

Abstract
Metabolic chemical reporters of glycosylation in combination with bioorthogonal reactions have been known for two decades and have been used by many different research laboratories for the identification and visualization of glycoconjugates. More recently, however, they have begun to see utility for the investigation of cellular metabolism and the tolerance of biosynthetic enzymes and glycosyltransferases to different sugars. Here, we take this concept one step further by using the metabolic chemical reporter 6-azido-6-deoxy-glucose (6AzGlc). We show that treatment of mammalian cells with the per-O-acetylated version of 6AzGlc results in robust labeling of a variety of proteins. Notably, the pattern of this labeling was consistent with O-GlcNAc modifications, suggesting that the enzyme O-GlcNAc transferase is quite promiscuous for its donor sugar substrates. To confirm this possibility, we show that 6AzGlctreatment results in the labeling of known O-GlcNAcylated proteins, that the UDP-6AzGlc donor sugar is indeed produced in living cells, and that recombinant OGT will accept UDP-6AzGlc as a substrate in vitro. Finally, we use proteomics to first identify several bona fide 6AzGlc-modifications in mammalian cells and then an endogenous O-glucose modification on host cell factor. These results support the conclusion that OGT can endogenously modify proteins with both N-acetyl-glucosamine and glucose, raising the possibility that intracellular O-glucose modification may be a widespread modification under certain conditions or in particular tissues.
Graphical Abstract:
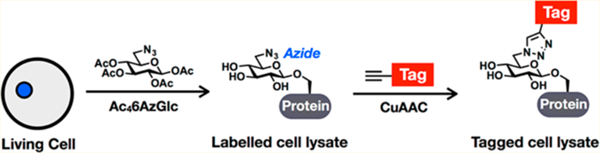
INTRODUCTION
O-GlcNAc modifications are the addition of the monosaccharide N-acetyl-glucosamine (GlcNAc) to proteins that are located throughout the cytosol, nucleus, and mitochondria in animal cells (Figure 1A).1,2 Unlike other forms of glycosylation, O-GlcNAcylation can be dynamically added and removed from substrate proteins through the actions of O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA).3 The modification is required for survival in mice and Drosophila and has been shown to regulate many critical areas of biology.4–6 As with other forms of protein glycosylation, a range of biorthogonal probes have been developed for the detection and characterization of O-GlcNAcylated proteins.7–9 We have contributed in this area through the generation of several metabolic chemical reporters (MCRs, Figure 1B).10–14 These MCRs are analogues of natural monosaccharides that contain azide- or alkyne-functionalities at different positions and complement the wide-range of reporters developed by the Bertozzi, Wittmann, Prescher, and other laboratories.15 Treatment of living cells with MCRs can result in their metabolism to the corresponding donor sugar and the subsequent enzymatic modification of proteins. During the creation and analysis of some of these MCRs, we observed unexpected tolerances for unnatural sugars by both the cellular biosynthetic machinery and OGT. For example, GlcNAc analogues containing modifications that replace the 6-hydroxyl group with an azide or alkyne can bypass the canonical GlcNAc salvage pathway to be biosynthetically transformed into the corresponding uridine diphosphate (UDP) sugar-donors.12 Additionally, OGT is able to transfer both of these analogues,12,14,16 as well as 2-azido-glucose,13,17 to protein substrates where they can be visualized using the copper-catalyzed azide−alkyne cycloaddition (CuAAC) in combination with appropriate tags. Together, these results suggest that proteins have the potential be enzymatically modified by OGT with a broad range of natural and unnatural monosaccharides in addition to GlcNAc. Here, we continue our exploration of these possibilities through the analysis of 6-azido-glucose (6AzGlc). We find that a variety of mammalian cells are robustly labeled by treatment with per-O-acetylated 6AzGlc (Ac46AzGlc, Figure 1B) and that this labeling can be reduced by a small molecule inhibitor of OGT. Notably, this labeling is not affected by the CRISPR-mediated knockout of the enzyme glutamine fructose-6-phosphate aminotransferase (GFAT),18 nor does it appear to be removed by OGA, both of the results indicating that 6AzGlc is not biosynthetically converted to 6AzGlcNAc. We then demonstrate that recombinant OGT can transfer 6AzGlc from chemically synthesized UDP-6AzGlc to a variety of protein substrates in prepared lysates from NIH3T3 cells, and we use isotope-targeted glycoproteomics (IsoTaG)19,20 in combination with Ac46AzGlc treated cells to directly observe several 6AzGlc modifications. Finally, we reanalyzed our existing proteomic data21 and found an endogenous O-glucose modification on the protein host cell factor (HCF). Taken together, these results further demonstrate that OGT has a high degree of promiscuity. Additionally, to our knowledge, our results are the first demonstration of intracellular O-glucose modification which may be significant on intracellular proteins under certain conditions that involve high UDP-glucose concentrations (i.e., glycogen synthesis). Importantly, these discoveries would not have been probable without the utility of MCRs not only for identifying modified proteins but also for probing the tolerance of cellular pathways and enzymes.
Figure 1.
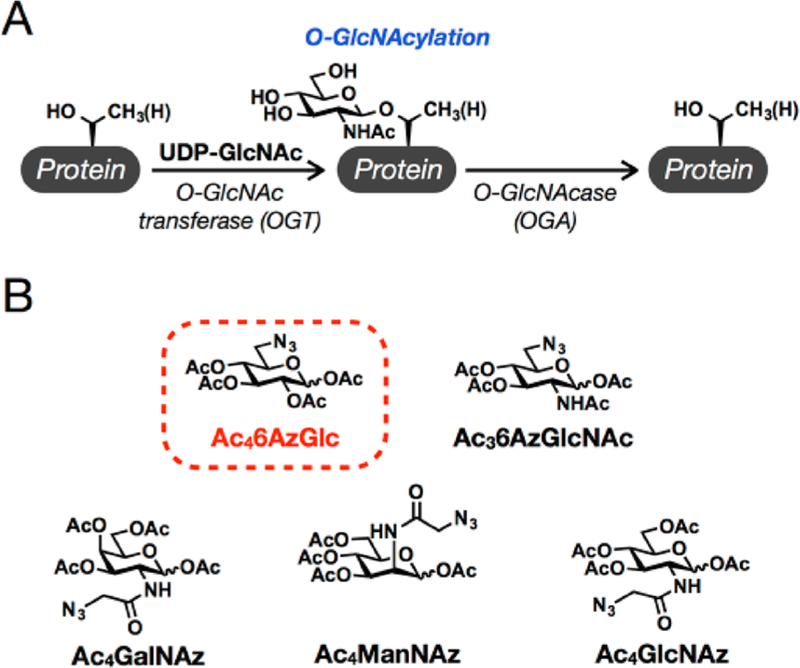
O-GlcNAc modification and metabolic chemical reporters (MCRs) of protein glycosylation. (A) O-GlcNAcylation is the dynamic addition of the monosaccharide N-acetyl-glucosamine to serine and threonine residues of intracellular proteins. (B) MCRs that are used in this study, including the new MCR described here, Ac46AzGlc, which is highlighted in red.
RESULTS AND DISCUSSION
The Reporter 6-Azido-6-deoxy-glucose Labels Proteins in a Range of Mammalian Cells.
Intrigued by the ability for carbohydrate analogues to label proteins in mammalian cells, we synthesized the acetylated glucose analogue 1,2 , 3,4-O-acetyl-6-azido-6-deoxy-glucose (Ac46AzGlc), and a variety of mammalian cells were then treated with this MCR (200 μM) for 16 h. The corresponding cell lysates were then subjected to CuAAC with an alkyne-TAMRA tag, and in-gel fluorescence scanning showed the labeling of proteins with varying intensities and patterns (Figure 2). Notably, the labeling pattern is different from the global protein-expression pattern as determined by Coomassie staining. To determine the concentration dependence of this labeling, NIH3T3 and H1299 cells were treated with a range of Ac46AzGlc concentrations for 16 h. Analysis by in-gel fluorescence shows a dose-dependence of labeling that is very similar to our other MCRs (Figure S1). Next, we examined the kinetics of protein labeling. More specifically, the same cell lines were treated with Ac46AzGlc (200 μM) for different lengths of time, followed by CuAAC and analysis by in-gel fluorescence (Figure S2). In both cell-lines, 6AzGlc-dependent labeling was detectable in as little as 2 h and peaked between 6 and 12 h. We then performed a pulse-chase experiment by first treating NIH3T3 and H1299 cells with Ac46AzGlc (200 μM) for 16 h. At this time, the media was exchanged for fresh media without MCR, and cells were collected after different lengths of time. CuAAC on the corresponding lysates and analysis by in-gel fluorescence showed fairly rapid loss of labeling, the majority of which occurs in the first 12−24 h (Figure S3).
