

Abstract
Aberrantly elevated expression levels of histone deacetylase 1 (HDAC1) and vimentin are closely associated with disease progression in hepatocellular carcinoma (HCC). It was previously demonstrated that knocking down expression of HDAC1 resulted in a concurrent decrease in the expression levels of vimentin. However, a causal link between these two proteins has not yet been demonstrated, to the best of our knowledge. In the present study, the association between HDAC1 and vimentin was investigated using an HDAC1 overexpression platform. HDAC1 and vimentin were significantly increased in HCC cells, and HDAC1 overexpression enhanced vimentin mRNA and protein expression levels in an HDAC1 dose-dependent manner. Subsequently, truncation and mutation of a vimentin promoter demonstrated that HDAC1-induced vimentin expression was dependent on a nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) binding site in the vimentin promoter sequence. Furthermore, HDAC1 induced vimentin expression by promoting NF-κB translocation between the cytoplasm and the nucleus, as opposed to modulating the total expression level of vimentin directly. The data in the present study demonstrated that HDAC1 is overexpressed in HCC and that HDAC1 may upregulate vimentin expression through the NF-κB signaling pathway, thus demonstrating a causal link between HDAC1 and vimentin in HCC, and may provide valuable information in understanding the pathogenesis of HCC.
Keywords: hepatocellular carcinoma, histone deacetylase 1, vimentin, nuclear factor κ-light-chain-enhancer of activated B cells, nuclear translocation
Introduction
Hepatocellular carcinoma (HCC) is one of the most common and frequently fatal malignances worldwide, with a life expectancy of ~6 months from diagnosis (1–3). Although treatment options for HCC have improved, at present, resection and transplantation remain the only curative approaches (4–6). Advances in curative treatment strategies for HCC are partially being held back by a lack of understanding of the molecular mechanisms surrounding the pathogenesis of HCC. Therefore, elucidating the mechanisms underlying HCC pathogenesis and progression is of significant importance in improving the therapeutic options for HCC.
HCC is typically caused by chronic hepatitis B or C virus infection and, to a lesser extent, through chronic exposure to toxins or hereditary liver diseases (7,8). There has been an increase in the identification of disease-associated risk factors, such as histone deacetylases (HDACs) and vimentin, as well as the signaling pathways involved, including activation of the Wnt signaling pathway and inactivation of the cellular tumor antigen p53 signaling pathway (9). However, the complex nature of this disease has complicated advances in understanding the pathological mechanisms of HCC.
HDACs and histone acetylases together regulate gene expression by histone acetylation and de-acetylation. Aberrant expression of these two enzymes may result in epigenetic alteration and, consequently, dysregulated gene expression (10,11). HDAC1 is the most extensively studied HDAC family member, and its expression is increased in a range of different types of cancer, including HCC, prostate cancer and gastric cancer (12–14). Furthermore, upregulated expression of HDAC1 has been associated with a decrease in tissue differentiation and decreased survival rates (12,15–18). Vimentin is an intermediate filament protein in the cytoplasm of mesenchymal cells, and has frequently been used as a marker to demonstrate epithelial-mesenchymal transition, which is associated with the progression of cancer (19). Previous studies have demonstrated that vimentin expression is associated with HCC progression and that the serum vimentin level may serve as an additional marker of HCC, suggesting a role of vimentin protein in HCC pathogenesis (20–22).
Although HDAC1 and vimentin are both important factors in HCC pathogenesis and progression, their roles in this disease have usually been studied separately, and whether there are interactions between HDAC1 and vimentin that affect HCC has not been investigated (12,21). A previous study demonstrated an association between HDAC1 dysregulation and vimentin expression in HCC (23); however, the underlying mechanisms have not been determined. Whether a causal relationship between HDAC1 and vimentin expression exists was investigated in the present study. The present data may provide valuable information regarding potential therapeutic targets.
Materials and methods
Cell lines, plasmids and small interfering (si)RNAs
The HCC cell line Hep3B and the normal human liver cell line THLE-3 were purchased from The Type Culture Collection of the Chinese Academy of Sciences, (Shanghai, China) and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (both Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), penicillin (100 U/ml), and streptomycin (100 mg/ml) at 37°C in a humidified incubator containing 5% CO2.
Human HDAC1, p65 and p50 gene sequences were amplified from human cDNA library and subcloned into a pcDNA3.1(+) vector (Thermo Fisher Scientific, Inc.). Gene sequences were verified by Sanger sequencing. The control Renilla luciferase reporter gene plasmid and the firefly luciferase reporter gene vector pGL3-Basic were purchased from Promega Corporation, (Madison, WI, USA). The vimentin promoter sequence was amplified from the genome of THLE-3 cells, subcloned into pGL3-Basic and termed ‘(−800/+72)Vimentin’. Vimentin promoter truncations and mutations at the NF-κB and PEA3 binding sites were performed on the full length (−800/+72)Vimentin and termed ‘(−725/+72)Vimentin’, ‘(−353/+72)Vimentin’, ‘(−261/+72)Vimentin’, (−200/+72)Vimentin, NF-κB MUT and PEA3 MUT, respectively. The sequences of the primers used for plasmid construction are listed in Table I. HDAC1 siRNA (cat. no. sc-29343) and control siRNA (cat. no. sc-37007) were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Table I.
