

Abstract
Drug‐induced kidney injury, a major cause of acute kidney injury, results in progressive kidney disease and is linked to increased mortality in hospitalized patients. Primary injury sites of drug‐induced kidney injury are proximal tubules. Clinically, kidney injury molecule‐1, an established tubule‐specific biomarker, is monitored to assess the presence and progression of injury. The ability to accurately predict drug‐related nephrotoxicity preclinically would reduce patient burden and drug attrition rates, yet state‐of‐the‐art in vitro and animal models fail to do so. In this study, we demonstrate the use of kidney injury molecule‐1 measurement in the kidney microphysiological system as a preclinical model for drug toxicity assessment. To show clinical relevance, we use quantitative systems pharmacology computational models for in vitro–in vivo translation of the experimental results and to identify favorable dosing regimens for one of the tested drugs.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Drug‐induced proximal tubule injury is a highly complex process, and current preclinical models fail to represent this complexity accurately. Microphysiological systems have the potential to bridge this gap, but more mechanistic and quantitative efforts are required to link in vitro results to clinical situations.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study addressed how drug‐induced proximal tubule damage can be recapitulated and measured in vitro, how in vitro findings can be translated to clinical outcomes, and how the optimal dosing regimen can be identified preclinically.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The study demonstrates the potential of the kidney microphysiological system as a preclinical model for drug toxicity testing. The developed workflow highlights how to integrate experimental data (microphysiological systems) and computational efforts (quantitative systems pharmacology models) to simulate clinically relevant situations.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ The integrated in vitro–in vivo translation approach combining experimental and computational efforts will help merge toxicity and effect studies, guide first‐in‐human drug studies, and identify potential drug candidate failure earlier in the preclinical stage.
Acute kidney injury (AKI) is characterized by progressive kidney disease and causes distant organ dysfunction (e.g., cardiovascular events), leads to infections, and is linked to increased mortality in hospitalized patients.1, 2 It may occur after exposure to environmental toxins3 or drugs,4 cardiac surgery5 or kidney transplantation.4 Pharmaceutically, various drugs can cause drug‐induced kidney injury (a contributor to AKI) after short‐term or long‐term exposure and are a major concern for patient safety.6 Failure to detect drug‐induced kidney injury or AKI and accurately assess the cause and extent of damage delays treatment and puts patients at unnecessary risk.
In the kidneys, proximal tubules are most affected by nephrotoxic compounds because of their involvement in modulating glomerular filtrate concentrations, drug transport, and metabolism.4, 7 Clinically established classification systems to stratify severity of AKI (e.g., Risk, Injury, Failure, Loss of function, End stage renal disease (RIFLE), Acute Kidney Injury Network (AKIN)4, 8) are based on measures such as increasing serum creatinine levels (1.5 times within 1–3 days) or low urine output (0.5 mL/kg/hour for 6 hours). Although these criteria reveal a loss of kidney function, they do not reflect damage specific to proximal tubules. In contrast, kidney‐injury molecule 1 (KIM‐1) is a biomarker of damage specifically to proximal tubules.9, 10, 11 The detection of KIM‐1 in the clinic is minimally invasive and can be used for the early assessment of AKI and proximal tubule damage.10, 12, 13, 14 Within 3 hours of tubular cell injury, KIM‐1 production increases and is shed from tubule cell membranes into the urine and plasma.15 Furthermore, proximal tubule epithelial cells undergo active dedifferentiation and proliferation, altering their morphology and conferring a semiprofessional phagocyte function used in tissue repair.16, 17 This process, among others, is probably activating immune cells residing in the kidneys, causing early inflammation.18 Secreted cytokines and chemokines then trigger further recruitment of circulating immune cells (e.g., neutrophils, T‐lymphocytes) to the site of injury, amplifying inflammation19 and promoting tissue repair.20 Predicting these drug‐related toxic responses prior to first‐in‐human pharmaceutical testing or during early clinical trials would be beneficial for the safe translation of promising compounds to clinical use.
Currently, animal models are the standard for drug toxicity assessment despite their limited prediction of human toxicity21 and failure to reduce attrition rates.22, 23 Similarly, available in vitro models for nephrotoxic drug testing lack both morphology and functionality of human kidneys and to date are poorly predictive of toxicity in humans.4, 21
Recent advances in microphysiological systems24, 25, 26, 27, 28 (MPSs), also known as tissue chips or organs‐on‐chips, have included the development of a kidney MPS29 that recapitulates the size, structure, and function of human renal proximal tubules in vitro. This kidney MPS has been tested for its ability to mimic drug‐related proximal tubule damage.21, 29, 30 However, it remains unclear how such in vitro findings may be used to predict drug‐induced nephrotoxicity in vivo. Validated methods are needed for in vitro–in vivo translation (IVIVT) using measurements of biomarkers and organ‐specific toxicities from such in vitro systems. The emerging field of quantitative systems pharmacology (QSP) may provide useful approaches to enable MPS‐to‐human translation31, 32 as well as to provide guidance for aspects of clinical study design such as dosing regimens and identify potential drug failure early.
In this study, the kidney MPS and a two‐dimensional (2D) system, comprising human renal proximal tubule cell (hRPTEC) cultures in a 96‐well plate,29 were used to assess drug toxicity. Toxicity was quantified by monitoring KIM‐1 and PrestoBlue (Invitrogen, Carlsbad, CA) after short‐term and long‐term exposure to the following three drugs known to cause AKI: cisplatin, rifampicin, and gentamicin. Subsequently, computational QSP models were used to predict human plasma and urine KIM‐1 time‐concentration profiles in a virtual patient population based on the in vitro KIM‐1 measurements. Finally, an approach for using the paired MPS–QSP framework was developed to identify favorable dosing regimens (interval, dose) to minimize kidney toxicity while maintaining therapeutic effect and was tested in a theoretical simulation study for one drug, rifampicin.
Through this work, we demonstrated the utility of the kidney MPS for preclinical drug toxicity–studies and the assessment of proximal tubule‐specific damage using a clinical biomarker (KIM‐1). Subsequent integration of experimental (MPS) and computational (QSP) efforts enabled the simulation of clinically relevant plasma and urine biomarker levels (KIM‐1).
The MPS–QSP workflow allows the adaptation to any drug compound and investigated MPSs to guide first‐in‐human drug studies, identify drug‐induced toxicities, and optimize a dosing regimen for novel compounds.
Methods
Kidney MPS: perfusion platform and HARV‐1 device
The kidney MPS is contained within the HARV‐1, a polydimethylsiloxane‐based microfluidic device developed by Nortis (Seattle, WA). The HARV‐1 attaches to a perfusion platform that contains media reservoirs and is then placed in a docking station within an incubator. These stations connect to the incubator gas pump, which drives media perfusion through the kidney MPS.