Figure 2.

A variety of proteins in different cell-lines are labeled upon 6AzGlc treatment. The indicated cell-lines were treated with Ac46AzGlc (200 μM) or DMSO vehicle for 16 h. At this time, the corresponding cell lysates were subjected to CuAAC with alkyne-TAMRA and the labeled proteins were visualized using in-gel fluorescence scanning. The difference between low and high intensity is simply an adjustment of the fluorescence image and the data is representative of two biological replicates.
Comparison of 6AzGlc to Other Metabolic Chemical Reporters.
Next, we compared the labeling of Ac46AzGlc with other previously characterized MCRs in NIH3T3 cells. First, these cells were treated with either Ac46AzGlc, Ac36AzGlcNAc,12 Ac4GalNAz,22 Ac4ManNAz, 23 or Ac4GlcNAz24 (all at 200 μM concentration) for 16 h. The cells were then collected by either typsinization or EDTA treatment to retain the cell-surface proteins. The cell lysates were again subjected to CuAAC with alkyne-TAMRA followed by in-gel fluorescence (Figure 3A). Treatment with all five MCRs resulted in the visualization of a wide-range of proteins. Notably, the pattern of labeling by 6AzGlc was most similar to 6AzGlcNAc, which we previously characterized as a highly selective O-GlcNAc reporter, and was largely the same in both the trypsin and EDTA cell-collection conditions. These data suggest that 6AzGlc may also be selective for intracellular proteins over cell-surface glycoproteins. To examine this possibility further, NIH3T3 cells were again individually treated with the same five MCRs, followed by collection with EDTA treatment. Any cell-surface labeling was then detected by first reacting the intact cells with a biotin-tag using the strain-promoted azide−alkyne cycloaddition (SPAAC), followed by incubation with FITC-labeled streptavidin and analysis by flow-cytometry (Figure 3B). As expected based on previous results, treatment with Ac4GalNAz, Ac4ManNAz, or Ac4GlcNAz all resulted in substantial labeling of cell-surface glycoproteins, while the O-GlcNAcylation selective MCR 6AzGlcNAc showed very little signal over background. Notably, 6AzGlc treatment resulted in similarly low levels of signal, indicating that the vast majority of proteins labeled by this reporter are intracellular.
Figure 3.
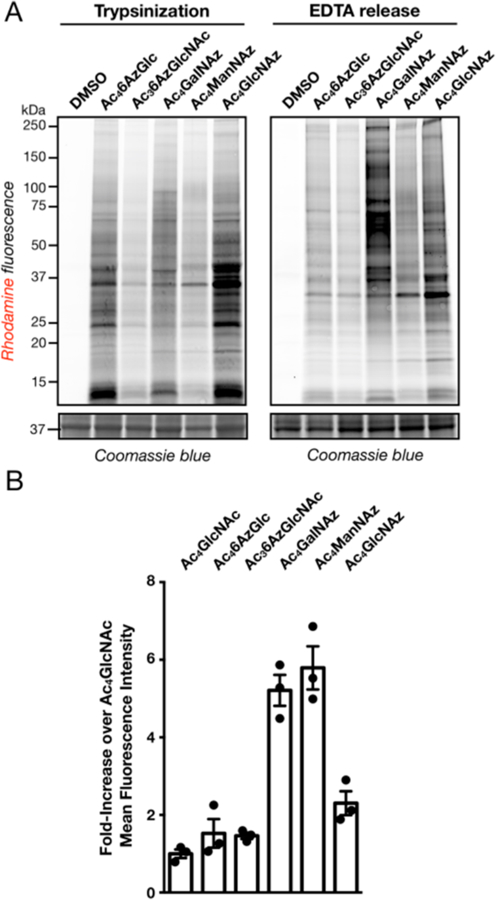
Majority of 6AzGlc-dependent labeling is intracellular. (A) Comparison of 6AzGlc to other azide-containing MCRs. NIH3T3 cells were incubated with one of the indicated MCRs (each at 200 μM) or DMSO vehicle for 16 h before CuAAC and in-gel florescence scanning. (B) 6AzGlc is largely excluded from cell-surface glycans. NIH3T3 cells were incubated in triplicate with the indicated MCRs (each at 200 μM) for 16 h. The cells were then harvested and reacted with DBCO-biotin. After incubation with FITC-streptavidin, cell-surface labeling was measured by flow cytometry. Error bars represent ± s.e.m. from the mean of biological replicates (n = 3). The data in all panels is representative of two biological replicates.
To further confirm this result, H1299 cells were treated with Ac46AzGlc (200 μM) for 16 h, and the corresponding lysates were subjected to CuAAC with a cleavable biotin tag, alkyne-azo-biotin. The modified proteins were enriched by incubation with streptavidin-coated beads, eluted, and subjected to separation by SDS-PAGE. Western blotting for the known O-GlcNAcylated proteins NEDD4, pyruvate kinase (PK), nucleoporin-62 (nup62) and nucleoporin-153 (nup153) showed enrichment upon 6AzGlc labeling, and the non-O-GlcNAcylated but abundant protein β-actin was not labeled by 6AzGlc, indicating that the MCR is not simply nonspecifically modifying intracellular proteins (Figure 4A). Finally, to determine whether the protein labeling by 6AzGlc is O-linked to proteins, we performed either β-elimination or enzymatic cleavage of N-linked glycans using PNGase-F. To perform β-elimination, H1299 cells were treated with Ac46AzGlc (200 μM) for 16 h, followed by CuAAC with alkyne-biotin, separation of SDS-PAGE and transfer to PVDF membranes. One set of membranes were treated under basic conditions (55 mM NaOH in H2O) for 24 h to induce β-elimination, and streptavidin blotting showed a notable loss in signal, albeit less than the loss of bona fide O-GlcNAc modifications detected by an anti-O-GlcNAc antibody (Figure 4B). Alternatively, cell lysates were treated with PNGase-F before CuAAC with alkyne-TAMRA and analysis by in-gel fluorescence (Figure 4C). We observed no loss of signal, despite loss of N-linked glycans as determined by Concanavalin A (ConA) blotting, demonstrating that the MCR is not incorporated into N-linked glycans. Together these data show that a notable fraction of the 6AzGlc labeling is O-linked in nature, but they do not rule out the possibility of some nonenzymatic or non-O-glycosylation modifications.
Figure 4.
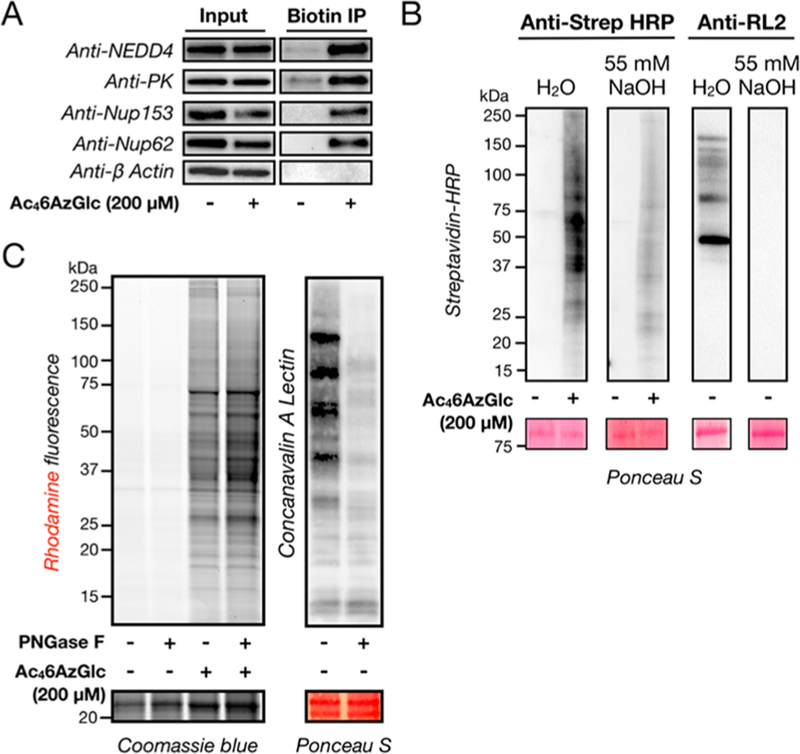
A significant fraction of 6AzGlc-dependent labeling is O-linked. (A) Known O-GlcNAcylated proteins are labeled by 6AzGlc. H1299 cells were treated with either Ac46AzGlc (200 μM) or DMSO for 16 h, followed by CuAAC with a cleavable alkyne-biotin tag. After enrichment on streptavidin beads, the labeled proteins were eluted and visualized by Western blotting. The nonglycosylated protein β-actin is a negative control. (B) A notable fraction of 6AzGlc-dependent signal is sensitive to β-elimination. NIH3T3 cells were treated with either Ac46AzGlc (200 μM) or DMSO vehicle for 16 h, followed by CuAAC with alkyne-biotin, separation by SDS-PAGE and transfer to a PVDF membrane. The indicated membranes were then treated for 24 h with either H2O or 55 mM NaOH before analysis by streptavidin or Western blotting. (C) 6AzGlc is not incorporated into N-linked glycans. NIH3T3 cells were treated with either Ac46AzGlc (200 μM) or DMSO vehicle for 16 h. The corresponding cell lysates were then incubated with either PNGase-F or H2O vehicle as indicated before CuAAC with alkyne TAMRA and analysis by in-gel fluorescence. A fraction of the treated lysate was separated before CuAAC and analyzed by Lectin blotting with Concanavalin A (ConA).