Primers used for plasmid construction.
| Primer name | Primer sequence | Plasmid name |
|---|---|---|
| V-800-F | 5′-TAGGTACCGCCCGTTAGGTCCCTCGACA-3′ | (−800/+72)Vimentin |
| V-725-F | 5′-TAGGTACCCGGGCCGGAGCAGCCCCCCT-3′ | (−725/+72)Vimentin |
| V-353-F | 5′-TAGGTACCCCCAGCCCAGCGCTGAAGTA-3′ | (−353/+72)Vimentin |
| V-261-F | 5′-TAGGTACCCCCCGCTTCTCGCTAGGTCC-3′ | (−261/+72)Vimentin |
| V-200-F | 5′-GGGACCCTCTTTCCTAACGG-3′ | (−200/+72)Vimentin |
| V-R | 5′-TCGGCCGGCTCGCGGTGCCC-3′ | Reverse primer for all the vimentin plasmids |
| N-F | 5′-TCCCTATTGGATAATGCGCTCCGCGG-3′ | NF-κB MUT |
| N-R | 5′-CCTAGCGAGAAGCGGGGA-3′ | |
| P-F | 5′-TTCCTAACGGAACCTTAAAAACAGCGCCCTCGG-3′ | PEA3 MUT |
| P-R | 5′-AGAGGGTCCCCTCCCACT-3′ | |
| H-F | 5′-TAGAATTCATGGCGCAGACGCAGGGCAC-3′ | HDAC1 |
| H-R | 5′-TACTCGAGTCAGGCCAACTTGACCTCCT-3′ | |
| p65-F | 5′-TAGGTACCATGGGACCCGAAAACGAGAG-3′ | p65 |
| p65-R | 5′-GAGAATTCTCAGGTCTGGGGACTTGCTT-3′ | |
| p50-F | 5′-GCGGTACCATGGCAGAAGATGATCCATA-3′ | p50 |
| p50-R | 5′ GCGAATTCCTAAATTTTGCCTTCTAGAG-3′ |
F, forward; R, reverse; N/NF-κB, nuclear factor κ-light-chain-enhancer of activated B cells; P, PEA3; H/HDAC1, histone deacetylase 1; p50, NF-κB p105 subunit; p65, transcription factor p65; MUT, mutant.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
RT-qPCR was performed as previously described, with certain modifications (24). The total RNA was extracted from cells using TRIzol® reagent (Thermo Fisher Scientific, Inc.) and reverse-transcribed into cDNA using Moloney murine leukemia virus reverse transcriptase (M-MLV RT) (Promega Corporation). Total RNA (2 µg) was first mixed with 0.5 µg Oligo(dT)18 primer and heated to 70°C for 5 min. After heating, the sample was immediately cooled on ice. Subsequently, 5 µl 5X reaction buffer, 1.25 µl 10 mM dATP, 1.25 µl 10 mM dCTP, 1.25 µl 10 mM dGTP, 1.25 µl 10 mM dTTP, 25 units recombinant RNasin ribonuclease inhibitor (Takara Biotechnology Co., Inc.) and 200 units M-MLV RT (Promega Corporation) were added and mixed, and incubated at 42°C for 60 min. Subsequently, the vimentin RNA level was quantified using SYBR Green (Bio-Rad Laboratories, Inc., Hercules, CA, USA), with GAPDH as an internal control. The following thermal cycling protocol was used: Polymerase activation and DNA denaturation (95°C, 1 min); 40 cycles of denaturation (95°C, 10 sec) and annealing/extension (60°C, 30 sec), followed by melt-curve analysis (65–95°C with 0.5°C increments, 2–5 sec/step). The following primers were used for RT-qPCR. The primers for vimentin were: Forward, 5′-AGGAAATGGCTCGTCACCTTCGTGAATA-3′; reverse, 5′-GGAGTGTCGGTTGTTAAGAACTAGAGCT-3′. The primers for GAPDH were: Forward, 5′-AGCCACATCGCTCAGACA-3′; reverse, 5′-TGGACTCCACGACGTACT-3′. The relative vimentin mRNA expression level was calculated using the 2−ΔΔCq method (25).
Subcellular fractionation
Cell nuclear and cytoplasmic fractions were prepared using NE-PER™ Nuclear and Cytoplasmic Extraction kit (Thermo Fisher Scientific, Inc.), according to the manufacturer's protocol. Cells were harvested and resuspended in DNA endonuclease I-Cre(I) by vortexing for 15 sec. Subsequently, the cell suspension was mixed with CRE II by vortexing for 10 sec and centrifuged at 16,000 × g for 5 min at 4°C to obtain the cytoplasmic fraction from the supernatant. The insoluble pellet, which contained the nuclear fraction, was resuspended in the nuclear extraction reagent and vortexed for 15 sec. After centrifugation at 16,000 × g for 10 min at 4°C, the nuclear fraction was obtained from the supernatant.