Culture and seeding of hRPTECs
Cryopreserved hRPTECs, provided by University of Washington, Seattle, were cultured in a T‐25 flask until confluent. To seed the kidney MPS, hRPTECs were injected into the HARV‐1 and mounted to the perfusion platform. After overnight incubation, the platforms were perfused at 0.5 μL/min.
The 2D system was created by seeding hRPTECs on a collagen IV coated 96‐well plate and grown to confluence.
The hRPTEC culture medium comprised DMEM/F12 (Dulbecco's Modified Eagle Medium: Nutrient Mixture F‐12) (Gibco (Gaithersburg, MD) catalog no. 11330‐032), 1% 100× insulin‐transferrin‐selenium‐sodium pyruvate (Gibco (Gaithersburg, MD) catalog no. 51300‐044), 1% anti‐anti (Gibco (Gaithersburg, MD) catalog no. 15240‐062), and 1.8% hydrocortisone (Sigma‐Aldrich (St. Louis, MO) catalog no. H6909). The hRPTECs were used between passages 1 and 2. Detailed methodologies (including immunohistochemistry) are provided in Methods S1 .
Experimental design and drug exposure
An overview of the experimental design and hardware specifications are presented in Table 1. The start of experiments, day 0, is 8 days after seeding hRPTECs in the MPS or 2D system. Cisplatin, rifampicin, and gentamicin were administered for 2 and for 10 days to the MPS and for 10 days to the 2D system, evaluating KIM‐1 every 48 hours. In addition, 6‐hour and 24‐hour KIM‐1 measurements were taken for the kidney MPS. The concentrations used for all drugs were selected based on a review of toxicity measurements in vitro in other assays as well as known clinical concentrations. Additional information about the experiments is provided in Methods S1 .
Table 1.
Overview of physical device specifications and experimental designs
| Parameter | Kidney microphysiological system | Two‐dimensional system | |
|---|---|---|---|
| Volume | 115 μL | 200 μL | |
| Matrix volume | 23 μL | N/A | |
| Lumen volume | 70 nL | N/A | |
| Flow rate | 0.5 μL/min | N/A | |
| Cell type | hRPTECs from donor “HIM‐31” | ||
| Estimated cell # | 5,000 | 10,000 | |
| Sampling schedule | 0, 6, 24 hours + every 48 hours | Every 48 hours | Every 48 hours |
| Drug exposure time | 2 days | 10 days | 10 days |
hRPTECs, human renal proximal tubule cells; min, minute; N/A, not applicable.
IVIVT
The translation of in vitro results to in vivo and clinical outcomes is a multistep process, and the developed workflow is presented in Figure 1. Population pharmacokinetics and biomarker toxicokinetics are incorporated in the QSP model to simulate clinically relevant biomarker profiles starting from in vitro measurements of KIM‐1 in the kidney MPS.
Figure 1.
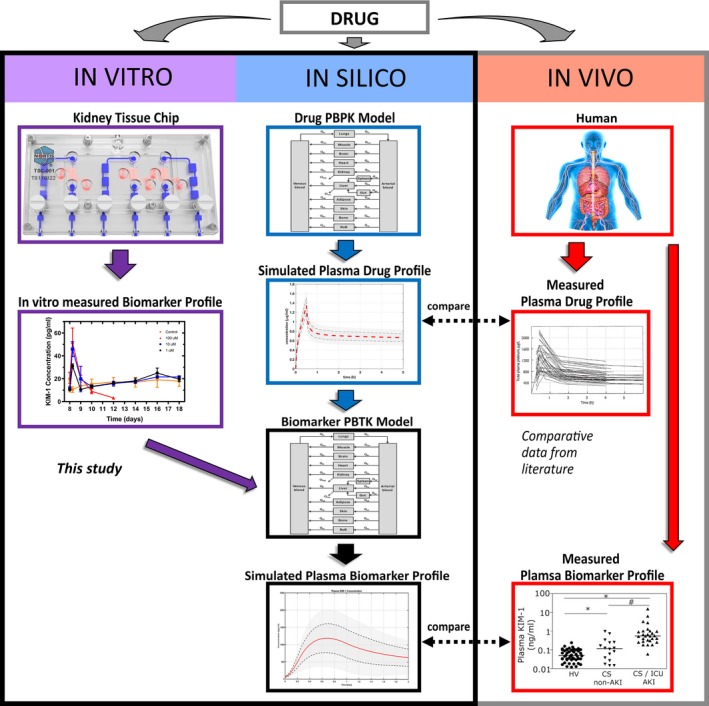
In vitro–in vivo translation workflow. Predicting drug‐related clinical biomarker profiles to assess organ‐specific toxicity from in vitro data is a multistep process. First, the toxic responses of drugs using clinical biomarkers are evaluated in the kidney microphysiological system (e.g., toxicity, viability; left panel, purple). Computational physiologically‐based pharmacokinetic (PBPK) models of drugs are simulated to predict drug exposure in human kidneys (middle panel, blue). These simulations are compared with measured drug profiles from patients to assess their validity (right panel, red). Then the quantitative systems pharmacology models relate the simulated drug human biodistribution profiles to the kidney injury biomarker profile using microphysiological system data (middle panel, black). Finally, the simulations of biomarkers are compared with clinically observed biomarker profiles. KIM‐1, kidney‐injury molecule 1; PBTK, physiologically based toxicokinetic.
Specifically, a whole‐body physiologically‐based pharmacokinetic (PBPK) model was developed in which major organs of the human body were represented by compartments. The connection between the compartments, i.e., blood flow, is based on cardiac output.33, 34 The model uses time‐dependent, mass‐balance, ordinary differential equations and assumes well‐mixed compartments. Using this model, first the pharmacokinetics of N = 100 virtual patients was simulated. The effect of interpatient variability was investigated by randomly sampling from normally distributed parameters, e.g., body weight, blood flow, and number of nephrons. Additional information as well as drug‐specific information is presented in Methods S2.
Second, KIM‐1 shedding rates (amount/time; Methods S2) were calculated from the measured extracellular KIM‐1 time‐concentration profile. Then, for each drug, these rates were linearly interpolated between the tested concentrations (Figure S1). The simulated drug biodistribution profiles in both kidneys were correlated to the interpolated grid of KIM‐1 shedding rates. The resulting shedding rate profile (a function of time and drug concentrations) was scaled up to represent the number of nephrons in a pair of human kidneys35 (Methods S2).
