

Abstract
Despite 25 years’ effort, serious questions remain regarding the mechanism(s) underlying electron transfer through (or from) electrode-bound double-stranded DNA. In part this is because a control experiment regarding the putatively critical role of guanine bases in the most widely proposed transport mechanism (hopping from guanine to guanine through the π-stack) appears to be lacking from the prior literature. In response, we have employed chronoamperometry, which allows for high-precision determination of electron transfer rates, to characterize transfer to a redox reporter appended onto electrode-bound DNA duplexes. Specifically, we have measured the effects of guanines and base mismatches on the electron transfer rate associated with such constructs. Upon doing so, we find that, counter to prior reports, the transfer rate is, to within relatively tight experimental confidence intervals, unaffected by either. Parallel studies of the dependence of the electron transfer rate on the length of the DNA suggest that transfer from this system obeys a “collision” mechanism in which the redox reporter physically contacts the electrode surface prior to the exchange of electrons.
Graphical Abstract
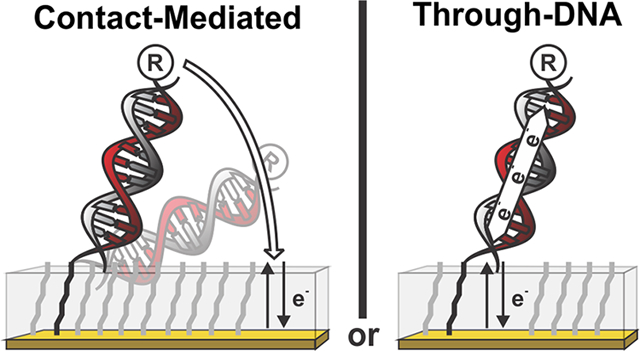
INTRODUCTION
Extensive scientific literature has described DNA duplexes as highly effient charge carriers, an effect claimed to arise from π−π interactions between their closely stacked base pairs, and has declared the effect potentially of great value in sensing, artificial photosynthesis, and molecular electronics (e.g., refs 1–11). Despite the large volume of this literature, however, the claim that charge transfer through DNA duplexes is rapid remains the subject of considerable debate, with some authors arguing in favor of rapid transfer1,2,12 and others claiming that DNA duplexes are effectively insulators.13–15
One of the most common approaches to measuring electron transfer from or through DNA duplexes, which was introduced by Barton and co-workers,16 employs thiol-modified DNA duplexes self-assembled onto gold electrodes and modified with redox-active reporters (e.g., Nile blue,12,17–21 methyleneblue,16,17,19,20,22,23 daunomycin,24–26 anthraquinone,8,19,23,27 pyrroloquinoline-quinone,25 Redmond Red,9 perylenediimide,28 TEMPO,27 or even whole bacteria29) that are either covalently attached to the DNA or intercalated within its base pairs. Using voltammetry to determine the electron transfer rates associated with these constructs, the community finds them to be on the order of ~ 1−100 s−1 (Table 1). There is also, we note, significant literature employing optical (refs 1, 30, and 31) and electronic (refs 32–34) means to monitor rates through DNA transfer or conductance; it is unclear, however, how to directly compare the results of these differing approaches.
Table 1.