Western blotting
For analysis using whole cell lysate, cells were harvested and lysed using lysis buffer (Thermo Fisher Scientific, Inc.) supplemented with protease inhibitors (Roche Applied Science, Penzberg, Germany), and mixed with SDS-PAGE loading buffer. For analysis using cytoplasmic and nuclear fractions, prepared fractions were mixed with SDS-PAGE loading buffer directly. Protein concentration was quantified using the Pierce BCA Protein Assay kit (Thermo Fisher Scientific, Inc.) and 20 µg whole cell lysate or 10 µg cytoplasmic and nuclear fractions were separated using a 12% SDS-PAGE gel and transferred onto a PVDF membrane. GAPDH was used as a loading control. Non-specific binding sites on the membrane were blocked with 5% skimmed milk for 1 h at room temperature. The membrane was sequentially incubated with primary and corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies for 2 and 1 h at room temperature, respectively. After extensive washes, immunobands were visualized using ECL substrate (Merck KGaA, Darmstadt, Germany,) under a charge-couple device camera (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The following primary antibodies were used in this study: Anti-HDAC1 antibody (cat. no. ab19845; Abcam, Cambridge, UK), anti-p65 antibody (cat. no. sc-372; Santa Cruz Biotechnology, Inc.), anti-vimentin antibody (cat. no. ab92547; Abcam) and anti-GAPDH antibody (cat. no. g8795; Sigma-Aldrich; Merck KGaA). HRP-conjugated secondary antibodies [Goat Anti-Mouse IgG (H+L)-HRP; cat. no. BA1050 and Goat Anti-Rabbit IgG (H+L)-HRP; cat. no. BA1056] were both obtained from Wuhan Boster Biological Technology, Ltd., (Wuhan, China). All the primary antibodies were used at a 1:1,000 dilution, and all the secondary antibodies were used at a 1:100,000 dilution.
Transfection and luciferase reporter gene activity assay
The luciferase reporter gene activity assay was performed as previously described with modifications (26). Cells were first transfected with firefly luciferase reporter gene plasmids with full length vimentin promoter (−800/+72)Vimentin, vimentin truncations (−725/+72)Vimentin, (−353/+72)Vimentin, (−261/+72)Vimentin, (−200/+72)Vimentin, or the vimentin mutations NF-κB MUT or PEA3 MUT, in addition to an HDAC1 plasmid and control Renilla luciferase reporter gene plasmid using Lipofectamine™ 2000, according to the manufacturer's protocol (Thermo Fisher Scientific, Inc.). Cells were incubated for 24 h post transfection, after which they were lysed with lysis buffer and firefly and Renilla luciferase activities were measured using Promega Dual Luciferase Assay kit and the GloMax®-Multi Detection System, according to the manufacturer's protocol (both from Promega Corporation). The transfection efficiency was normalized to Renilla luciferase activity. For the siRNA knockdown assay, p65 siRNA (sc-29410; Santa Cruz Biotechnology, Inc.) or control siRNA (sc-37007; Santa Cruz Biotechnology, Inc.) at a final concentration of 100 nM was introduced into cells using X-tremeGENE siRNA Transfection Reagent (Roche Applied Science) 24 h prior to plasmid transfection. For the signaling pathway inhibition assay, an NF-κB inhibitor (Celastrol; 300 nM) or Janus kinase (JNK) inhibitor (SP600125; 10 µM; both InvivoGen. Inc., Toulouse, France) was added to the cell culture 4–6 h post transfection. For p65 + p50 co-transfection, plasmids encoding p65 and p50 at a ratio of 1:1 were mixed and transfected into cells using Lipofectamine® 2000 according to the manufacturer's protocol (Thermo Fisher Scientific. Inc.).
Chromatin immunoprecipitation (ChIP) assay
ChIP analysis was performed using a Pierce™ Magnetic ChIP kit (Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. All reagents used were included in the kit unless otherwise stated. Cells were first crosslinked with 1% formaldehyde, lysed with membrane extraction buffer and digested with MNase, and the chromatin smear was obtained by sonication (3×20 sec pulses at 3 W power on ice with a 20-sec incubation on ice between pulses). The obtained chromatin smear was then sequentially incubated with 5 µg of either anti-p65 antibody (cat. no. sc-372; Santa Cruz Biotechnology, Inc.) or normal rabbit IgG overnight at 4°C and magnetic protein A/G beads for 2 h at 4°C. Subsequent to washings, precipitated DNA was recovered from magnetic beads with elution buffer and a PCR was performed with Q5® High-Fidelity DNA Polymerase (New England BioLabs, Inc., Ipswich, MA, USA) using vimentin promoter-specific primers: Forward primer, 5′-GGGCTCCATGAGTCATATCC-3′ and reverse primer, 5′-ATCTGGCTCAAGACCTTTGC-3′. The following thermal cycling conditions were used: Initial denaturation, 98°C, 30 sec; 35 of cycles of denaturation (98°C, 10 sec), annealing (55°C, 10 sec) and elongation (72°C, 30 sec); and final extension (72°C, 2 min).