Published studies suggest a recruitment of circulating immune cells to the kidneys upon injury.18, 20 Because the investigated in vitro systems are immunodeficient, we added an immune component into the QSP model, focusing on the effects of neutrophils as early responders. Lacking quantitative human data, the recruitment of neutrophils and their effect on KIM‐1 shedding in vivo was based on an ischemia‐reperfusion injury mouse model36 measured at 2, 24, 48, and 72 hours with an increase in neutrophils of 2.6‐fold, 3‐fold, 2.7‐fold, and 2‐fold, respectively, over baseline. A separate in vitro model showed that activated neutrophils in a cell model increased KIM‐1 shedding 3.25‐fold.37 This information was implemented in the QSP model assuming comparable effects in humans.
Finally, the simulated biomarker profiles from plasma and urine (with and without the immune effect) were compared with reports of plasma and urine KIM‐1 measurements from clinical AKI cases (Table S5).
Identifying optimal dosing regimen using preclinical data
Various doses and dosing intervals of rifampicin were investigated to identify those that would provide efficacy while avoiding kidney toxicity. Specifically, the impact of multiple dosing regimens on kidney toxicity and therapeutic effects was simulated for 2‐day and 10‐day exposures of rifampicin. In accordance with clinically accepted dosing regimen guidelines,38 the dosing intervals simulated were 6, 12, 24, 48, and 72 hours; doses ranged from 100–1200 mg in 100 mg steps.
The area under the simulated drug concentration–time profile in the kidneys was calculated and correlated to drug exposures from the kidney MPS experiments. Toxic exposures were defined as exposures after which KIM‐1 levels were near detection limits and lower than control, indicating a reduced cell number because of cell death, whereas safe exposures were defined as exposures after which KIM‐1 levels were comparable with control levels.
The expected drug concentration–effect relationship (pharmacodynamics) of rifampicin was simulated using a maximum effect sigmoidal model (E max). For each dosing regimen, the effect E of rifampicin on inhibiting the growth of mycobacteria tuberculosis39 was calculated as a function of mean drug concentration after 2 and 10 days:
| (1) |
where EC50 is the drug concentration leading to 50% inhibition, C is the drug concentration, and the steepness of the curve (Hill coefficient; Methods S2). Subsequently, the area under the effect curve was calculated and normalized to maximum effect to assess the efficacy differences of the simulated dosing regimens.
Data analysis and plotting
All simulations, parameter estimations, and data analyses were performed in Matlab (R2017a; The MathWorks, Natick, MA. Plotting was performed in Prism (version 7.0c; GraphPad Software, La Jolla, CA). Wilcoxon t‐tests were performed, and P values < 0.05 were considered statistically significant.
Results
The kidney MPS mimics human physiology and sheds less KIM‐1 than the 2D system
Nephrotoxic drug testing was conducted with the kidney MPS and 2D system. An overview of both is provided in Table 1 and Figure 2. Live/dead staining showed no evidence of dead cells in a confluent proximal tubule in the kidney MPS.
Figure 2.
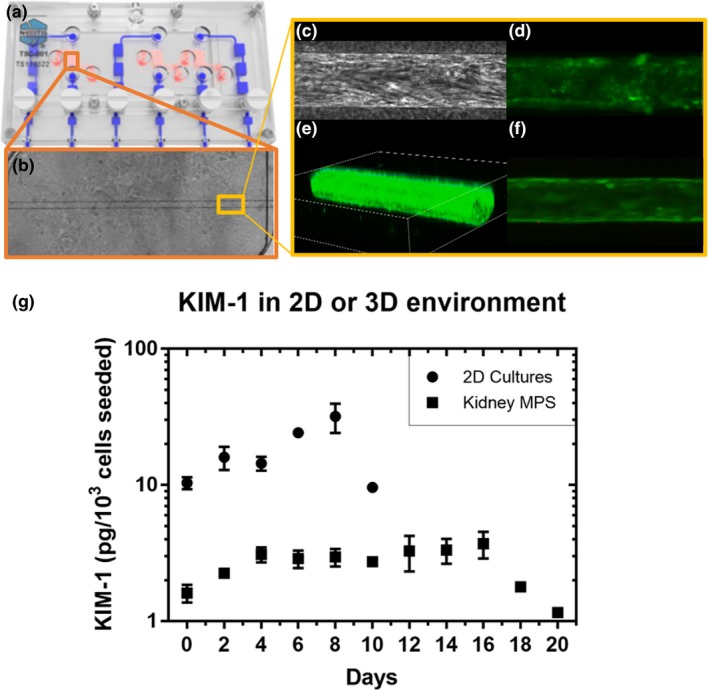
Overview of the kidney microphysiological system (MPS) hardware and biology. (a) The Nortis (Seattle, WA) HARV‐1 triple single channel. In red is the chamber for cell culture and in blue is the perfusion pathway for media. (b) A phase contrast image of a fully confluent proximal tubule in the collagen I chamber of the HARV‐1. (c) A phase contrast image of a segment of the proximal tubule showing human renal proximal tubule cells organized in the direction of flow. (d) Staining using fluorescein‐conjugated lotus lectin to mark the brush border of the human renal proximal tubule cells. (e) A three‐dimensional projection of the confluent proximal tubule in the kidney MPS. (f) A live/dead stain showing a healthy proximal tubule segment. (g) The difference in kidney‐injury molecule 1 (KIM‐1) secretion between cells in the two‐dimensional system and kidney MPS, representing two‐dimensional and three‐dimensional microenvironments, respectively, measured during a 2‐day interval. Data were corrected for sample volume and cell number differences.
In untreated conditions, shed KIM‐1 concentration was ~10‐fold lower in the MPS Compared with the 2D system (measured every 48 hours for 10 days; Figure 2), indicating a lower injury or stress state. In addition, the MPS remained viable and shed low amounts of KIM‐1 (~1–3 picogram/103 cells seeded) for 10 days after the 2D system had died (Figure 2).
Kinetics of short‐term drug‐induced KIM‐1 shedding in the kidney MPS
In a 48‐hour exposure experiment, the relationship between drug‐induced proximal tubule injury and KIM‐1 shedding in the kidney MPS was characterized using three doses of the following known nephrotoxic drugs: cisplatin, rifampicin, and gentamicin (Methods S2). KIM‐1 at 6, 24, and 48 hours after drug dosing was quantified to assess early AKI responses to exposure (Table 1), with the experimental time points determined from clinical sampling schedules (Table S5). After 6 hours of drug exposure, for all drugs and dose concentrations, an increase in KIM‐1 shedding (Figure 3) was statistically significant (P < 0.0001). The timing is consistent with observations of drug‐induced kidney injury in vivo (Table S5; e.g., acute paraquat intoxication at 6 hours). Shed KIM‐1 increased proportionally to dose in the MPS treated with cisplatin but was not dose dependent for rifampicin or gentamicin.