A Survey of the Prior Experimental Literature Concerning Through-DNA Transfer As Measured via Electrochemical Interrogation of Surface-Attached DNAs
| ref | guanine-free controls | testing disruption of the π–π interaction via mismatches or abasic sites | length dependence studies | length of DNA sequences | packing density (pmol cm−2) | observed transfer rates | conclusions |
|---|---|---|---|---|---|---|---|
| 3 | no | no | no | 24 | not reported | not reported | through-DNA transfer |
| 5 | no | yes | no | 55 | 5.3 | 3–15 s−1 for methylene blue; 118–445 s−1 for ferrocene | through-DNA transfer |
| 7 | no | yes | no | 15 | not reported | 1–10 s−1 | through-DNA transfer |
| 8 | no | yes | no | 20 | not reported | not reported | through-DNA transfer |
| 9 | no | yes | no | 15, 21 | not reported | not reported | through-DNA transfer |
| 10 | no | yes | no | 20 | not reported | not reported | through-DNA transfer |
| 12 | no | yes | yes | 17 | 0.5–12 | 3–8 s−1 | through-DNA transfer |
| 13 | no | no | no | 20 | 1–2 | reported in cm s−1 | contact-mediated transfer |
| 14 | no | yes | no | 20 | 2–4 | reported in cm s−1 | contact-mediated transfer |
| 15 | no | no | qualitative | 17 | not reported | 4–21 s−1 | contact-mediated transfer |
| 16 | no | no | no | 15 | 41 | 60 s−1 | through-DNA transfer |
| 17 | no | yes | no | 16–60 | 40 | not reported | through-DNA transfer |
| 18 | no | yes | no | 17 | 1–3 | 1–3 s−1 | through-DNA transfer |
| 19 | no | yes | no | 17 | 3–5 | 2–4 s−1 | through-DNA transfer |
| 20 | no | yes | no | 17 | 0.8–3.2 | 0.6–40 s−1 | through-DNA transfer at higher packing density |
| 21 | no | yes | yes | 17, 100 | 1 | 25–40 s−1 | through-DNA transfer |
| 22 | no | no | no | 20 | 1.5–8.5 | not reported | through-DNA transfer |
| 23 | no | no | no | 20 | 1.1–5.8 | 1.3–3.5 s−1 | through-DNA transfer at higher packing density |
| 24 | no | no | no | 16 | 33–39 | extrapolated to 108–109 s−1 when no surface linker from measured rates of 4.4–733 s−1 | through-DNA transfer |
| 26 | no | yes | yes | 15 | 60–75 | ~100 s−1 | through-DNA transfer |
| 28 | no | yes | yes | 17 | not reported | 3–40 s−1 | through-DNA transfer |
| 29 | no | yes | no | 20 | not reported | not reported | through-DNA transfer |
| 35 | no | no | no | 15 | not reported | not reported | through-DNA transfer |
| 36 | no | no | no | 15 | 30–50 | not reported | through-DNA transfer |
| 37 | no | no | no | 15, 20 | not reported | “too large to be distinguished” | through-DNA transfer |
| 38 | no | yes | no | 15 | not reported | not reported | through-DNA transfer |
| 39 | no | yes | no | 14–16 | not reported | 70–500 s−1 | contact-mediated transfer |
| 40 | no | yes | no | 20 | 7.3 | not reported | through-DNA transfer |
| 41 | no | yes | no | 17 | 20 | 0.4–12 s−1 | through-DNA transfer |
| 42 | no | yes | no | 20 | 75–350 | reported in cm s−1 | through-DNA transfer |
| 43 | no | yes | no | 20 | 1–7 | 59–450 s−1 | through-DNA transfer |
| 44 | no | no | no | 20 | 3–5 | reported in cm s−1 | contact-mediated transfer |
| 45 | no | yes | no | 20 | 0.9–4.1 | 88–259 s−1 | through-DNA transfer |
| 46 | no | no | no | 20–33 | 1–13.8 | reported in cm s−1 | contact-mediated transfer |
| 47 | no | no | no | 20 | 2–2.5 | not reported | contact-mediated transfer |
| this work | yes | yes | yes | 13–50 | 1–12 | 1.6–4.8 s−1 | contact-mediated transfer |
The community claiming effcient through-DNA transfer has largely argued that charges move via “hops” through an intact π-stack from one guanine to the next (see refs 1, 12, 28), with this base playing a critical role due to its low reduction potential (which is below that of the other three bases48). Support regarding the necessity of an intact π-stack comes from studies reporting significantly slower through-DNA transfers upon introduction of mismatches or abasic sites that are thought to disrupt this stacking.12,16,21,26 Although the presence of guanines is central to the hypothesized rapid transfer, there is an unexpected lack of control experiments in the relevant literature testing the putatively critical role of this base (see Table 1). A second concern regarding much of the prior literature is its often untested assumption that all of the electrode-bound DNA molecules are in their double-stranded state (e.g., refs 12, 19–22, 36, 41); because the transfer rates associated with single-stranded DNA are as much as an order of magnitude greater than those of double-stranded DNA,20,21,49,50 the unnoticed presence of incomplete hybridization would cause an overestimation of the transfer rates of the double-stranded state. In an effort to fill these gaps, here we have used a combination of chronoamperometry and chronocoulometry to precisely quantify electron transfer rates from guanine-free, guanine-rich, and mismatch-containing constructs that we confirmed experimentally to be fully hybridized (Figure 1). Following this, we then used the approach to characterize the electron transfer kinetics of duplex constructs as a function of their length, a powerful means of discriminating between competing transfer models.