Statistical analysis
All experiments were repeated three times. Data were presented as mean ± standard deviation. A Student's t test or a one-way analysis of variance with a Student-Newman-Keuls post hoc were used to compare the differences between two groups or three or more groups, respectively. P<0.05 was considered to indicate a statistically significant difference. All statistical analysis was performed with GraphPad PRISM 4.0.3 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
HDAC1 overexpression results in increased vimentin expression
A previous study demonstrated that HDAC1 downregulation may result in a decrease in vimentin expression in HCC (23). In the present study, to confirm the association between HDAC1 and vimentin expression in HCC, the expression of HDAC1 and vimentin in THLE-3 normal liver cells and Hep3B HCC cells were determined. HDAC1 protein expression levels were markedly increased in Hep3B cells compared with THLE-3 cells (Fig. 1A). Notably, similar to HDAC1, the expression of vimentin also markedly increased in Hep3B cells (Fig. 1A).
Figure 1.

HDAC1 overexpression upregulates vimentin expression in HCC. (A) HDAC1 protein expression in the THLE-3 normal liver cell line and the Hep3B HCC cell line. (B) HDAC-1 mRNA expression levels in THLE-3 cells transfected with increasing quantities of a plasmid containing HDAC1. NS, not significant; *P<0.05; **P<0.01. (C) HDAC-1 protein expression levels in THLE-3 cells transfected with increasing quantities of a plasmid containing HDAC1. HCC, hepatocellular carcinoma; HDAC1, histone deacetylase 1.
To further investigate whether vimentin expression was associated with HDAC1 expression, THLE-3 cells were transfected with increasing quantities of HDAC1, and the mRNA and protein levels of vimentin were measured. Vimentin mRNA expression levels were increased significantly when ≥1 µg of HDAC1 was transfected (P<0.05; Fig. 1B). Western blotting demonstrated that vimentin protein expression levels were additionally increased in an HDAC1 dose dependent manner (Fig. 1C). Together, these data suggested that HDAC1 and vimentin are increased in HCC cells, and HDAC1 expression may upregulate vimentin expression.
An NF-κB transcription factor-binding site in the vimentin promoter is associated with HDAC1-induced vimentin expression
To determine whether HDAC1 upregulated vimentin expression through the transactivation of a vimentin promoter, luciferase reporter gene plasmids were constructed with gene expression under the control of a full length or a 5-flanking region of a truncated vimentin promoter sequence (Fig. 2A), and the activation of these plasmids by HDAC1 was determined. HDAC1 overexpression significantly activated the vimentin promoter (P<0.001 vs. pcDNA3.1; Fig. 2B). Through the use of truncation mutants, it was demonstrated that HDAC1-mediated activation of the vimentin promoter was abrogated when the sequence between −261 and −200 was removed; suggesting that the sequence in this region was necessary for HDAC1 induced activation (Fig. 2C).
Figure 2.

HDAC1 transactivates the vimentin promoter. (A) A schematic illustration of the vimentin promoter and 5′ end truncations. (B) Luciferase activity in THLE-3 cells transfected with (−800/+72) Vimentin with or without HDAC1. ***P<0.001 vs. pcDNA3.1. (C) Luciferase activity in THLE-3 cells transfected with (−800/+72) Vimentin or 5′ end serial truncations with or without HDAC1. **P<0.01. HDAC1, histone deacetylase 1; NF-κB, nuclear factor κ-light-chain-enhancer of activated B cells; LUC, luciferase.
Based on the bioinformatics analysis performed previously (26), there is a NF-κB binding site between −261 and −200 (Fig. 2). To determine whether the NF-κB binding site was involved in the HDAC-1 overexpression-induced vimentin promoter activation, the ability of HDAC1 to activate a full-length vimentin promoter with a mutated NF-κB binding site was determined (Fig. 3A). As shown in Figure. 3B, HDAC1 overexpression was unable to activate the NF-κB mutated vimentin promoter; however, HDAC1 mediated activation of the vimentin promoter was still significantly increased when the NF-κB neighboring domain, PEA3 was mutated, with a potency similar to (−800/+72)Vimentin. The data presented in Fig. 2C suggest that the NF-κB binding site between-261 and −200 in vimentin promoter is involved in the HDAC1-induced vimentin expression.
Figure 3.

NF-κB binding site in the vimentin promoter is involved in HDAC1 overexpression-induced vimentin expression. (A) A schematic illustration of vimentin promoter mutations. (B) Luciferase activity in THLE-3 cells transfected with (−800/+72) Vimentin, NF-κB MUT or PEA3 MUT with or without HDAC1. NS, not significant; ***P<0.001. HDAC1, histone deacetylase 1; NF-κB, nuclear factor κ-light-chain-enhancer of activated B cells; LUC luciferase.