Figure 3.
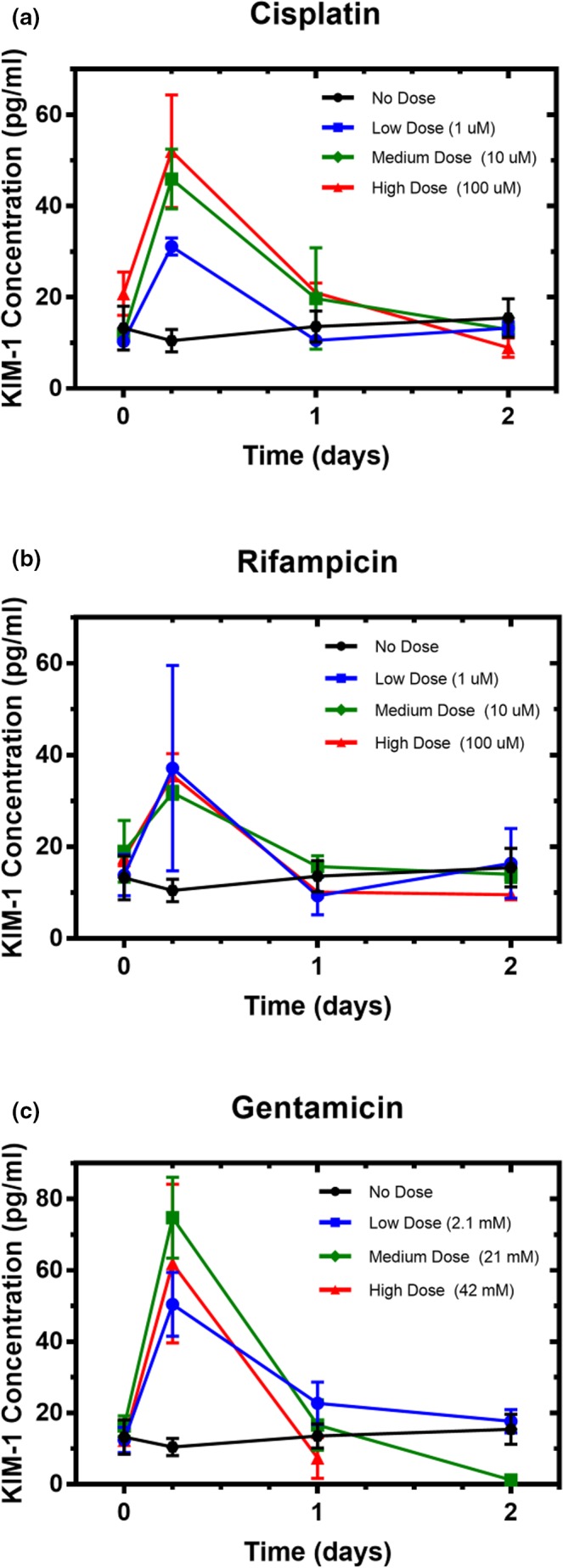
Shed kidney‐injury molecule 1 (KIM‐1) in the kidney microphysiological system effluent during exposure to drug. Cells in the kidney microphysiological system were continuously exposed to (a) cisplatin, (b) rifampicin, or (c) gentamicin for 2 days. The dose‐dependent response peaked as early as 6 hours, with rapid decline to baseline within 24 hours.
By 24 hours in most treatment groups, shed KIM‐1 returned to or below baseline levels and remained at that level through the end of the experiment (48 hours). In medium and high doses of gentamicin, shed KIM‐1 at 48 hours was significantly below baseline levels of <10–12 pg/mL (P < 0.001; Figure 3 c), coinciding with a significant decrease in metabolic activity (P < 0.0001; Figure 4 a), suggesting that cells had died in these conditions and thereby were no longer shedding KIM‐1. The only other condition found to significantly reduce metabolic activity as measured by PrestoBlue was the high dose of cisplatin (P < 0.0001; Figure 4 a).
Figure 4.
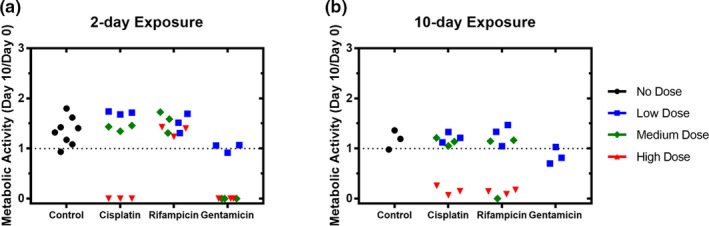
Metabolic activity of the kidney microphysiological system after short or long drug exposure. Cells in the kidney microphysiological system were continuously exposed to each of three drugs for (a) 2 days or (b) 10 days. Measures of PrestoBlue (Invitrogen, Carlsbad, CA), an indicator of metabolic activity, at day 10 were normalized to day 0 measures of individual replicates within each group.
Assessment of effects of nephrotoxic drugs with long‐term exposure
Both 2D cultures and the kidney MPS were exposed to drug for either 2 or 10 days and monitored for 10 days, by shed KIM‐1 every 2 days and metabolic activity via PrestoBlue on days 0 and 10. In the MPS, the greatest difference found between short‐term and long‐term exposure was for rifampicin. Specifically, 10‐day exposure to high‐dose rifampicin caused a significant reduction in metabolic activity (P < 0.0001; Figure 4 b) and shed KIM‐1 (P < 0.001; Figure S3. right) at day 10 when compared with day 0, whereas 2‐day exposure had no effect on either measure (Figures 4 a, S3 , left). Furthermore, in contrast to the 2‐day exposure, a 10‐day exposure to a medium dose of rifampicin also reduced shed KIM‐1 (Figure S3) but did not significantly reduce metabolic activity (Figure 4) when compared with controls. Together, these results suggest that rifampicin reduces metabolic activity in the MPS in an exposure‐driven manner.
Another major difference was observed in the KIM‐1 profiles of the low‐dose gentamicin groups in the MPS. After 10‐day exposure to low‐dose gentamicin, KIM‐1 declined to near zero (Figure S3), whereas metabolic activity did not differ from the 2‐day exposure (Figure 4). However, KIM‐1 measures after 2 days of exposure declined below baseline before returning to normal 6 days after removal of the drug (Figure S3, left). This dip below baseline and return to normal levels is also observed in medium and high doses of rifampicin in the 2‐day exposures (Figures 3, S3, left). Given that these doses did not cause a reduction in metabolic activity (Figure 4), the return of KIM‐1 levels to baseline is possibly a result of the role of KIM‐1 in tubular repair40 among other repair processes.40, 41
Cisplatin was observed to be toxic at the highest dose in both systems, causing a reduction in KIM‐1 secretion 48 hours after dosing (Figures S3 S4). Furthermore, viability measures were near zero in the MPS but only reduced ~50% in the 2D system (Figures 4, S5 ). The KIM‐1 profiles of the other two doses trended similar to controls (Figures S3, S4), and viability measures after both 2‐day and 10‐day exposures were not reduced (Figures 4, S5 ).