6AzGlc Can Be Transferred to Proteins by OGT but Not Removed by OGA.
The experiments above suggest that some fraction 6AzGlc is being metabolized to a donor sugar that is then transferred by OGT to substrate proteins. One possibility is that 6AzGlc is being transformed to the corresponding 2-acetamido sugar, 6AzGlcNAc, which can then be transformed to UDP-6AzGlcNAc and transferred by OGT. The rate determining enzyme that would be responsible for this transformation is glutamine fructose-6-phosphate aminotransferase (GFAT), which generates glucosamine-6-phosphate and glutamate from fructose-6-phosphate and glutamine. We used the CRISPR/Cas9 system to target GFAT1 in H1299 cells to generate clones that are null for the enzyme. Notably, the loss of GFAT in these cells can be rescued by treatment with exogenous GlcNAc to maintain glycosylation and the propagation of this cell line. Comparison of 6AzGlc labeling in the wild-type H1299 and a GFAT knockout cell line showed no reduction in protein labeling (Figure 5A), demonstrating that protein modification is not dependent on the enzymatic generation of 6AzGlcNAc from 6AzGlc. Notably, we removed GlcNAc from the media during 6AzGlc treatment to prevent any potential competition for labeling. These data suggest that 6AzGlc may be biosynthesized to UDP-6AzGlc and then transferred to proteins by OGT. To test this possibility, we first synthesized UDP-6AzGlc (Figure S4). Soluble protein lysates were prepared from H1299 cells by polyethylene glycol precipitation25 and then incubated with different combinations of recombinant OGT and UDP-6AzGlc for 3 h. These reactions were precipitated, resuspended, and subjected to reaction with phosphine-biotin before visualization by streptavidin blotting (Figure 5B). Signal was observed in the reactions containing both OGT and UDP-6AzGlc, demonstrating that OGT is able to transfer UDP-6AzGlc to mammalian proteins. Given this result, we then ask whether OGT is responsible for the modification of proteins in living cells. Accordingly, NIH3T3 or H1299 cells were treated with DMSO vehicle or the OGT inhibitor 5SGlcNAc26 (150 μM) for 24 h, which had the expected effect on overall O-GlcNAcylation levels (Figure S5A). At this time, fresh media containing Ac46AzGlc (200 μM) was added for 16 h. Analysis by in-gel fluorescence showed decreased labeling in the presence of 5SGlcNAc (Figure 5C), demonstrating that OGT inhibition reduces the modification of proteins by 6AzGlc. We feel that this incomplete loss of labeling could be due to three overlapping reasons. First, the antibody used to detect loss of endogenous O-GlcNAc, termed RL2, was actually raised against nucleoporin proteins and not O-GlcNAc27 and does not visualize all O-GlcNAcylated proteins.28 This raises the small possibility that the RL2-detectable O-GlcNAc modifications are more reversible and therefore susceptible to OGT inhibition. Second, the labeling of proteins by 6AzGlc is not reversed by OGA (see directly below and Figure 6A), potentially resulting in increased kinetics of O-GlcNAc modification removal compared to 6AzGlc after OGT inhibition. Finally, it is likely that some of the protein labeling by MCRs is “off target”. For example, once deacetylated the anomeric aldehyde of the reporter could nonenzymatically form schiff-bases or other glycation products. Alternatively, these reporters may be converted to other reactive metabolites. Chen, Wang, and co-workers also recently published data showing that high concentrations (2 mM) of acetylated sugars in cell lysate, or prolonged treatment times (200 μM for 48 h) in cells, can result in nonenzymatic modification of cysteine residues.29 However, the same authors previously demonstrated that under the standard labeling conditions used here (200 μM for 16 h) acetylated-MCR labeling is essentially all enzymatically mediated, suggesting that this issue may not be a widespread problem.30
Figure 5.
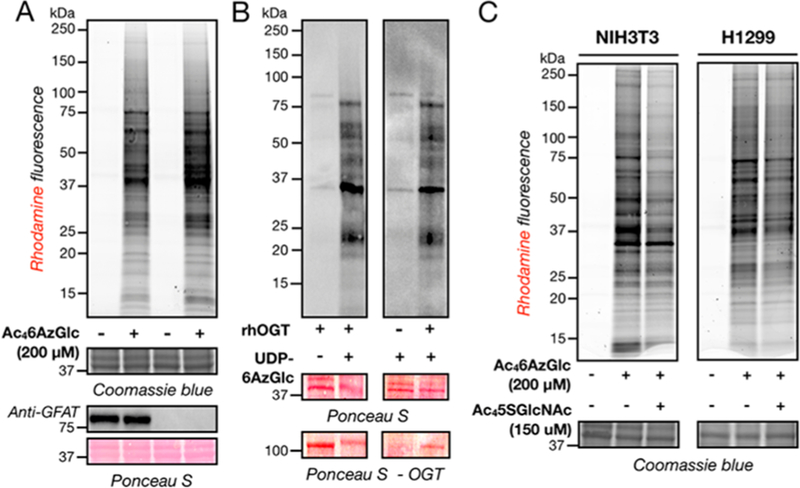
Some labeling of proteins upon 6AzGlc treatment is likely direct and O-GlcNAc transferase dependent. (A) Protein labeling is not dependent on the potential conversion of 6AzGlc to 6AzGlcNAc. Wild-type or GFAT-null H1299 cells were treated with Ac46AzGlc (200 μM) or DMSO vehicle for 16 h. They were then subjected to CuAAC and analysis by in-gel fluorescence. (B) O-GlcNAc transferase can directly transfer 6AzGlc to mammalian proteins. Recombinant O-GlcNAc transferase (rOGT) and/or synthetic UDP-6AzGlc were incubated with mammalian cell lysates for 3 h. The proteins were subjected to a precipitation/resuspension cycle and reacted with phosphine-biotin before visualization by streptavidin blotting. (C) Protein labeling is reduced by O-GlcNAc transferase inhibition. NIH3T3 or H1299 cells were first treated with either Ac45SGlcNAc (150 μM) or DMSO for 24 h. At this time, fresh media containing Ac46AzGlc (200 μM) was added for an additional 16 h before CuAAC and analysis by in-gel fluorescence scanning.
Figure 6.