HDAC1 induces vimentin expression through the NF-κB signaling pathway
To determine whether HDAC1 overexpression could promote NF-κB binding with the vimentin promoter, a ChIP assay was performed. The vimentin promoter sequence was not pulled down by an anti-p65 antibody when HDAC1 was not overexpressed. However, following HDAC1 overexpression, the vimentin promoter sequence was detected in the anti-p65 antibody pull-down, indicating that HDAC1 overexpression could result in the binding of NF-κB with the vimentin promoter (Fig. 4A). The control immunoglobulin G antibody did not result in a pull-down of the vimentin promoter sequence irrespective of HDAC1 expression (Fig. 4A).
Figure 4.

HDAC1 overexpression upregulates vimentin expression via the NF-κB signaling pathway. (A) Chromatin immunoprecipitation assay using an anti-p65 antibody to detect NF-κB binding to vimentin promoter. (B) THLE-3 cells were mock transfected or transfected with control siRNA or p65 siRNA and p65 expression was determined by western blotting. (C) THLE-3 cells were treated with p65 siRNA or control siRNA, and were further transfected with (−800/+72) Vimentin together with or without HDAC1 and the luciferase activity was measured. ns, not significant; *P<0.05, ***P<0.001. (D) THLE-3 cells were first transfected with (−800/+72) Vimentin together with sham vector or a combination of p65 and p50 and the luciferase activity was measured. **P<0.01 vs. pcDNA3.1. (E) THLE-3 cells were transfected with (−800/+72) Vimentin with or without HDAC1 for 4–6 h, then treated with NF-κB or JNK signaling pathway inhibitors and the luciferase activity was measured. ns, not significant; ***P<0.001. HDAC1, histone deacetylase 1; NF-κB, nuclear factor κ-light-chain-enhancer of activated B cells; p50, NF-κB p105 subunit; p65, transcription factor p65; siRNA, small interfering RNA.
To confirm the participation of NF-κB in HDAC1 overexpression-induced vimentin expression, a p65 knockdown by siRNA interference was performed. p65 siRNA knockdown efficiency was first confirmed by western blotting (Fig. 4B). p65 knockdown abrogated the activation of the vimentin promoter by HDAC1 overexpression (P<0.05), whereas control siRNA interference had no effect (P>0.05; Fig. 4C). NF-κB overexpression may increase the protein expression levels of all the NF-κB forms, including the unphosphorylated form in the cytoplasm and the phosphorylated form in the nucleus (27), and the increase of phosphorylated NF-κB may enhance vimentin expression. Therefore, the role of NF-κB overexpression on the transactivation of vimentin was investigated. The results demonstrated that NF-κB (p65+p50) overexpression could activate the vimentin promoter (P<0.01; Fig. 4D).
To investigate the involvement of the NF-κB signaling pathway in HDAC1 overexpression-induced vimentin expression, a signaling pathway inhibition assay was performed. Vimentin promoter activation was significantly decreased when NF-κB inhibitor (P<0.001), but not JNK inhibitor, was added (Fig. 4E). Taken together, these data indicated that HDAC1 overexpression-induced vimentin upregulation via the NF-κB signaling pathway.
HDAC-1 induces NF-κB translocation from the cytoplasm into the nucleus
As the NF-κB signaling pathway was modulated in HDAC1-induced vimentin expression, the impact of HDAC1 overexpression on NF-κB expression and translocation was subsequently investigated. First, the whole cell lysate of THLE-3 cells with or without HDAC1 overexpression were tested for p65 expression. As presented in Fig. 5A, p65 expression did not demonstrate any apparent alterations in expression before or after HDAC1 overexpression. The cytoplasmic and nucleic fractions were further separated and the expression levels of p65 in both fractions were determined. p65 was present in both fractions when HDAC1 was not overexpressed (Fig. 5B). However, when HDAC1 was overexpressed, a near complete translocation of p65 from the cytoplasm into the nucleus was observed, indicating that HDAC1 overexpression modulates the NF-κB signaling pathway by promoting NF-κB translocation from the cytoplasm into the nucleus.
Figure 5.
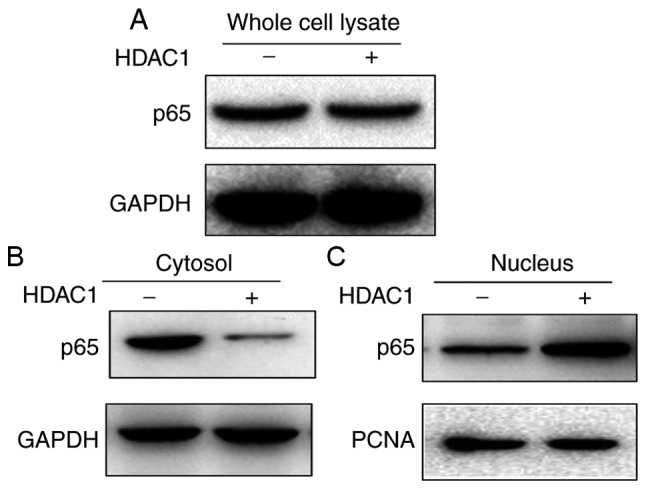
HDAC1 overexpression results in NF-κB translocation from cytoplasm into nucleus. THLE-3 cells transfected with or without HDAC1 for 24 h, and then (A) whole cell lysate (B) cytoplasmic and (C) nucleic fractions were separated for western blotting to determine p65 expression. HDAC1, histone deacetylase 1; NF-κB, nuclear factor κ-light-chain-enhancer of activated B cells; p65, transcription factor p65.
