., and Griffin, J.D. (2004). Modulation of Notch signaling by mastermind-like (MAML) transcriptional co-activators and their involvement in tumorigenesis. Semin. Cancer Biol. 14, 348-356. [DOI] [PubMed]
- 34.Siddiqui W.A., Ahad A., Ahsan H. The mystery of BCL2 family: Bcl-2 proteins and apoptosis: an update. Arch. Toxicol. 2015;89:289–317. doi: 10.1007/s00204-014-1448-7. [DOI] [PubMed] [Google Scholar]; Siddiqui, W.A., Ahad, A., and Ahsan, H. (2015). The mystery of BCL2 family: Bcl-2 proteins and apoptosis: an update. Arch. Toxicol. 89, 289-317. [DOI] [PubMed]
- 35.Niu M., Xu R., Wang J., Hou B., Xie A. MiR-133b ameliorates axon degeneration induced by MPP(+) via targeting RhoA. Neuroscience. 2016;325:39–49. doi: 10.1016/j.neuroscience.2016.03.042. [DOI] [PubMed] [Google Scholar]; Niu, M., Xu, R., Wang, J., Hou, B., and Xie, A. (2016). MiR-133b ameliorates axon degeneration induced by MPP(+) via targeting RhoA. Neuroscience 325, 39-49. [DOI] [PubMed]
- 36.Zhou W., Bi X., Gao G., Sun L. miRNA-133b and miRNA-135a induce apoptosis via the JAK2/STAT3 signaling pathway in human renal carcinoma cells. Biomed. Pharmacother. 2016;84:722–729. doi: 10.1016/j.biopha.2016.09.074. [DOI] [PubMed] [Google Scholar]; Zhou, W., Bi, X., Gao, G., and Sun, L. (2016). miRNA-133b and miRNA-135a induce apoptosis via the JAK2/STAT3 signaling pathway in human renal carcinoma cells. Biomed. Pharmacother. 84, 722-729. [DOI] [PubMed]
- 37.Stewart K.S., Zhou Z., Zweidler-McKay P., Kleinerman E.S. Delta-like ligand 4-Notch signaling regulates bone marrow-derived pericyte/vascular smooth muscle cell formation. Blood. 2011;117:719–726. doi: 10.1182/blood-2010-05-284869. [DOI] [PMC free article] [PubMed] [Google Scholar]; Stewart, K.S., Zhou, Z., Zweidler-McKay, P., and Kleinerman, E.S. (2011). Delta-like ligand 4-Notch signaling regulates bone marrow-derived pericyte/vascular smooth muscle cell formation. Blood 117, 719-726. [DOI] [PMC free article] [PubMed]
- 38.Xiao W., Gao Z., Duan Y., Yuan W., Ke Y. Downregulation of miR-19a exhibits inhibitory effects on metastatic renal cell carcinoma by targeting PIK3CA and inactivating Notch signaling in vitro. Oncol. Rep. 2015;34:739–746. doi: 10.3892/or.2015.4041. [DOI] [PubMed] [Google Scholar]; Xiao, W., Gao, Z., Duan, Y., Yuan, W., and Ke, Y. (2015). Downregulation of miR-19a exhibits inhibitory effects on metastatic renal cell carcinoma by targeting PIK3CA and inactivating Notch signaling in vitro. Oncol. Rep. 34, 739-746. [DOI] [PubMed]
- 39.Wen P., Song D., Ye H., Wu X., Jiang L., Tang B., Zhou Y., Fang L., Cao H., He W. Circulating MiR-133a as a biomarker predicts cardiac hypertrophy in chronic hemodialysis patients. PLoS ONE. 2014;9:e103079. doi: 10.1371/journal.pone.0103079. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wen, P., Song, D., Ye, H., Wu, X., Jiang, L., Tang, B., Zhou, Y., Fang, L., Cao, H., He, W., et al. (2014). Circulating MiR-133a as a biomarker predicts cardiac hypertrophy in chronic hemodialysis patients. PLoS ONE 9, e103079. [DOI] [PMC free article] [PubMed]
- 40.Wang L., Li X., Zhou Y., Shi H., Xu C., He H., Wang S., Xiong X., Zhang Y., Du Z. Downregulation of miR-133 via MAPK/ERK signaling pathway involved in nicotine-induced cardiomyocyte apoptosis. Naunyn Schmiedebergs Arch. Pharmacol. 2014;387:197–206. doi: 10.1007/s00210-013-0929-1. [DOI] [PubMed] [Google Scholar]; Wang, L., Li, X., Zhou, Y., Shi, H., Xu, C., He, H., Wang, S., Xiong, X., Zhang, Y., Du, Z., et al. (2014). Downregulation of miR-133 via MAPK/ERK signaling pathway involved in nicotine-induced cardiomyocyte apoptosis. Naunyn Schmiedebergs Arch. Pharmacol. 387, 197-206. [DOI] [PubMed]
- 41.National Research Council . Eighth Edition. National Academy of Sciences; 2011. Guide for the Care and Use of Laboratory Animals. [Google Scholar]; National Research Council (2011). Guide for the Care and Use of Laboratory Animals, Eighth Edition (National Academy of Sciences).