In the 2D system, measures of shed KIM‐1 in response to the three drugs at three doses showed less discernable dose relationships when compared with the MPS. Day‐to‐day measures varied widely in both control and drug treatment arms (Figure S4 ). Notably, the high dose of rifampicin did not cause a reduction in metabolic activity in the 2D system (Figure S5 ) but did in the MPS (Figure 4). The reductions in metabolic activity caused by high cisplatin and medium and high gentamicin doses were also more modest in the 2D system than in the MPS. Taken together, these results suggest a lack of responsiveness to drug insult of the 2D system for toxicity assessment in comparison with the kidney MPS and indicate that the MPS is likely a more useful model to assess toxicity.
IVIVT, including the impact of neutrophil recruitment on predicting clinical toxicity
Experimental KIM‐1 profiles after 2‐day drug exposure (Figure 3) were correlated to drug exposure, and this correlation was implemented in the QSP model and scaled by the number of nephrons in both human kidneys (Methods S2). Drug pharmacokinetics using the full‐body PBPK model were simulated for cisplatin, rifampicin, and gentamicin (Methods S2), and the resulting plasma and urine KIM‐1 time‐concentration profiles are in Figure 5 both without (left) and with (center and right) a putative immune system response added to the PBPK model.
Figure 5.
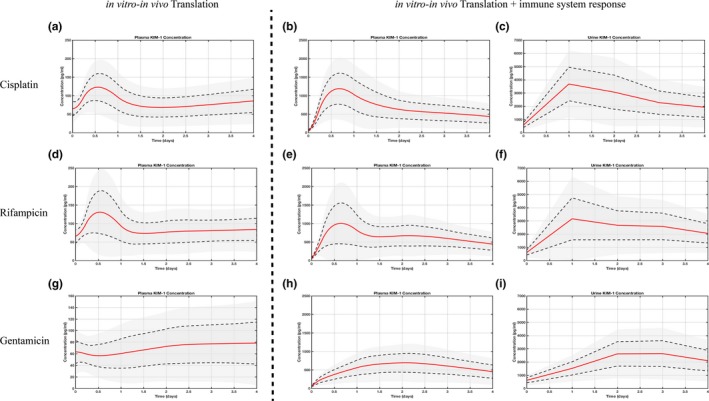
Simulation of clinical biomarker levels without and with immune effect. Simulated plasma biomarker profiles (left panel) for cisplatin (a), rifampicin (d), and gentamicin (g) using kidney‐injury molecule 1 (KIM‐1) profiles from the three‐dimensional kidney microphysiological system. Early in vitro biomarker assessment translated into distinct peaks for cisplatin and rifampicin (16 hours postinjury), but not gentamicin. Accounting for the immune system response in silico, both plasma (middle panel: b, e, h) and urine (right panel: c, f, i) concentrations are comparable with clinically reported KIM‐1 values and time course. Red line indicates a population mean of 100 virtual patients, gray area within dashed line one standard deviation, and outer gray area two standard deviations.
The early assessment of KIM‐1 (6, 24 hours) in vitro translated to a twofold increase over baseline for cisplatin and rifampicin but not for gentamicin (although a peak was observed in vitro), possibly caused by the considerable difference in tested (~2.1 mM) and clinically used (~10 μM) drug concentrations. Peaks of KIM‐1 concentration were observed between 12 and 24 hours for the former two, and ~100 hours after drug exposure for the latter.
The effects of neutrophil recruitment on KIM‐1 shedding were incorporated to the QSP model to evaluate how that would affect simulated in vivo plasma and urine KIM‐1. Consequently, distinct plasma KIM‐1 peaks (~1,000 pg/mL) in vivo were predicted between 12 and 24 hours after exposure to cisplatin and rifampicin, followed by a slow decline toward baseline similar to clinically observed time courses and concentrations of KIM‐1 (Table S5). Likewise, urine concentrations were elevated after 24 hours (~3,000 pg/mL) and declined slowly.
In contrast, KIM‐1 plasma and urinary levels after gentamicin exposure still lack the distinct peak within 24 hours of drug exposure but show comparable time course and levels thereafter.
Combining the kidney MPS data and QSP modeling to identify optimal dosing regimens
Another application of IVIVT is to identify optimal dosing regimens to guide first‐in‐human studies. To demonstrate this, the effects of varying dosing intervals (6–72 hours) and administered doses (100–1,200 mg) on toxicity (assessed by KIM‐1 and PrestoBlue) and therapeutic effect (assessed by inhibition of mycobacteria tuberculosis growth) for rifampicin were simulated using the QSP model. The results in Figure 6 a,b indicate that after 2 days of drug exposure, a range of dose and dose intervals would provide substantial treatment effect (Figure 6 b), and none would result in major toxicity in human kidneys (Figure 6 a). Varying either dose interval or administered dose level while the other is constant can modulate the treatment effect, e.g., the inhibition of bacterial growth.
Figure 6.
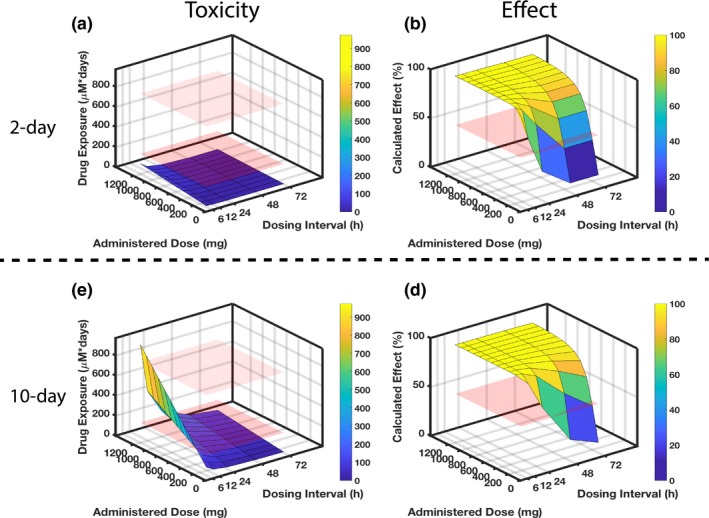
Identifying optimal rifampicin clinical dosing regimen. Combining experimental and computational efforts allows identification of optimal dosing regimens with respect to organ‐specific toxicity and drug effect after 2‐day (top) and 10‐day (bottom) drug exposure. Regarding the toxicity in parts (a) and (c), the red planes indicate rifampicin exposure levels that correspond to known experimental toxicity outcomes: the lower plane indicates no adverse effect observed in either kidney‐injury molecule 1 or viability (measured by metabolic activity with PrestoBlue (Invitrogen, Carlsbad, CA)) after 2‐day exposure to the 100 μM drug; the upper plane indicates the exposure at which adverse effects become apparent, with both kidney‐injury molecule 1 and viability declines for the 10‐day exposure to the 100 μM drug. Regarding the efficacy in parts (b) and (d), the red plane sits at 50% calculated effect as a comparison to the dose‐dependent and dose interval–dependent surfaces that are simulated for results. (a) The 2‐day rifampicin exposure for a range of drug doses and dosing intervals reveals no dosing regimen that would lead to kidney toxicity in a clinical setting as all exposures are below the “no adverse effect” lower plane. To be effective at killing bacteria within 2 days, however, requires frequent dosing, e.g., every 12 hours at 600 mg. (c) When extending the drug exposure to 10 days (lower panel), less frequent and lower dosing is needed to minimize toxicity. (d) Concomitantly, the longer dosing intervals and reduced doses will negatively impact effectiveness.