Together, these data demonstrated that HDAC1 is overexpressed in HCC and this HDAC1 overexpression could upregulate vimentin expression through the NF-κB signaling pathway. The present study demonstrated a causal relationship between HDAC1 and vimentin in HCC, providing valuable information for understanding the pathogenesis of HCC, as well as serving as potential targets for the treatment of HCC.
Discussion
HDACs are enzymes involved in the transcriptional regulation of genes; the expression of HDACs is increased in a number of different types of tumors, including HCC, prostate cancer and gastric cancer (12–14) and HDAC inhibitors are effective in suppressing tumor growth at an early stage (28). In HCC, the best studied HDAC family member is HDAC1, which is expressed at aberrantly high levels in HCC cells (29,30). Vimentin, one of the most widely expressed type III intermediate filament proteins, is significantly increased in a number of different types of solid tumors and is regularly used as a marker for epithelial-mesenchymal transition, a process closely associated with tumor progression (31). In HCC, the association of elevated vimentin expression with tumor development, especially metastasis, has been widely studied and the serum vimentin expression level is used as a marker for HCC (21,22,32,33). Although HDAC1 and vimentin have been widely studied in HCC and other tumors, the association between them has not been investigated, to the best of our knowledge. A previous study demonstrated that downregulation of HDAC1 resulted in a decrease of vimentin expression, indicating a potential link between these two proteins. In the present study a causal relationship was confirmed between HDAC1 and vimentin. Furthermore, the signaling pathway responsible for the HDAC1 overexpression-induced vimentin expression was identified. The findings of our current study would not only provide information for the understanding of the pathogenesis of HCC, but also provide potential targets for this disease or possibly other solid tumors.
There are 18 known HDACs in mammals and the majority of them have been associated with tumor progression, and HDACs have been demonstrated to function distinctively in cancer cells (34,35). For example, HDAC2, but not HDAC1, has a suppressive effect on cell growth in breast cells (34). Additionally, knockdown of HDAC1 and HDAC2 may exert differing effects on cell survival (35). In HCC, HDAC1 and HDAC2 are both associated with tumor growth (28). However, HDAC1 and HDAC2 possess a high degree of resemblance in their genomic sequence (36). Therefore, it may be interesting to investigate, albeit beyond the scope of the present study, whether HDAC2 has an effect on vimentin expression and whether the effect is different from that of HDAC1, as well as the potential mechanisms.
Regulation of vimentin expression has been investigated by other studies and different signaling pathways are involved under different circumstances. In IL-6- promoted head and neck tumor metastasis, vimentin expression is activated through the JAK/STAT3/SNAIL signaling pathway (37), while in tubular epithelial cells, hypoxia-induced vimentin expression is mediated through the Notch signaling pathway (38). TGF-β1 regulates vimentin expression in skeletal myogenic cells through a signaling pathway involving Smad/AP1/SP1 (24). In the present study, HDAC1 overexpression-induced vimentin expression in HCC cells was through the NF-κB signaling pathway. Given the complex regulation network surrounding vimentin expression (39), whether the NF-κB signaling pathway is also responsible for vimentin expression in other tumors remains to be determined.
Together, the present study demonstrated that HDAC1 is overexpressed in HCC and that HDAC1 overexpression could upregulate vimentin expression through the NF-κB signaling pathway in HCC cells. Furthermore a causal relationship between HDAC1 and vimentin in HCC was identified, which may improve our understanding of HCC pathogenesis, as well as identifying potential targets for HCC treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by Natural Science Foundation of Guangdong Province, Initiation Project for PhD (grant no. S2012040007235).