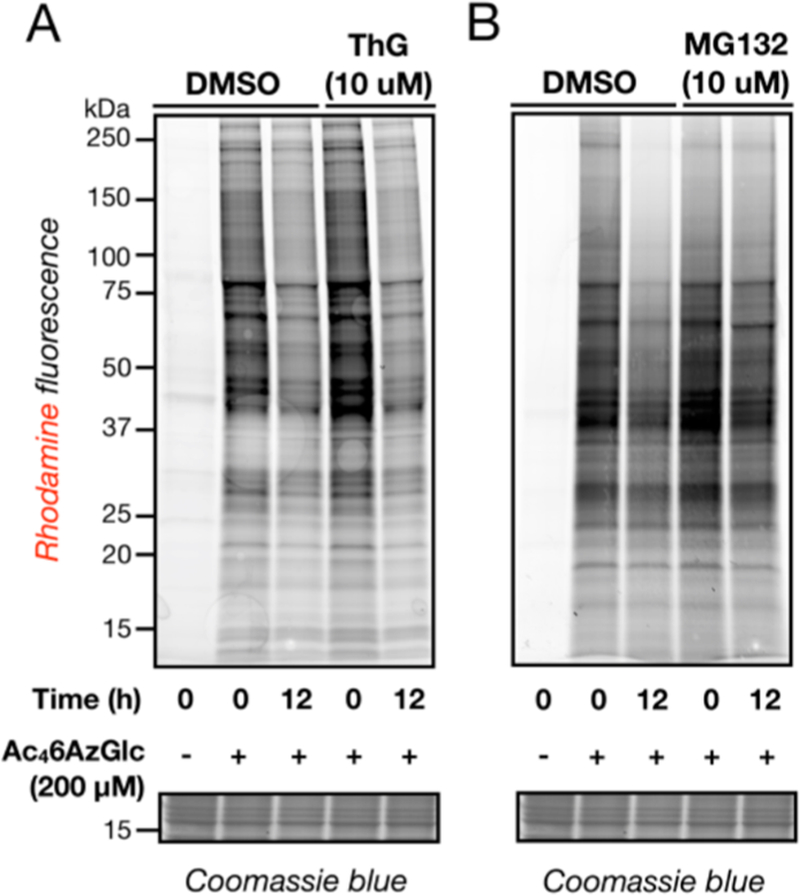
O-GlcNAcase does not remove 6AzGlc labeling. (A) H1299 cells were treated with 200 μM Ac46AzGlc for 5 h, at which time media was exchanged for fresh media containing 10 μM of the OGA inhibitor Thiamet-G or DMSO. Cells were harvested at the times indicated and subjected to CuAAC before being analyzed by in-gel fluorescence scanning. (B) Inhibition of the proteasome stabilizes some 6AzGlc signal. H1299 cells were treated with 200 μM Ac46AzGlc for 5 h, at which time media was exchanged for fresh media containing 10 μM of the proteasome inhibitor MG132 or DMSO. Cells were harvested at the times indicated and subjected to CuAAC before being analyzed by in-gel fluorescence scanning.
We then examined whether the 6AzGlc-dependent protein labeling was reversible by OGA by performing a pulse-chase experiment in the absence or presence of the selective OGA inhibitor Thiamet-G.31 More specifically, H1299 cells were first treated with Ac46AzGlc (200 μM) for 5 h. The media on these cells was then exchanged for fresh media containing either DMSO vehicle or Thiamet-G (10 μM), which again had the expected effect of raising O-GlcNAc levels (Figure S5B). After 12 h, the corresponding cell lysates were subjected to CuAAC with alkyne-TAMRA and analysis by in-gel fluorescence (Figure 6A). Treatment with Thiamet-G had no effect on the loss of 6AzGlc labeling, showing that OGA is not responsible for the removal of the modification from proteins. To determine if the reduction in labeling was due to protein turnover, we repeated this pulse-chase experiment in the presence of the proteasome inhibitor MG132 (10 μM). Analysis by in-gel fluorescence showed some stabilization in the signal upon MG132 treatment, demonstrating that the modified proteins are degraded. These data argue for a model where OGT can transfer glucose-analogues to protein substrates that then cannot be removed by OGA, which is consistent with the established anchimeric-assistance dependent mechanism of OGA. They also support the possibility that 6AzGlc-modified proteins are preferentially degraded by the proteasome, a possibility that we are currently exploring.
The Reporter 6AzGlc and Endogenous O-Glucose Can Be Directly Identified on Substrate Proteins.
Finally, we set out to directly determine if 6AzGlc modifications could be detected on proteins from living cells. To accomplish this, we took advantage of the IsoTaG system developed by the Bertozzi lab.19,20 Briefly, NIH3T3 cells were treated with Ac46AzGlc (200 μM) followed by lysis and CuAAC with a mixture of isotopically labeled, acid-cleavable affinity tags. After enrichment using streptavidin beads and on-bead trypsinolysis, the 6AzGlc-labeled peptides were eluted with weak acid to yield isotopically recoded glycopeptides. Analysis of these peptides by LC−MS was assisted by the IsoStamp v2.0 software for selection and validation of isotopically recoded glycopepties. Using IsoTaG, we were able to directly observe 8 different glycopeptides with O-linked site identifications by HCD and/or EThcD (Table S1), including a peptide corresponding to residues 50−92 of the protein 60S acidic ribosomal protein P1 (RPLP1, Figure S6) and residues 506−519 of the protein AHNAK (Figure S7), demonstrating that this modification can indeed directly occur in living cells. Importantly, both of these proteins have been previously identified as a potential O-GlcNAcylated proteins in previous proteomics experiments.21,32 Because of the concerns raised by the potential nonenzymatic labeling of cysteine residues,29 we also searched for this modification, and were able to find 5 peptides with a modification assigned to cysteine (Table S1). Coupled with the fact that ∼20 endogenous S-GlcNAc modifications have been identified by proteomics,33 we believe that this type of nonenzymatic modification is not the major contributor to the signal that we observe. This result prompted us to ask whether O-glucose could be identified as an endogenous modification in mammalian cells. On several substrates, O-GlcNAc occurs in clusters that are in close proximity on the primary sequence of the protein. Therefore, we probed our previously collected data sets of O-GlcNAcylated peptides that had been characterized using the IsoTaG methodology for the presence of an O-glucose residue by adding O-linked hexose as a variable modification. Exactly as we hypothesized, we identified a glycopeptide from primary human T cells corresponding to residues 612−637 of the host cell factor (HCF) protein, a highly O-GlcNAcylated substrate, upon treatment with an MCR that results in O-GlcNAz labeling.21,34 Specifically, both IsoTaG-modified GlcNAz and hexose were found on this peptide (Figures S8 and S9). Importantly, this endogenous O-glucose modification was identified using HCD and EthCD ionization, as well as visualization of the “double” neutral-ion loss of both carbohydrates by CID fragmentation. Together these results demonstrate that O-glucose is an endogenous modification of at least HCF in human cells.
CONCLUSION
Over the course of essentially two decades, the development of MCRs has contributed to the ability to identify and visualize a variety of glycoproteins. In our own studies on MCRs, we have also become interested in using these molecules to interrogate the promiscuity of cellular biosynthetic pathways and glycosyltransferases.12,13,35 Here, we characterize the cellular modification of proteins by the MCR 6AzGlc and find that treatment of several different mammalian cells with the per-O-acetylated analogue results in robust labeling (Figure 2A). Additionally, we show that the vast majority of this labeling occurs intracellularly (Figure 3) and much of it is O-linked to proteins (Figure 4 and Table S1). We also provide evidence that labeling is not dependent on the transformation of 6AzGlc to 6AzGlcNAc, as knocking out the enzyme responsible for the rate-determining step in this transformation, GFAT, does not result in a reduction of labeling (Figure 5A). We then show that OGT can transfer 6AzGlc to proteins and that inhibition of OGT results in reduced 6AzGlc labeling (Figure 5). This ability for OGT to accept UDP-6AzGlc as a substrate is fairly surprising given previous results that indicate that OGT is unable to transfer glucose from UDP-glucose to a peptide substrate in vitro,36 which was interpreted by the authors to show the key importance of the 2-acetamido functionality. However, a second enzymatic and structural study called into question the importance of the electronics of the 2-acetamide by showing that OGT is able to transfer the GlcNAcF3 (2-N-trifluoroacetamide).37 Additionally, OGT has been shown to display different kinetic properties when it is modifying full-length proteins when compared to peptide substrates,38 as it is in our recombinant experiments, and the Vocadlo lab demonstrated that OGT could transfer UDP-glucose to nucleoporin 62 but not some other O-GlcNAcylated proteins.17
We currently do not know the extent or functional significance of this modification in living cells or in vivo, but this is yet another demonstration of the interesting substrate promiscuity of OGT. We hope that our data encourage a more detailed kinetic analysis of recombinant OGT with UDP-glucose and purified protein substrates, as this will illuminate the concentrations of UDP-glucose that could potentially compete with UDP-GlcNAc in cells. It is also unclear whether glucose modification can substitute for O-GlcNAc in cells, as our GFAT knockout cells will not survive without GlcNAc supplementation. However, the loss of the biosynthesis of UDP-GlcNAc will also affect important cell-surface glycoproteins that could contribute to the cell-death phenotype. Interestingly, in our pulse-chase analysis with 6AzGlc labeling we found that the signal is lost relatively rapidly with kinetics that are similar to other reversible MCRs like GlcNAz and 6AzGlcNAc.12 OGA does not appear to be responsible for this removal (Figure 6A), which is consistent with its enzymatic mechanism that uses anchimeric assistance of the 2-acetamido group. Some of this signal loss is due to proteasomal degradation of the proteins (Figure 6B), but not all, suggesting that another unidentified enzyme may exist that can remove glucose-like modifications from proteins. Using these MCR-based discoveries as a catalyst, we then re-examined proteomics data in a new light and found an endogenous O-glucose modification on the protein host cell factor (HCF). Notably, this discovery was made in primary human T cells and not simply a cultured cell-line. This demonstrates that O-glucose is indeed an intracellular modification, which was previously only known as a modification of cell-surface proteins at specific serine residues within epidermal growth factor domains.39 The levels and physiological relevance of intracellular protein O-glucosylation remain to be defined, but this study has interesting implications for OGT as a sensor for the metabolism of multiple carbohydrate donor sugars in addition to UDP-GlcNAc. Importantly, this discovery was initiated through the use of an MCR, highlighting their utility for exploring cellular enzymology in addition to their well-established uses in glycoprotein analysis.