After 10 days of drug exposure (Figure 6 c,d), simulated toxicity and effect outcomes are consistent with a commonly used clinical dosing regimen38 for the treatment of tuberculosis (600 mg every 24 hours). With that regimen, drug exposure remains below the safe exposure threshold we determined from KIM‐1 and PrestoBlue assessments (i.e., below the lower red plane; Figure 6 c) while the calculated drug effect remains high (100%; Figure 6 d). The results also illustrate that extended exposure for 10 days increases the likelihood of toxicity, as the simulated surface rises above the lower red plane for a range of dose‐interval pairs (Figure 6 c).
This approach demonstrates the possibility to link in vitro experiments through QSP simulations to clinical dosing regimens to minimize nephrotoxicity while maintaining therapeutic effect.
Discussion
The aims of this work were to recapitulate drug‐induced kidney injury in vitro and to present a framework for translation of in vitro findings to relevant clinical outcomes. The presented integrated IVIVT approach combined quantitative MPS toxicodynamic biomarker profiles and drug pharmacokinetics to simulate drug responses in humans, investigate the effect of neutrophil recruitment on KIM‐1 shedding, and assess the effects of varying dosing regimens on therapeutic effect and nephrotoxicity.
Initial experimental investigations revealed the potential to recapitulate drug‐induced proximal tubule damage in vitro with the kidney MPS but less well with the 2D system. Notably, much lower amounts of the injury biomarker KIM‐1 were observed in the untreated kidney MPS when compared with the untreated 2D system. A possible explanation may be the dedifferentiation of renal epithelial cells in the static 2D system to a mesenchymal phenotype42 and the loss of essential metabolic and transport functions (e.g., albumin uptake, Alkaline Phosphatase (ALP) release, and aquaporin activity).29, 43, 44 This in vitro dedifferentiation process is analogous to renal proximal tubule remodeling in vivo, where dedifferentiated hRPTECs rapidly proliferate and replenish cells at the site of injury. In addition, dedifferentiated cells are the predominant source of KIM‐1 in urine.42 In contrast, fluid shear stress in a three‐dimensional environment such as in the kidney MPS restores relevant functions and maintains the epithelial cell phenotype.29, 43, 44 This maintenance of function and phenotype under fluid shear stress conditions may explain the continually low levels of shed KIM‐1 seen throughout our studies in the kidney MPS (indicating a low‐stress or injury state).
For toxicity assessment with long‐term drug exposure, our results indicated that the kidney MPS may be more sensitive to drug insult than the 2D system. PrestoBlue, an assay for metabolic activity, indicated that 10 days of exposure to a high dose of rifampicin reduced viability in the kidney MPS to near zero (Figure 4 b), whereas in the 2D system, viability was unaffected relative to controls (Figure S5). The epithelial‐to‐mesenchymal transition of 2D cultures likely contributes to this observed lack of response to drug insult. Other authors have reported similar findings, stating that the fluid shear stress in the MPS ensures the maintenance of a healthy epithelial cell population that is more sensitive to toxic insult.43 When assessing acute responses, the flow‐through nature of the kidney MPS is especially important for capturing the kinetic profile of the shed KIM‐1 response. After flowing through the MPS, spent media accumulates in the waste compartment, enabling early biomarker assessment at 6 and 24 hours after exposure to cisplatin, rifampicin, and gentamicin and showed distinct increases in KIM‐1 levels (Figure 3). The initial magnitude of the increase of KIM‐1 in the MPS coincides with clinical observations in the first 6 to 24 hours. For example, patients who developed AKI after heart bypass surgery saw a ~3.5‐fold increase in urinary KIM‐1 at 6 hours (Table S5). However, KIM‐1 remains elevated some days after a proximal tubule injury in vivo (Table S5). The return to baseline of KIM‐1 in the MPS after 24‐hour drug exposure in many treatment arms could be attributed to the lack of immune response in this in vitro system.45
Although all three investigated drugs—cisplatin, gentamicin, and rifampicin—are highly effective in treating bacterial infections and cancer, they also contribute to drug‐induced kidney injury.38, 46 Following treatment, renal impairment and dysfunction are common problems limiting the use of these drugs and emphasizing the need for better predictive preclinical models of nephrotoxicity.
Our study highlights the potential of the kidney MPS as an in vitro tool for the assessment of drug‐induced proximal tubular injury. However, it remains unclear how these findings are relevant in a clinical setting. Therefore, we performed MPS‐to‐human translation (IVIVT) of biomarker profiles by correlating data obtained from the kidney MPS to drug exposure in the kidneys in vivo, calculated by the population‐PBPK model (Figure 1). The mathematical QSP model linked simulated drug pharmacokinetics to the measured toxicodynamic KIM‐1 profiles, resulting in plasma and urine KIM‐1 profile simulations as a function of clinically relevant drug concentrations (Figure 5). Although the kidney MPS is immunodeficient, we could predict clinically relevant KIM‐1 levels5, 8, 12, 46 in silico when we accounted for the known activity of neutrophils in the process (Table S5, Figure 5). Future efforts to combine an in vitro immune system with the kidney MPS might further elucidate the role of activated neutrophils in KIM‐1 shedding and kidney inflammation and tissue repair. In our theoretical study, we employed a QSP model for rifampicin to identify favorable dose regimens that would minimize kidney toxicity while maintaining high drug effect as a function of drug exposure (Figure 6). The simulation results retrospectively indicated that the clinical dosing regimen for rifampicin (600 mg per day) had lower nephrotoxic liability when compared with a higher dose (>600 mg) or more frequent drug administration, whereas the efficacy would not be further improved either by higher dose or a more frequent dosing regimen. These findings support the potential to apply this translational workflow prospectively for drugs under development.