Figure 1.

To investigate the electron transfer kinetics of DNA duplexes, we designed redox-reporter-modified guanine-free, guanine-rich, and mismatch-containing constructs attached via one end to a mercaptohexanol monolayer on a gold electrode. As the redox reporter (labeled “R”), we use methylene blue, which is commonly used to monitor through-DNA electron transfer.16,17,19,20,22,35,36,43,51 To measure the rate of transfer fromsuch constructs, we use chronoamperometry, an approach that determines electron transfer kinetics with improved precision than can generally be achieved by the previously employed voltammetric techniques.52 Shown, for example, are current decays measured for a 16-base DNA sequence in its single- and double-stranded forms. Fitting these to a single-exponential decay returns kapp = 150 ± 3 s−1 for the single-stranded constructcontaining only adenine and thymine and kapp = 2.8 ± 0.9 s−1 when its complement is added (at 100 nM) and the system is allowed to hybridize. (The “error bar” intervals quoted for these numbers as well as those reported or illustrated elsewhere in this paper reflect 95% confidence intervals estimated from replicate measurements conducted on 12 independently fabricated electrodes.)
RESULTS AND DISCUSSION
Cyclic voltammetry has historically been employed to determine transfer rates in DNA duplexes.14,21,24 This approach relies on Laviron’s analysis, which only indirectly estimates rates from the peak separation observed in voltammograms.53 It appears, however, that for DNA duplexes the precision of this approach is relatively poor; for example, rates of 25 and 39 s−1 are deemed experimentally indistinguishable,21 and it is claimed that determined rates “can only be assessed with confidence as order of magnitude estimates”.19 Chronoamperometry, in contrast, enables the direct determination of transfer rates, greatly improving on the precision with which they can be measured (Figure 1). Thusly motivated, we have used this approach to measure electron transfer from a methylene blue at the distal terminus of DNA duplexes. These are attached via a thiol anchor on 6- mercaptohexanol self-assembled monolayers and deposited on a gold electrode, a strategy similar to that employed in many previous studies of through-DNA charge transfer (e.g., refs 1, 8, 12, 54).
To measure transfer rates, chronoamperometry employs a dual potential pulse in which we first apply a reductive potential and monitor the current decay resulting from the reduction of methylene blue [Figures 1 and S1A (Supporting Information, SI)]. This is followed by the application of an oxidative potential that regenerates the redox reporter (Figure S1A, SI). Determining the apparent electron transfer rate, kapp, of this population can simply be achieved by fitting the current decay trace to an exponential function.50,52 We use the “apparent” nomenclature to distinguish this rate from the true, equilibrium electron transfer rate, k0, since the measured rate combines this with the rate of diffusion of the redox reporter to the surface (i.e., mass transport).
Because the transfer rate of the single-stranded state is faster than that of the double-stranded state (50 fold more rapid for a 16-base construct; Figure 1B), surface contamination with unhybridized single-stranded DNA leads to overestimation of the transfer rate of the double-stranded state. Fortunately, however, in addition to providing rate information, chronoamperometry also provides information regarding the relative populations of double-stranded and single-stranded molecules, allowing us to quantitatively assess the extent of hybridization (Figure 2A). Specifically, we can determine the relative populations of the two states by examining the chronoamperograms in their chronocoulommogram form (i.e., as a plot of transferred charge as a function of inverse time), which reports the number of redox reporters that have a transfer rate corresponding to the maximum of the trace (Figure 2A). Using this to characterize our system, we find that attaching single-stranded DNA to the electrode and then adding its complement in solution (at 100 nM) leads to effectively complete hybridization within 5 min, as reported by the disappearance of the more rapidly transferring population (Figure 2A). In contrast, the commonly employed approach21,41,43 of hybridizing the DNA prior to surface deposition produces a large population of single-stranded molecules, leading, in this case, to a 40-fold overestimation of the transfer rate of the double-stranded state [Figures 2B and S2 (SI)].
Figure 2.