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
HZ and JX designed the experiment. HZ and CZ performed the assays. HZ and YW analyzed the data. HZ wrote the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.World-Health-Organization, corp-author. Cancer (fact sheet) 2018. https://www.who.int/en/news-room/fact-sheets/detail/cancer. [Sep 12;2018 ]; [Google Scholar]
- 2.Ito Y, Takeda T, Higashiyama S, Sakon M, Wakasa KI, Tsujimoto M, Monden M, Matsuura N. Expression of heparin binding epidermal growth factor-like growth factor in hepatocellular carcinoma: An immunohistochemical study. Oncol Rep. 2001;8:903–907. doi: 10.3892/or.8.4.903. [DOI] [PubMed] [Google Scholar]
- 3.Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–1255. doi: 10.1016/S0140-6736(11)61347-0. [DOI] [PubMed] [Google Scholar]
- 4.Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: Worldwide incidence and trends. Gastroenterology. 2004;127:S5–S16. doi: 10.1053/j.gastro.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 5.Waly Raphael S, Yangde Z, YuXiang C. Hepatocellular carcinoma: Focus on different aspects of management. ISRN Oncol. 2012;2012:421673. doi: 10.5402/2012/421673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 7.Morgan RL, Baack B, Smith BD, Yartel A, Pitasi M, Falck-Ytter Y. Eradication of Hepatitis C virus infection and the development of hepatocellular carcinomaa: A meta-analysis of observational studies. Ann Intern Med. 2013;158:329–337. doi: 10.7326/0003-4819-158-5-201303050-00005. [DOI] [PubMed] [Google Scholar]
- 8.Gordon SC, Lamerato LE, Rupp LB, Li J, Holmberg SD, Moorman AC, Spradling PR, Teshale EH, Vijayadeva V, Boscarino JA, et al. Antiviral therapy for chronic hepatitis B virus infection and development of hepatocellular carcinoma in a US population. Clin Gastroenterol Hepatol. 2014;12:885–893. doi: 10.1016/j.cgh.2013.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirstein MM, Vogel A. The pathogenesis of hepatocellular carcinoma. Dig Dis. 2014;32:545–553. doi: 10.1159/000360499. [DOI] [PubMed] [Google Scholar]
- 10.Kaliman P, Álvarez-López MJ, Cosín-Tomás M, Rosenkranz MA, Lutz A, Davidson RJ. Rapid changes in histone deacetylases and inflammatory gene expression in expert meditators. Psychoneuroendocrinology. 2014;40:96–107. doi: 10.1016/j.psyneuen.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shankar S, Srivastava RK. Histone deacetylase inhibitors: mechanisms and clinical significance in cancer: HDAC inhibitor-induced apoptosis. Adv Exp Med Biol. 2008;615:261–298. doi: 10.1007/978-1-4020-6554-5_13. [DOI] [PubMed] [Google Scholar]
- 12.Rikimaru T, Taketomi A, Yamashita Y, Shirabe K, Hamatsu T, Shimada M, Maehara Y. Clinical significance of histone deacetylase 1 expression in patients with hepatocellular carcinoma. Oncology. 2007;72:69–74. doi: 10.1159/000111106. [DOI] [PubMed] [Google Scholar]
- 13.Halkidou K, Gaughan L, Cook S, Leung HY, Neal DE, Robson CN. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate. 2004;59:177–189. doi: 10.1002/pros.20022. [DOI] [PubMed] [Google Scholar]
- 14.Choi JH, Kwon HJ, Yoon BI, Kim JH, Han SU, Joo HJ, Kim DY. Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn J Cancer Res. 2001;92:1300–1304. doi: 10.1111/j.1349-7006.2001.tb02153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quint K, Agaimy A, Di Fazio P, Montalbano R, Steindorf C, Jung R, Hellerbrand C, Hartmann A, Sitter H, Neureiter D, Ocker M. Clinical significance of histone deacetylases 1, 2, 3, and 7: HDAC2 is an independent predictor of survival in HCC. Virchows Arch. 2011;459:129–139. doi: 10.1007/s00428-011-1103-0. [DOI] [PubMed] [Google Scholar]
- 16.Seo J, Min SK, Park HR, Kim DH, Kwon MJ, Kim LS, Ju YS. Expression of Histone Deacetylases HDAC1, HDAC2, HDAC3, and HDAC6 in invasive ductal carcinomas of the breast. J Breast Cancer. 2014;17:323–331. doi: 10.4048/jbc.2014.17.4.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007;1:19–25. doi: 10.1016/j.molonc.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng L, Seto E. Deacetylation of nonhistone proteins by HDACs and the implications in cancer. Handb Exp Pharmacol. 2011;206:39–56. doi: 10.1007/978-3-642-21631-2_3. [DOI] [PubMed] [Google Scholar]
- 19.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nature Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 20.Hu L, Lau SH, Tzang CH, Wen JM, Wang W, Xie D, Huang M, Wang Y, Wu MC, Huang JF, et al. Association of Vimentin overexpression and hepatocellular carcinoma metastasis. Oncogene. 2004;23:298–302. doi: 10.1038/sj.onc.1206483. [DOI] [PubMed] [Google Scholar]