EXPERIMENTAL PROCEDURES
General Information.
All reagents used for chemical synthesis were purchased from Sigma-Aldrich or Alfa Aesar. All solvents were purchased from EMD Millipore. Reagents and solvents were used as received. All reaction were conducted under a nitrogen atmosphere using anhydrous solvents. Analytical thin-layer chromatography (TLC) was conducted on EMD Silica Gel 60 F254 plates and detected by ceric ammonium molybdate or UV. 60 A silica gel (EMD) for flash chromatography. 1H spectra were obtained at 400 MHz on a Varian spectrometer Mercury 400, and chemical shifts are recorded in ppm (δ) relative to solvent, and coupling constants (J) are reported in Hz.
Cell Culture.
H1299 were grown in RPMI media (Corning) supplemented with 10% Fetal Bovine Serum (Altanta Biologicals). NIH3T3 and MEF were grown in DMEM high glucose (Corning) supplemented with 10% Fetal Calf Serum (HyClone, Thermo Scientific). Cos7, Hela, HEK 293, MCF7 were grown in DMEM high glucose supplemented with 10% Fetal Bovine Serum. MCF7 were supplemented with 1× insulin (Thermo Scientific). GFAT knockout H1299 were grown in RPMI media supplement with 10% Fetal Bovine Serum containing 10 mM GlcNAc. All Cell lines were grown at 37 °C and 5.0% CO2.
Metabolic Labeling.
To cells at 80−85% confluency, media was exchanged for fresh media containing Ac46AzGlc, Ac36AzGlcNAc,12 Ac4GalNAz,19 Ac4ManNAz,20 Ac4GlcNAz,21 Thiamet-G,24 Ac45SGlcNAc.23 (1000× stock in DMSO), or DMSO vehicle was added as indicated. GFAT knockout H1299 were not supplemented with 10 mM GlcNAc during treatment.
Analysis by In-Gel Fluorescence.
Cells were collected by trypsinization or EDTA (1 mM in PBS) and pelleted by centrifugation for 4 min at 2000g, followed by washing 2× with PBS (1 mL). Cell pellets were then resuspended in 100 μL of 1% NP-40 lysis buffer [1% NP-40, 150 mM NaCl, 50 mM triethanolamine (TEA) pH 7.4] with Complete, Mini, EDTA-free Protease Inhibitor Cocktail Tablets (Roche) for 20 min and then centrifuged for 10 min at 10 000g at 4 °C. The supernatant (soluble cell lysate) was collected and the protein concentration was determined by BCA assay (Pierce, ThermoScientific). Protein concentration was normalized to 1 μg μL−1, and to 200 μg of protein, newly made click chemistry cocktail (12 μL) was added [Alkyne-TAMRA tag (Click Chemistry tools, 100 μM, 10 mM stock solution in DMSO); tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (1 mM, 50 mM freshly prepared stock solution in water); tris[(1-benzyl-1-H-1,2,3-triazol-4-yl)methyl]amine (TBTA) (100 μM, 10 mM stock solution in DMSO); CuSO4·5H2O (1 mM, 50 mM freshly prepared stock solution in water)]. The reaction was gently vortexed and allowed to sit at room temperature for 1 h. Upon completion, 1 mL of ice cold methanol was added to the reaction, and it was placed at −20 °C for 2 h to precipitate proteins. The reactions were then centrifuged at 10 000g for 10 min at 4 °C. The supernatant was removed, the pellet was allowed to air-dry for 15 min, and then 50 μL 4% SDS buffer (4% SDS, 150 mM NaCl, 50 mM TEA pH 7.4) was added to each sample. The mixture was sonicated in a bath sonicator to ensure complete dissolution, and 50 μL of 2× SDS-free loading buffer (20% glycerol, 0.2% bromophenol blue, 1.4% β-mercaptoethanol, pH 6.8) was then added. The samples were boiled for 5 min at 97 °C, and 40 μg of protein was then loaded per lane for SDS-PAGE separation. Following SDS-PAGE separation, gels were scanned on a Typhoon 9400 Variable Mode Imager (GE Healthcare) using a 532 nm for excitation and 30 nm bandpass filter centered at 610 nm for detection.
Flow Cytometry of Cell-Surface Labeling with DBCO-Biotin.
NIH3T3 cells grown in 6-well plates at 80−85% confluency were treated with 200 μM Ac46AzGlc, Ac46AzGlcNAc, Ac4GalNAz, Ac4ManNAz, Ac4GlcNAz or GlcNAc in triplicate for 16 h at which time media was removed and cells were gently washed with PBS before being detached from the plate with 1 mM EDTA in PBS. Cells were collected by centrifugation (5 min, 300g at 4 °C) and were washed three times with PBS (5 min, 300g at 4 °C). Cells were then resuspended in 200 μL PBS containing DBCO-biotin (Click Chemistry Tools, 60 μM) for 1 h, after which time they were washed three times with PBS (5 min, 300g at 4 °C) before being resuspended in ice-cold PBS containing fluorescein isothiocynate (FITC) conjugated avidin (Sigma, 5 μg μL−1, 30 min at 4 °C). Cells were then washed three times in PBS (5 min, 300g at 4 °C) before being resuspended in 400 μL PBS for flow-cytometry analysis. A total of 10 000 cells [dead cells were excluded by treatment with propidium iodide (2.5 μg mL−1 in water, 30 min)] were analyzed on a BD SORP LSRII Flow Cytometer using the 488 nm argon laser.
Biotin Enrichment.
H1299 cell-pellets labeled with Ac46AzGlc or DMSO for 16 h were resuspended in 52 μL H2O, and 100 μL of 0.05% SDS buffer (0.05% SDS, 10 mM TEA pH 7.4, 150 mM NaCl) with Complete Mini protease inhibitor cocktail (Roche Biosciences). To this was added 2 μL benzonase (Sigma), and the cells were incubated on ice for 30 min. Then, 4% SDS buffer (400 μL) was added, and the cells were briefly sonicated in a bath sonicator followed by centrifugation (10 000g for 10 min at 15 °C). Soluble protein concentration was normalized by BCA assay (Pierce, ThermoScientific) to 1 mg mL−1, and 2 mg of total protein was subjected to the appropriate amount of click chemistry cocktail containing Alkyne-Azo-Biotin (5 mM, Click Chemistry Tools) for 1 h, after which time 4 volumes of ice-cold MeOH were added. Precipitation proceeded 2 h at −20 °C. Precipitated proteins were centrifuged at 5200g for 30 min at 0 °C and washed 3 times with 10 mL of ice-cold MeOH, with resuspension of the pellet each time. The pellet was then air-dried for 15 min. To capture the biotinylated proteins by streptavidin beads, the air-dried protein pellet was resuspended in 1600 μL of resuspension buffer (6 M urea, 2 M thiourea, 10 mM HEPES pH 8.0) by bath sonication. Streptavidin beads (50 μL of a 50% slurry, Thermo) were washed 2× with 1 mL PBS and 1× with 1 mL of resuspension buffer and resuspended in resuspension buffer. Each sample was combined with streptavidin beads and incubated on a rotator for 2 h. Beads where then washed with 2× resuspension buffer (1 mL), 2× PBS (1 mL), 2× with 1% SDS in PBS (1 mL). Each sample was then incubated with 25 mM sodium hydrosulfite for 30 min, and the supernatant was collected, and repeated once more. The supernatant was pooled and 4× volume of MeOH was added. Precipitation proceeded for 2 h at −20 °C. Precipitated proteins were centrifuged at 10 000g for 10 min at 4 °C. The supernatant was removed, the pellet was allowed to air-dry for 15 min, and then 15 μL 4% SDS buffer (4% SDS, 150 mM NaCl, 50 mM TEA pH 7.4) was added to each sample. The mixture was sonicated in a bath sonicator to ensure complete dissolution, and 15 μL of 2× SDS-free loading buffer was then added. The samples were boiled for 5 min at 97 °C, and 30 μL of solution was then loaded per lane for SDS-PAGE separation. After proteins were separated by SDS-PAGE (Criterion TGX 4−20% Gel, Bio-Rad), proteins were transferred to a PVDF membrane (Bio-Rad) using manufacturer’s protocols. All Western blots were blocked in TBST (0.1% Tween-20, 150 mM NaCl, 10 mM Tris pH 8.0) containing 5% nonfat milk for 1 h at rt. The blots were then incubated with the appropriate primary antibody in blocking buffer for 1 h at rt. The anti-NEDD4 antibody (Millipore) was used at a 1:10 000 dilution, the anti-Pyruvate kinase antibody (Abcam) was used at 1:1000, anti-Nuclear pore “153” (Covance) was used at 1:1000 and anti-Nuclear pore 62 (BioLegend) was used at 1:5000. The blots were then washed three times in TBST for 10 min and incubated with the horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h in blocking buffer at rt. HRP-conjugated antimouse, antirabbit, and antigoat (Jackson ImmunoResearch) were used at 1:10 000 dilutions. After being washed three more times with TBST for 10 min, the blots were developed using ECL reagents (Bio-Rad) and the ChemiDoc XRS+ molecular imager (Bio-Rad).