The adoption of the presented workflow for toxicity assessment or dosing regimen investigations would be supported by testing a broader set of drugs. Quantification of total and unbound drug and secreted protein loss, such as KIM‐1, in the kidney MPS because of binding to platform and media components may further improve the translational predictions of the in vivo responses using in vitro results. In addition, studying donor–donor variability by culturing tissue biopsies from different patient populations or individuals would elucidate the need to tailor drug investigations to specific patients or patient groups. The PBPK models may be improved by implementing more mechanistic details, e.g., active drug transport or the loss of metabolic functions in the kidneys. The biomarker QSP model may be improved by recognizing and implementing the involvement of KIM‐1 in varying processes. For example, upon injury, KIM‐1 is shed at an accelerated rate47 causing local inflammation and epithelial cellular dedifferentiation that initiates tubular repair. Furthermore, KIM‐1 plays a role as a receptor for phosphatidyl serine, whereby KIM‐1–displaying hRPTECs convert into semiprofessional phagocytes that capture and clear necrotic and apoptotic cell bodies from the injured tubular lumen as a means of managing local inflammation.48 In tandem, other systemic factors (e.g., natural killer T cells, bone marrow stromal cells, activated macrophages) aggregate in the peritubular space at injury sites.15 These are not yet considered in the mathematical models.
From a clinical standpoint, the impact of a sampling schedule (i.e., when to measure a biomarker) on treatment planning accuracy or population parameter estimation is recognized49, 50 and the presented results illustrated that this is also an important phenomenon in vitro. In all, our results indicate the importance of selecting the appropriate in vitro system, biomarker sampling schedule, and method for translation to in vivo pharmacology to accurately predict drug‐related clinical toxicity.
Funding
The research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Numbers NIH‐U24TR001951, NIH‐5UH3TR000504, NIH‐UG3TR002158 and NIH‐P30ES007033. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
The authors declared no competing interests for this work.
Author Contributions
C.M., M.C., C.L.S., N.B.S., J.H., and E.J.K. wrote the manuscript. C.M., M.C., C.L.S., and N.B.S. designed the research. N.B.S. and C.M. performed the research. C.M. and N.B.S. analyzed the data.
Supporting information
Supplementary Figures. Figures S1–S5.
Supplementary Methods S1. Experimental model details.
Supplementary Methods S2. Computational model details and code, Figure S6, Figure S7, Tables S1–S5.
Acknowledgments
The authors would like to thank Nortis Inc. and Elijah Weber for their valuable discussions and input.
References
- 1. Hoste, E.A. & De Corte, W. Clinical consequences of acute kidney injury. Contrib. Nephrol. 174, 56–64 (2011). [DOI] [PubMed] [Google Scholar]
- 2. Parr, S.K. & Siew, E.D. Delayed consequences of acute kidney injury. Adv. Chronic Kidney Dis. 23, 186–194 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vervaet, B.A. , D'Haese, P.C. & Verhulst, A. Environmental toxin–induced acute kidney injury. Clin. Kidney J. 10, 747–758 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tiong, H.Y. , et al Drug‐induced nephrotoxicity: clinical impact and preclinical in vitro models. Mol. Pharm. 11, 1933–1948 (2014). [DOI] [PubMed] [Google Scholar]
- 5. Rosner, M.H. & Okusa, M.D. Acute kidney injury associated with cardiac surgery. Clin. J. Am. Soc. Nephrol. 1, 19–32 (2006). [DOI] [PubMed] [Google Scholar]
- 6. Awdishu, L. & Mehta, R.L. The 6R's of drug induced nephrotoxicity. BMC Nephrol. 18, 124 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Redahan, L. & Murray, P.T. Biomarkers of drug‐induced kidney injury. Curr. Opin. Crit. Care 23, 463–469 (2017). [DOI] [PubMed] [Google Scholar]
- 8. Ho, J. , et al Urinary, plasma, and serum biomarkers' utility for predicting acute kidney injury associated with cardiac surgery in adults: a meta‐analysis. Am. J. Kidney Dis. 66, 993–1005 (2015). [DOI] [PubMed] [Google Scholar]
- 9. Bonventre, J.V. Kidney injury molecule‐1 (KIM‐1): a urinary biomarker and much more. Nephrol. Dial. Transplant. 24, 3265–3268 (2009). [DOI] [PubMed] [Google Scholar]
- 10. Han, W.K. , Bailly, V. , Abichandani, R. , Thadhani, R. & Bonventre, J.V. Kidney injury molecule‐1 (KIM‐1): a novel biomarker for human renal proximal tubule injury. Kidney Int. 62, 237–244 (2002). [DOI] [PubMed] [Google Scholar]
- 11. Bailly, V. , et al Shedding of kidney injury molecule‐1, a putative adhesion protein involved in renal regeneration. J. Biol. Chem. 277, 39739–39748 (2002). [DOI] [PubMed] [Google Scholar]
- 12. Sabbisetti, V.S. , et al Blood kidney injury molecule‐1 is a biomarker of acute and chronic kidney injury and predicts progression to ESRD in type I diabetes. J. Am. Soc. Nephrol. 25, 2177–2186 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van Meer, L. , Moerland, M. , Cohen, A.F. & Burggraaf, J. Urinary kidney biomarkers for early detection of nephrotoxicity in clinical drug development. Br. J. Clin. Pharmacol. 77, 947–957 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vaidya, V.S. , et al Kidney injury molecule‐1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat. Biotechnol. 28, 478–485 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bonventre, J.V. & Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Invest. 121, 4210–4221 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Timmeren, M.M. , et al Tubular kidney injury molecule‐1 (KIM‐1) in human renal disease. J. Pathol. 212, 209–217 (2007). [DOI] [PubMed] [Google Scholar]
- 17. Gebremichael, Y. , et al Multi‐scale mathematical model of drug‐induced proximal tubule injury: linking urinary biomarkers to epithelial cell injury and renal dysfunction. Toxicol. Sci. 162, 200–211 (2017). [DOI] [PubMed] [Google Scholar]
- 18. Lee, S.A. , Noel, S. , Sadasivam, M. , Hamad, A.R.A. & Rabb, H. Role of immune cells in acute kidney injury and repair. Nephron 137, 282–286 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deng, B. , et al The leukotriene B4‐leukotriene B4 receptor axis promotes cisplatin‐induced acute kidney injury by modulating neutrophil recruitment. Kidney Int. 92, 89–100 (2017). [DOI] [PubMed] [Google Scholar]