- 21.Mitra A, Satelli A, Xia X, Cutrera J, Mishra L, Li S. Cell-surface Vimentin: A mislocalized protein for isolating csVimentin(+) CD133(−) novel stem-like hepatocellular carcinoma cells expressing EMT markers. Int J Cancer. 2015;137:491–496. doi: 10.1002/ijc.29382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun S, Poon RT, Lee NP, Yeung C, Chan KL, Ng IO, Day PJ, Luk JM. Proteomics of hepatocellular carcinoma: Serum vimentin as a surrogate marker for small tumors (≤2 cm) J Proteome Res. 2010;9:1923–1930. doi: 10.1021/pr901085z. [DOI] [PubMed] [Google Scholar]
- 23.Zhou H, Wang J, Peng G, Song Y, Zhang C. A novel treatment strategy in hepatocellular carcinoma by down-regulation of histone deacetylase 1 expression using a shRNA lentiviral system. Int J Clin Exp Med. 2015;8:17721–17729. [PMC free article] [PubMed] [Google Scholar]
- 24.Wu Y, Zhang X, Salmon M, Lin X, Zehner ZE. TGFbeta1 regulation of vimentin gene expression during differentiation of the C2C12 skeletal myogenic cell line requires Smads, AP-1 and Sp1 family members. Biochim Biophys Acta. 2007;1773:427–439. doi: 10.1016/j.bbamcr.2006.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.livak kj, schmittgen td. analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Huang W, Hu K, Luo S, Zhang M, Li C, Jin W, Liu Y, Griffin GE, Shattock RJ, Hu Q. Herpes simplex virus type 2 infection of human epithelial cells induces CXCL9 expression and CD4+ T cell migration via activation of p38-CCAAT/enhancer-binding protein-β pathway. J Immunol. 2012;188:6247–6257. doi: 10.4049/jimmunol.1103706. [DOI] [PubMed] [Google Scholar]
- 27.Lizzul PF, Aphale A, Malaviya R, Sun Y, Masud S, Dombrovskiy V, Gottlieb AB. Differential expression of phosphorylated NF-kappaB/RelA in normal and psoriatic epidermis and downregulation of NF-kappaB in response to treatment with etanercept. J Invest Dermatol. 2005;124:1275–1283. doi: 10.1111/j.0022-202X.2005.23735.x. [DOI] [PubMed] [Google Scholar]
- 28.Ler SY, Leung CH, Khin LW, Lu GD, Salto-Tellez M, Hartman M, Iau PT, Yap CT, Hooi SC. HDAC1 and HDAC2 independently predict mortality in hepatocellular carcinoma by a competing risk regression model in a Southeast Asian population. Oncol Rep. 2015;34:2238–2250. doi: 10.3892/or.2015.4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun TY, Xie HJ, Li Z, Kong LF, Ding YZ. Analysis of miRNAs related to abnormal HDAC1 expression in hepatocellular carcinoma. Int J Clin Exp Med. 2016;9 [Google Scholar]
- 30.Yoo YG, Na TY, Seo HW, Seong JK, Park CK, Shin YK, Lee MO. Hepatitis B virus X protein induces the expression of MTA1 and HDAC1, which enhances hypoxia signaling in hepatocellular carcinoma cells. Oncogene. 2008;27:3405–3413. doi: 10.1038/sj.onc.1211000. [DOI] [PubMed] [Google Scholar]
- 31.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salvi A, Bongarzone I, Ferrari L, Abeni E, Arici B, De Bortoli M, Scuri S, Bonini D, Grossi I, Benetti A, et al. Molecular characterization of LASP-1 expression reveals vimentin as its new partner in human hepatocellular carcinoma cells. Int J Oncol. 2015;46:1901–1912. doi: 10.3892/ijo.2015.2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dong Q, Zhu X, Dai C, Zhang X, Gao X, Wei J, Sheng Y, Zheng Y, Yu J, Xie L, et al. Osteopontin promotes epithelial-mesenchymal transition of hepatocellular carcinoma through regulating vimentin. Oncotarget. 2016;7:12997–13012. doi: 10.18632/oncotarget.7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harms KL, Chen X. Histone deacetylase 2 modulates p53 transcriptional activities through regulation of p53-DNA binding activity. Cancer Res. 2007;67:3145–3152. doi: 10.1158/0008-5472.CAN-06-4397. [DOI] [PubMed] [Google Scholar]
- 35.Lei WW, Zhang KH, Pan XC, Wang DM, Hu Y, Yang YN, Song JG. Histone deacetylase 1 and 2 differentially regulate apoptosis by opposing effects on extracellular signal-regulated kinase 1/2. Cell Death Dis. 2010;1:e44. doi: 10.1038/cddis.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Ruijter AJ, Van Gennip AH, Caron HN, Stephan K, Van Kuilenburg AB. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/bj20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yadav A, Kumar B, Datta J, Teknos TN, Kumar P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res. 2011;9:1658–1667. doi: 10.1158/1541-7786.MCR-11-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du R, Sun W, Xia L, Zhao A, Yu Y, Zhao L, Wang H, Huang C, Sun S. Hypoxia-induced down-regulation of microRNA-34a promotes EMT by targeting the Notch signaling pathway in tubular epithelial cells. PLoS One. 2012;7:e30771. doi: 10.1371/journal.pone.0030771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarria AJ, Nordeen SK, Evans RM. Regulated expression of vimentin cDNA in cells in the presence and absence of a preexisting vimentin filament network. J Cell Biol. 1990;111:553–565. doi: 10.1083/jcb.111.2.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.