β-Elimination.
Cells were collected by trypsinization and pelleted by centrifugation for 4 min at 2000g, followed by washing 2× with PBS (1 mL). Cell pellets were then resuspended in 100 μL of 1% NP-40 lysis buffer with Complete, Mini, EDTA-free Protease Inhibitor Cocktail Tablets (Roche) for 20 min and then centrifuged for 10 min at 10 000g at 4 °C. The supernatant (soluble cell lysate) was collected and the protein concentration was determined by BCA assay (Pierce, ThermoScientific). Either the protein concentration was normalized to 1 μg μL−1, and to 200 μg of protein, newly made click chemistry cocktail (12 μL) was added [Alkyne-biotin tag (Click Chemistry tools, 100 μM, 10 mM stock solution in DMSO); tris(2-carboxyethyl)-phosphine hydrochloride (TCEP) (1 mM, 50 mM freshly prepared stock solution in water); tris[(1-benzyl-1-H-1,2,3-triazol-4-yl)methyl]- amine (TBTA) (100 μM, 10 mM stock solution in DMSO); CuSO4· 5H2O (1 mM, 50 mM freshly prepared stock solution in water)] for streptavidin horseradish peroxidase (Strep-HRP), or 200 μg was diluted to 150 μg μL−1, and 50 μL of 4× loading buffer (200 mM Tris, 8% SDS, 40% glycerol, 0.4% bromophenol blue, 2.8% β-mercaptoethanol, pH 6.8) was added for RL2 analysis. The click reaction was gently vortexed and allowed to sit at room temperature for 1 h. Upon completion, 1 mL of ice cold methanol was added to the reaction, and it was placed at −20 °C for 2 h to precipitate proteins. The reactions were then centrifuged at 10 000g for 10 min at 4 °C. The supernatant was removed, the pellet was allowed to air-dry for 15 min, and then 100 μL 4% SDS buffer was added to each sample. The mixture was sonicated in a bath sonicator to ensure complete dissolution, and 100 μL of 2× SDS-free loading buffer was then added. The samples were boiled for 5 min at 97 °C, and 5 μg protein solution for Strep-HRP analysis and 15 μg of protein for RL2 analysis was then loaded per lane for SDS-PAGE separation. After proteins were separated by SDS-PAGE (Criterion TGX 4−20% Gel, Bio-Rad), proteins were transferred to a PVDF membrane (Bio-Rad) using manufacturer’s protocols. The blot was then washed once with TBS for 10 min, and then incubated with H2O or 55 mM NaOH for 24 h at 40 °C. The blots were then washed 3× with TBST, and then blocked with TBST containing 5% BSA for 16 h at rt. The blots were then incubated with the appropriate primary antibody in blocking buffer for 16 h at rt. The anti-RL2 antibody (Thermo Scientific) was used at a 1:1000 dilution, the Strep-HRP (Jackson Immuno Research Laboratories, Inc.) was used at 1:5000. The blots were then washed three times in TBST for 10 min. The Strep-HRP blots were then developed together using ECL reagents. The RL2 blots were incubated with the horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h in blocking buffer at rt. After being washed three more times with TBST for 10 min, the blots were developed together using ECL reagents.
PNGase F Treatment.
PNGase F (Glycerol-Free) was obtained from New England BioLabs. Cells were collected by trypsinization and pelleted by centrifugation for 4 min at 2000g, followed by washing 2× with PBS (1 mL). Cell pellets were then resuspended in “1× NP-40”, and “1× GlycoBuffer 2” supplemented with with Complete, Mini, EDTA-free Protease Inhibitor Cocktail Tablets for 20 min at 4 °C followed by tip sonication (35% amplitude, 5 s pulse duration, 5 s off, 3 times) while on ice, then centrifuged for 20 min at 10 000g at 4 °C. The supernatant (soluble cell lysate) was collected and the protein concentration was determined by BCA assay (Pierce, ThermoScientific). Protein concentration was normalized to 2 μg μL−1 using “1× NP-40”, and “1× GlycoBuffer 2”, and to 100 μg of protein 3 μL of PNGase F or H2O was added and incubated for 6 h at 37 °C. Afterward the PNGase F was quenched using equal volume of 4× SDS, and proteins were precipitated by adding a 3× volume of methanol, a 0.75× volume of chloroform, and 2× volume of H2O followed by vortexing and centrifugation (5 min, 5000g, rt). The aqueous phase was discarded without disturbing the interface layer before adding a 2.5 × volume of methanol, vortexing, and pelleting protein (10 min, 5000g, rt). The resulting protein pellet was allowed to air-dry for 5−10 min. Either the samples were suspended in 25 μL 4% SDS buffer and sonicated in a bath sonicator to ensure complete dissolution, and 25 μL of 2× SDS-free loading buffer was then added and used directly for lectin blots, or they were resuspended in 1% SDS (1% SDS, 10 mM TEA pH 7.4, 150 mM NaCl), to which 6 μL newly made click chemistry cocktail was added. The reaction was gently vortexed and allowed to sit at room temperature for 1 h. Upon completion, 500 μL of ice cold methanol was added to the reaction, and it was placed at −20 °C for 2 h to precipitate proteins. The reactions were then centrifuged at 10 000g for 10 min at 4 °C. The supernatant was removed, the pellet was allowed to air-dry for 15 min, and then resuspended as above. The samples were boiled for 5 min at 97 °C, and 20 μg protein solution for lectin analysis and 40 μg of protein for in-gel fluorescence analysis was then loaded per lane for SDS-PAGE separation. After proteins were separated by SDS-PAGE (Criterion TGX 4−20% Gel, Bio-Rad), the blot was either analyzed for fluorescence or proteins were transferred to a PVDF membrane (Bio-Rad) using manufacturer’s protocols. Then blot was then blocked with TBST containing 5% BSA for 1 h at rt. The blots washed three times in TBST for 5 min and incubated with biotin-conjugated Concanavalin A (Vector Lab), diluted 1:1000 in TBST, for 1 h. The blot was then washed 3× with TBST for 10 min. Then the blot was then incubated with Strep-HRP at 1:1000 in blocking buffer for 1 h. After being washed 3× with TBST for 10 min, the blot was developed using ECL reagents.
OGT Expression.
BL21(DE3) chemically competent E. coli (Novagen) were transformed with a pET24b plasmid encoding 6His-tagged ncOGT (nucleocytoplasmic OGT) by heat shock and plated on selective LB agar plates containing 50 μg mL−1 kanamycin (LB-kan). A single colony was then selected and use to inoculate 50 mL LB-kan for 16 h. Eight mL of culture was then used to inoculate 300 mL of Terrific broth (EMD). The culture was then grown to a OD600 of 0.8 by shaking at 250 rpm at 37 °C. Then culture was then cooled to room temperature and expression was induced with a final concentration of 0.5 mM IPTG and left at 20 °C shaking at 250 rpm for 20 h. Bacteria were harvested by centrifugation (6000g, 30 min, 4 °C). The cell pellets were lysed by resuspending the pellet in 10 mL of 100 mM Tris-HCl, 1 M NaCl, 1 mM Trition X-100, 5 mg/mL lysozyme, pH 7.5 with Complete, Mini, EDTA-free Protease Inhibitor Cocktail Tablets for 30 min, followed by tip sonication (35% amplitude, 10 s pulse duration, 30 s off, 3 times) while on ice. The crude cell lysate was cleared by centrifugation (42 000g, 30 min, 4 °C) and was transferred into a new tube and 2.5 mL of prewashed Ni-NTA Agarose (Qiagen) was added and placed onto a rotator for 1 h, 4 °C. The solution was then transferred to a gravity-flow columns and allowed to drain, and washed with an additional 100 mL of 25 mM Tris HCl, 0.5 M NaCl, 1 mM DTT, 20 mM imidazole, pH 7.5 and then eluted using elution buffer (wash buffer, 200 mM imidazole). Elution fractions were then concentrated using spin-column concentrators to 1 mL (Amicon Ultra 50 kDa MW cutoff, Millipore) and exchanged 4× to storage buffer (25 mM Tris-HCl, 1 mM EDTA, 150 mM NaCl, 1 mM TCEP, pH 7.5). After the final exchange the solution was concentrated to 2 mL and 500 μL of 50% glycerol was added.