We have used chronoamperometry to monitor the electron transfer rates associated with surface-attached, redox-reporter-modified DNAs. (A) To ensure that the surface-attached DNA is fully in its hybridized, double-stranded state, we record chronoamperograms every second after the addition of 100 nM of the complementary strand. A plot of the resulting chronoamperograms in their “chronocoulommogram” format (charge transferred versus 1/time) clearly illustrates the evolving relative populations of single- and double-stranded molecules. Specifically, the overlaid chronocoulommograms exhibit an “isosbestic” point (point at which all the curves cross), indicating that the system only populates two states: single-stranded DNA transitions into fully double-stranded DNA without ever significantly populating any partially hybridized states. (B) If, instead, we hybridize the DNA prior to attaching it to the surface,21,41,43 we find that the transfer rate we observe (112 ± 30 s−1) is intermediate between those of the single-stranded (150 ± 3 s−1) and fully duplex (2.8 ± 0.9 s−1) states (see chronocoulommograms in Figure S2, SI), suggesting that single-stranded DNA is formed during the deposition process.55,56 Shown are data for a 16-base construct lacking guanine bases at a packing density of 2.9 pmol cm−2.
A central argument in the prior through-DNA-transfer literature is that rapid charge transport requires both thepresence of guanines and the integrity of π-stacking.1 As we note above, however, our exploration of the literature suggests that at least the former argument has never been tested (Table 1). To do so we have compared the rate of transfer from two double-stranded, 40-base-pair constructs, one of which is comprised entirely of A−T base pairs and a second comprised of 24 G−C base pairs and 16 A−T base pairs, with no more than three bases in a row excluding guanine (constructs 7 and 8 in Table 2). Doing so we find that, in contrast to theoretical and experimental expectations that guanine-containing DNA duplexes should transfer electrons more rapidly,2,48,57 the ratesassociated with these two constructs are effectively indistinguishable (2.8 ± 0.9 s−1 and 2.5 ± 0.5 s−1, respectively; Figure 3) under the conditions that we have employed. To test the second argument, we have introduced a C−A mismatch into each of these same two constructs. Again in contrast toprevious reports,8,20,25,38,54 we find that the introduction of this mismatch leaves the electron transfer kinetics of the two constructs effectively unchanged (2.6 ± 0.8 and 3.2 ± 0.5 s−1, respectively; Figure 3).
Table 2.
Anchor and Corresponding Complement Nucleotide Sequences Employeda
| construct label | 5′-termini | DNA sequence | 3′-termini |
|---|---|---|---|
| 1 | HS | ATTAT TTTTT ATT | MB |
| 1c | AAT AAAAA ATAAT | ||
| 2 | HS | ATTAT TTTTT ATTTA T | MB |
| 2c | A TAAAT AAAAA ATAAT | ||
| 3 | HS | ATTAT TTTTT ATTTA TTTTT | MB |
| 3c | AAAAA TAAAT AAAAA ATAAT | ||
| 4 | HS | ATTAT TTTTT ATTTA TTTTT ATTTT A | MB |
| 4c | TAAAA TAAAA ATAAA TAAAA AATAA T | ||
| 5 | HS | ATTAT TTTTT ATTTA TTTTT ATTTT ATTTT | MB |
| 5c | AAAAT AAAAT AAAAA TAAAT AAAAA ATAAT | ||
| 6 | HS | ATTAT TTTTT ATTTA TTTTT ATTTT ATTTT ATTTT TT | MB |
| 6c | AAAAA ATAAA ATAAA ATAAA AATAA ATAAA AAATA AT | ||
| 7 | HS | ATTAT TTTTT ATTTA TTTTT ATTTT ATTTT ATTTT TTATT | MB |
| 7c | AATAA AAAAT AAAAT AAAAT AAAAA TAAAT AAAAA ATAAT | ||
| 7m | AATAA AAAAT AAAAT AAAAT AAAAA TAAAC AAAAA ATAAT | ||
| 8 | HS | ATGGC TTGGC ATGGC TTGGC ATGGC ATGGC ATGGC TTGGC | MB |
| 8c | GCCAA GCCAT GCCAT GCCAT GCCAA GCCAT GCCAA GCCAT | ||
| 8m | GCCAA GCCAT GCCAT GCCAT GCCAA GCCAC GCCAA GCCAT | ||
| 9 | HS | ATTAT TTTTT ATTTA TTTTT ATTTT ATTTT ATTTT TTATT TATTT TTATT | MB |
| 9c | AATAA AAATA AATAA AAAAT AAAAT AAAAT AAAAA TAAAT AAAAA ATAAT |
HS represents a SH(CH2)6 hexanethiol chain that was used to anchor the chain on a 6-mercapto-1-hexanol monolayer, and MB represents a covalently attached methylene blue at the phosphate backbone terminus. The synthesis and modifications of all constructs were performed using standard phosphoramidite solid-phase chemistry by BioSearch Technologies Inc. and constructs were dual-HPLC purified.