- 20. Bonavia, A. & Singbartl, K. A review of the role of immune cells in acute kidney injury. Pediatr. Nephrol. 33, 1629–1639 (2018). [DOI] [PubMed] [Google Scholar]
- 21. Adler, M. , et al A quantitative approach to screen for nephrotoxic compounds in vitro . J. Am. Soc. Nephrol. 27, 1015–1028 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Denayer, T. , Stöhr, T. & Van Roy, M. Animal models in translational medicine: validation and prediction. New Horizons Translat. Med. 2, 5–11 (2014). [Google Scholar]
- 23. McGonigle, P. & Ruggeri, B. Animal models of human disease: challenges in enabling translation. Biochem. Pharmacol. 87, 162–171 (2014). [DOI] [PubMed] [Google Scholar]
- 24. Maass, C. , Stokes, C.L. , Griffith, L.G. & Cirit, M. Multi‐functional scaling methodology for translational pharmacokinetic and pharmacodynamic applications using integrated microphysiological systems (MPS). Integr. Biol. (Camb) 9, 290–302 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ewart, L. , et al Application of microphysiological systems to enhance safety assessment in drug discovery. Annu. Rev. Pharmacol. Toxicol. 58, 65–82 (2018). [DOI] [PubMed] [Google Scholar]
- 26. Watson, D.E. , Hunziker, R. & Wikswo, J.P. Fitting tissue chips and microphysiological systems into the grand scheme of medicine, biology, pharmacology, and toxicology. Exp. Biol. Med. (Maywood) 242, 1559–1572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Edington, C.D. , et al Interconnected microphysiological systems for quantitative biology and pharmacology studies. Sci. Rep. 8, 4530 (2018). 10.1038/s41598-018-22749-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maass, C. , et al Establishing quasi‐steady state operations of microphysiological systems (MPS) using tissue‐specific metabolic dependencies. Sci. Rep. 8, 8015 (2018). 10.1038/s41598-018-25971-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weber, E.J. , et al Development of a microphysiological model of human kidney proximal tubule function. Kidney Int. 90, 627–637 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wilmer, M.J. , et al Kidney‐on‐a‐chip technology for drug‐induced nephrotoxicity screening. Trends Biotechnol. 34, 156–170 (2016). [DOI] [PubMed] [Google Scholar]
- 31. Cirit, M. & Stokes, C.L. Maximizing the impact of microphysiological systems with in vitro‐in vivo translation. Lab Chip 18, 1831–1837 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stokes, C.L. , Cirit, M. & Lauffenburger, D.A. Physiome‐on‐a‐chip: the challenge of “scaling” in design, operation, and translation of microphysiological systems. CPT Pharmacometrics Syst. Pharmacol. 4, 559–562 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Valentin, J. Basic anatomical and physiological data for use in radiological protection: reference values. Ann. ICRP 32, 5–265 (2002). [PubMed] [Google Scholar]
- 34. Brown, R.P. , Delp, M.D. , Lindstedt, S.L. , Rhomberg, L.R. & Beliles, R.P. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health 13, 407–484 (1997). [DOI] [PubMed] [Google Scholar]
- 35. Scotcher, D. , Jones, C. , Posada, M. , Rostami‐Hodjegan, A. & Galetin, A. Key to opening kidney for in vitro‐in vivo extrapolation entrance in health and disease: part I: in vitro systems and physiological data. AAPS J. 18, 1067–1081 (2016). [DOI] [PubMed] [Google Scholar]
- 36. Awad, A.S. , et al Compartmentalization of neutrophils in the kidney and lung following acute ischemic kidney injury. Kidney Int. 75, 689–698 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lingadahalli, S . Activated neutrophils mediate KIM‐1 shedding and renal remodelling. ETD Archive Paper 558 (2013).
- 38. Norton, B.L. & Holland, D.P. Current management options for latent tuberculosis: a review. Infect. Drug Resist. 5, 163–173 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Clewe, O. , Wicha, S.G. , de Vogel, C.P. , de Steenwinkel, J.E.M. & Simonsson, U.S.H. A model‐informed preclinical approach for prediction of clinical pharmacodynamic interactions of anti‐TB drug combinations. J. Antimicrob. Chemother. 73, 437–447 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bonventre, J.V. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J. Am. Soc. Nephrol. 14, 55S–61S (2003). [DOI] [PubMed] [Google Scholar]
- 41. Humphreys, B.D. , et al Repair of injured proximal tubule does not involve specialized progenitors. Proc Natl Acad Sci U S A. 108, 9226–9231 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Waanders, F. , van Timmeren, M.M. , Stegeman, C.A. , Bakker, S.J. & van Goor, H. Kidney injury molecule‐1 in renal disease. J. Pathol. 220, 7–16 (2010). [DOI] [PubMed] [Google Scholar]
- 43. Jang, K.J. , et al Human kidney proximal tubule‐on‐a‐chip for drug transport and nephrotoxicity assessment. Integr. Biol. (Camb) 5, 1119–1129 (2013). [DOI] [PubMed] [Google Scholar]
- 44. Frohlich, E.M. , Zhang, X. & Charest, J.L. The use of controlled surface topography and flow‐induced shear stress to influence renal epithelial cell function. Integr. Biol. (Camb) 4, 75–83 (2012). [DOI] [PubMed] [Google Scholar]
- 45. Yang, L. , et al KIM‐1‐mediated phagocytosis reduces acute injury to the kidney. J. Clin. Invest. 125, 1620–1636 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tekce, B.K. , et al Does the kidney injury molecule‐1 predict cisplatin‐induced kidney injury in early stage? Ann. Clin. Biochem. 52, 88–94 (2015). [DOI] [PubMed] [Google Scholar]
- 47. Gandhi, R. , et al Accelerated receptor shedding inhibits kidney injury molecule‐1 (KIM‐1)‐mediated efferocytosis. Am. J. Physiol. Renal Physiol. 307, F205–F221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ichimura, T. , et al Kidney injury molecule‐1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J. Clin. Invest. 118, 1657–1668 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maaß, C. , et al Dependence of treatment planning accuracy in peptide receptor radionuclide therapy on the sampling schedule. EJNMMI Res. 6, 30 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tam, V.H. , Preston, S.L. & Drusano, G.L. Optimal sampling schedule design for populations of patients. Antimicrob. Agents Chemother. 47, 2888–2891 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures. Figures S1–S5.
Supplementary Methods S1. Experimental model details.
Supplementary Methods S2. Computational model details and code, Figure S6, Figure S7, Tables S1–S5.