UDP-6AzGlc rhOGT Transfer.
One 100 mm × 20 mm dish of H1299 cells were collected by trypsinization and pelleted by centrifugation for 4 min at 2000g, followed by washing 2× with PBS (1 mL). Cell pellets were then resuspended in 400 μL of reaction buffer (25 mM Tris-HCl, 1 mM EDTA, 150 mM NaCl, 12.5 mM MgCl2 2.5 mM TCEP, pH 7.5) with Complete, Mini, EDTA-free Protease Inhibitor Cocktail Tablets and lysed by tip sonication (35% amplitude, 5 s pulse duration, 5 s, 5 times) while on ice and then centrifuged for 10 min at 10 000g at 4 °C. The supernatant (soluble cell lysate) was collected and 800 μL of 30% PEG 8000 (in reaction buffer) was added, vortexed, and then centrifuged for 10 min at 10 000g at 4 °C. The precipitate was then resuspended in 300 μL of reaction buffer, centrifuged for 10 min at 10 000g at 4 °C. The supernatant (soluble cell lysate) was collected and the protein concentration was determined by BCA assay (Pierce, ThermoScientific). A typical transfer was conducted by mixing 100 μg protein, 1 μM ncOGT, 10 or 100 μM nucleotide sugar in 100 μL of reaction buffer. The mixture was then incubated for 3 h at 37 °C and the reaction was quenched with chloroform/methanol precipitation. The supernatant was removed and the pellet was allowed to air-dry for 15 min, and then 25 μL 4% SDS buffer was added to each sample. The mixture was sonicated in a bath sonicator to ensure complete dissolution, and then 73 μL of H2O was added, and 2 μL of 10 mM EZ-Link Phosphine-PEG3-Biotin (10 mM in DMSO, Thermo Scientific) was added and then incubated for 3 h at 37 °C and quenched with chloroform/ methanol precipitation. The pellet was allowed to air-dry for 15 min, and then 50 μL 4% SDS buffer was added to each sample. The mixture was sonicated in a bath sonicator to ensure complete dissolution, and 50 μL of 2× SDS-free loading buffer was then added. The samples were boiled for 5 min at 97 °C, and 10 or 5 μg of protein was then loaded per lane for SDS-PAGE separation. After proteins were separated by SDS-PAGE (Criterion TGX 4−20% Gel), proteins were transferred to a PVDF membrane (Bio-Rad) using manufacturer’s protocols. The blot was then blocked with TBST containing 5% BSA for 16 h at 4 °C, washed 3× with TBST. Then the blot was incubated with Strep-HRP (Jackson Immuno Research Laboratories, Inc.) in blocking buffer for 16 h at 4 °C. After being washed 3× with TBST for 10 min, the blot was developed using ECL reagents.
Calcium Phosphate Transfection.
One day before transfection, 5 × 105 target cells were plated. One hour before transfection, media was exchanged for fresh media. To an Eppendorf tube was added water (398 μL, autoclaved), 2 M CaCl2 62 μL (filter sterilized), and 40 μL DNA (500 ng/μL, 20 μg total, phenol/chloroform extracted, EtOH precipitated). Solution was mixed by pipetting, then DNA solution is added dropwise to a falcon tube containing 500 μL 2× HBS (500 mM HEPES, 1.5 mM Na2HPO4, 280 mM NaCl, 10 mM KCl, 12 mM dextrose, pH 7.05, filter sterilized) while pumping bubbles. The resulting solution was added dropwise to target cells while swirling slowly. A fine black precipitate was observed after 30 min and the media was opaque in color. Seven hours after transfection, media was exchanged for fresh media.
GFAT Knockout with CRISPR.
Expression vector pSpCas9(BB)-2A-Puro (PX459) (Addgene no. 48139) was digested with BbsI, and a pair of annealed oligonucleotides corresponding the first exon of human GFAT1 (5′-CACCGCTTCAGAGACTGGAGTACAG-3′ and 5′-AAACCTGTACTCCAGTCTCTGAAGC-3′), was ligated into the guide RNA to generate SpCas9-GFAT, which was confirmed by sequencing (Laragen, Culver City, CA). H1299 cells were then transiently transfected with SpCas9-GFAT1 using calcium phosphate transfection methods as described above. 48 h post-transfection, 1 μg mL−1 puromycin was added to the media for the next 72 h. Western blot analysis showed that the mixed population of cells had essentially no detectable GFAT expression. More specifically, after NP-40 lysis and BCA for analysis by in-gel fluorescence, 25 μL of 4× loading was added to 200 μg of proteins in 75 μL of NP-40 lysis buffer. The samples were boiled for 5 min at 97 °C, and 20 μg of protein was then loaded per lane for SDS-PAGE separation. After proteins were separated by SDS-PAGE (Criterion TGX 4−20% Gel), proteins were transferred to a PVDF membrane (Bio-Rad) using manufacturer’s protocols. The blot was then blocked with TBST containing 5% nonfat milk 16 h at 4 °C and then the blot was incubated with Anti-GFAT1 (IBL-America, 1:500) in blocking buffer for 16 h at 4 °C. The blot was then washed with 3× TBST for 10 min. The blot was then incubated with the horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h in blocking buffer at rt. Afterward it was washed 3× with TBST for 10 min, and the blot was developed using ECL reagents.
Direct Detection of 6AzGlc-Labeling in Living Cells.
NIH3T3 cell-pellets labeled with Ac46AzGlc for 16 h were resuspended in 52 μL H2O, and 100 μL of 0.05% SDS buffer with Complete Mini protease inhibitor cocktail (Roche Biosciences). To this was added 2 μL Benzonase (Sigma), and the cells were incubated on ice for 30 min. Then, 4% SDS buffer (400 μL) was added, and the cells were briefly sonicated in a bath sonicator followed by centrifugation (10 000g for 10 min at 15 °C). Soluble protein concentration was normalized by BCA assay (Pierce, ThermoScientific) to 1 mg mL−1, and 3 mg of total protein was subjected to the appropriate amount of click chemistry cocktail containing IsoTag V2 Saline Probe mixture (10 mM) for 1 h, after which time 4 volumes of ice-cold MeOH were added. Precipitation proceeded 2 h at −20 °C. Precipitated proteins were centrifuged at 5200g for 30 min at 0 °C and washed 3 times with 10 mL of ice-cold MeOH, with resuspension of the pellet each time. Reduction, alkylation, biotin enrichment, and elution were performed analogously to those previously described.20,21
Mass Spectrometry Procedures.
The desalted samples were resuspended in 0.1% formic acid in water (15 μL). The sample (4.0 μL) was loaded onto a C18 trap column (3 cm, 3 μm particle size C10 Dr. Maisch 150 μm I.D) and then separated on an analytical column (Thermo Scientific Acclaim PepMap 100, 2 μm particle size, 250 mm length, 75 μm internal diameter) at 150 nL/min with a Thermo Scientific EASY-nLC 1000 system connected in line to a Thermo Scientific Orbitrap Fusion Tribrid. The column temperature was maintained at 50 °C. The glycopeptides were separated via a stepwise gradient from 5% to 98% of 0.1% formic acid in acetonitrile over 90 min (0−1 min, 0−5%; 1−61 min, 5−28%; 61−80 min, 28−98%; 80−90 min, 98%−0%). Survey scans of peptide precursors were performed at 120 K fwhm resolution (m/z = 200). Tandem MS was performed on the most abundant precursors exhibiting a charge state from 2 to 5 at a resolving power setting of 15 K and fragmentation energy of 36 V. Tandem MS was performed on the most abundant precursors exhibiting a charge state from 2 to 5 at a resolving power settings of 15 K and an ionization energy of 36 V. HCD fragmentation was applied with 37% collision energy, CID fragmentation was applied with 35% collision energy, and EThcD applied with 50/40% collision energy, and resulting fragments detected using the normal scan rate in the ion trap.
Data Analysis Procedures.
The raw data was processed using Proteome Discoverer 2.2 (Thermo Fisher Scientific) and searched against the mouse-specific SwissProt-reviewed database downloaded on Jan 21, 2014. The data was searched using Byonic v1.0.362 as a node in Proteome Discoverer 2.2 for glycopeptide searches. Indexed databases for tryptic digests were created with full cleavage specificity at K and R. The database allowed up to three missed cleavages with variable modifications (methionine oxidation, +15.995 Da; carbamidomethylcysteine, +57.021 Da; and others as described below). Precursor ion mass tolerances for spectra acquired using the Orbitrap were set to 10 ppm. The fragment ion mass tolerance for spectra acquired using the Orbitrap were set to 20 ppm. The fragment ion mass tolerance for spectra acquired using the ion trap were set to 0.6 Da. Glycopeptide searches allowed for tagged O-glycan or C-glycan variable modifications (GlcSi02 on serine, threonine, and cysteine, +289.124). Glycopeptide spectral assignments passing a false discovery rate (FDR) of 1% at the spectrum level based on a target decoy database were manually validated for an isotope precursor pattern.
Identification of Endogenous O-Glucose.
Primary human T-cells treated with 40 μM Ac4GalNAz were enriched and analyzed by IsoTaG as previously described.21 When O-linked hexose (+162) was added as a variable modification, a glycopeptide from HCFC1 modified by both O-GlcNAz and an O-Hexose was confidently identified (Byonic, FDR < 1%).
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Y.H.L. for creation of the GFAT knock-out cell line. This research was supported by the National Science Foundation (CHE-1506503 to M.R.P.), the Damon Runyon Cancer Research Foundation (DDR-19–12 to M.R.P.), Susan G. Komen for the Cure (CCR14299333 to M.R.P.), the American Cancer Society (RSG-14–225-01-CCG to M.R.P.), the National Institutes of Health (R01GM125939 to M.R.P.), Burroughs Wellcome Fund CASI (to C.M.W.), Harvard University (to C.M.W.), and in part by the National Cancer Institute of the US National Institutes of Health (CCSG P30CA014089).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b13488.
Supporting figures, experimental methods, NMR characterization (PDF)
Proteomic data tables (XLSX)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Bond MR; Hanover JA J. Cell Biol 2015, 208, 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Yang X; Qian K Nat. Rev. Mol. Cell Biol 2017, 18, 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Vocadlo DJ Curr. Opin. Chem. Biol 2012, 16, 488. [DOI] [PubMed] [Google Scholar]
- (4).Shafi R; Iyer SP; Ellies LG; O’Donnell N; Marek KW; Chui D; Hart GW; Marth JD Proc. Natl. Acad. Sci. U. S. A 2000, 97, 5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).O’Donnell N; Zachara NE; Hart GW; Marth JD Mol. Cell. Biol 2004, 24, 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sinclair DAR; Syrzycka M; Macauley MS; Rastgardani T; Komljenovic I; Vocadlo DJ; Brock HW; Honda BM Proc. Natl. Acad. Sci. U. S. A 2009, 106, 13427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Banerjee PS; Hart GW; Cho JW Chem. Soc. Rev 2013, 42, 4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chuh KN; Pratt MR Curr. Opin. Chem. Biol 2015, 24, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chuh KN; Batt AR; Pratt MR Cell Chem. Biol 2016, 23, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zaro BW; Yang Y-Y; Hang HC; Pratt MR Proc. Natl. Acad. Sci. U. S. A 2011, 108, 8146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bateman LA; Zaro BW; Chuh KN; Pratt MR Chem. Commun 2013, 49, 4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chuh KN; Zaro BW; Piller F; Piller V; Pratt MR J. Am. Chem. Soc 2014, 136, 12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zaro BW; Batt AR; Chuh KN; Navarro MX; Pratt MR ACS Chem. Biol 2017, 12, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chuh KN; Batt AR; Zaro BW; Darabedian N; Marotta NP; Brennan CK; Amirhekmat A; Pratt MR J. Am. Chem. Soc 2017, 139, 7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chuh KN; Pratt MR Glycoconjugate J 2015, 32, 443. [DOI] [PubMed] [Google Scholar]
- (16).Mayer A; Gloster TM; Chou WK; Vocadlo DJ; Tanner ME Bioorg. Med. Chem. Lett 2011, 21, 1199. [DOI] [PubMed] [Google Scholar]
- (17).Shen DL; Liu T-W; Zandberg W; Clark T; Eskandari R; Alteen MG; Tan HY; Zhu Y; Cecioni S; Vocadlo D ACS Chem. Biol 2017, 12, 206. [DOI] [PubMed] [Google Scholar]
- (18).Watzele G; Tanner WJ Biol. Chem 1989, 264, 8753. [PubMed] [Google Scholar]
- (19).Woo CM; Iavarone AT; Spiciarich DR; Palaniappan KK; Bertozzi CR Nat. Methods 2015, 12, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Woo CM; Felix A; Byrd WE; Zuegel DK; Ishihara M; Azadi P; Iavarone AT; Pitteri SJ; Bertozzi CR J. Proteome Res 2017, 16, 1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Woo CM; Lund PJ; Huang AC; Davis MM; Bertozzi CR; Pitteri S Mol. Cell. Proteomics 2018, 17, 764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hang HC; Yu C; Kato DL; Bertozzi CR Proc. Natl. Acad. Sci. U. S. A 2003, 100, 14846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Saxon E; Bertozzi C Science 2000, 287 (5460), 2007. [DOI] [PubMed] [Google Scholar]
- (24).Vocadlo D; Hang H; Kim E; Hanover J; Bertozzi C Proc. Natl. Acad. Sci. U. S. A 2003, 100, 9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Griffin ME; Jensen EH; Mason DE; Jenkins CL; Stone SE; Peters EC; Hsieh-Wilson LC Mol. BioSyst 2016, 12, 1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Gloster TM; Zandberg WF; Heinonen JE; Shen DL; Deng L; Vocadlo DJ Nat. Chem. Biol 2011, 7, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Snow CM; Senior A; Gerace LJ Cell Biol 1987, 104, 1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Teo CF; Ingale S; Wolfert MA; Elsayed GA; Nöt LG; Chatham JC; Wells L; Boons G-J Nat. Chem. Biol 2010, 6, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Qin W; Qin K; Fan X; Peng L; Hong W; Zhu Y; Lv P; Du Y; Huang R; Han M; Cheng B; Liu Y; Zhou W; Wang C; Chen X Angew. Chem., Int. Ed 2018, 57, 1817. [DOI] [PubMed] [Google Scholar]
- (30).Qin W; Lv P; Fan X; Quan B; Zhu Y; Qin K; Chen Y; Wang C; Chen X Proc. Natl. Acad. Sci. U. S. A 2017, 114, E6749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Yuzwa SA; Macauley MS; Heinonen JE; Shan X; Dennis RJ; He Y; Whitworth GE; Stubbs KA; McEachern EJ; Davies GJ; Vocadlo DJ Nat. Chem. Biol 2008, 4, 483. [DOI] [PubMed] [Google Scholar]
- (32).Hahne H; Sobotzki N; Nyberg T; Helm D; Borodkin VS; van Aalten DMF; Agnew B; Kuster BJ Proteome Res 2013, 12, 927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Maynard JC; Burlingame AL; Medzihradszky KF Mol. Cell. Proteomics 2016, 15, 3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Boyce M; Carrico IS; Ganguli AS; Yu S-H; Hangauer MJ; Hubbard SC; Kohler JJ; Bertozzi CR Proc. Natl. Acad. Sci. U. S. A 2011, 108, 3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Zaro BW; Chuh KN; Pratt MR ACS Chem. Biol 2014, 9, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lazarus MB; Jiang J; Gloster TM; Zandberg WF; Whitworth GE; Vocadlo DJ; Walker S Nat. Chem. Biol 2012, 8, 966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Schimpl M; Zheng X; Borodkin VS; Blair DE; Ferenbach AT; ttelkopf AWSU; Navratilova I; Aristotelous T; Albarbarawi O; Robinson DA; Macnaughtan MA; van Aalten DM F. Nat. Chem. Biol 2012, 8, 969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Shen DL; Gloster TM; Yuzwa SA; Vocadlo DJ J. Biol. Chem 2012, 287, 15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Takeuchi H; Kantharia J; Sethi MK; Bakker H; Haltiwanger RS J. Biol. Chem 2012, 287, 33934. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.