Figure 3.

Rate of electron transfer associated with duplex DNA is not a significant function of its sequence composition, presence or absence of a mismatch, or even its relative surface coverage on the electrodes (i.e., packing density), a result that holds at both (A) lower and (B) higher packing densities. For example, the rate associated with a fully complementary, 40-base-pair guanine-free construct is effectively indistinguishable from that of a construct in which we have replaced more than half of the A−T base pairs by G−C base pairs. The introduction of a C−A mismatch (at base pair 11) in either construct likewise does not measurably alter the transfer rate. The “error bars” shown again reflect 95% confidence intervals estimated from replicate measurements conducted on 12 independently fabricated electrodes.
In the work described above, we employed packing densities (number of DNA strands per unit area) of 1−3 pmol cm−2, arange that overlaps well with the packing densities employed in half of the 18 previous papers that characterized this parameter (Table 1). Some authors, however, have argued that the through-DNA transfer occurs only at higher packing densities (refs 20, 23, 43, 45, but see, also, ref 5, which reports no dependence on packing density). To explore this, we measured the transfer kinetics associated with our various constructs when they are deposited at densities of 9−12 pmol cm−2. We find, however, that even under these conditions all four of our constructs (guanine-rich and guanine-free, perfectly matched and mismatched) again exhibit identical transfer rates (Figure 3).
The length dependence of transfer kinetics can help delineate between possible charge transfer mechanisms. Guanine hopping, for example, is predicted to exhibit a power law dependence on DNA length with an exponent of between −1 and −2 (refs 12, 18). The length dependence of tunneling-mediated transfer, in contrast, is predicted to be exponential.12 Given the potential utility of such studies in elucidating the transfer mechanism, we were surprised to find only limited discussion of the length dependence of DNA-associated transfer in the prior literature (Table 1). Specifically, we find only five earlier studies exploring this effect with only modest precision. In one, Kraatz and co-workers15 usedscanning electrochemical microscopy to determine that systems with a reporter located at the terminus of a 17-base-pair construct “exhibit faster electron transfer” than systems in which the reporter is placed closer to the electrode, but their analysis does not go beyond this qualitative statement. In a second, Barton and co-workers26 used cyclic voltammetry to measure transfer rates using daunomycin placed at positions 1, 3, 5, and 12 of five different 15-base-pair constructs. They report, however, only the semiqualitative result that all five constructs transfer with rates of ~100 s−1, arguing that “difference in the rates (…) of only one order of magnitude (…) may not be detectable by our analysis.” In a third, Barton and co-workers21 used cyclic voltammetry to measure transfer rates from the Nile blue placed on the termini of constructs of either 100 or 17 base pairs. They report that the rates they observe (39 and 25 s−1, respectively) are “indistinguishable”, but as they do not provide any error analysis, it is diffcult to ascertain the confidence that can be assigned to this claim. In two follow up papers Slinker and co-workers12,28 used cyclic voltammetryto determine the length dependence of transfer from sets of 17-base-pair constructs with either Nile blue or perylenediimide placed at position 4, 9, 13, or 17. However, the rates reported for the first three constructs in the first set and all four constructs in the second set are within experimental error of one another; thus, these data provide little evidence in favor of any length dependence. In short, the few prior reports on the length-dependence of electron transfer from electrode-bound DNA provide no significant evidence in support of (or against) either the hopping or tunneling mechanisms.
In response to the above observations, we used chronoamperometry to measure the length-dependence of electron transfer from a set of eight fully double-stranded constructs ranging in length from 13 (the shortest construct estimated to be stable under our experimental conditions60) to 50 basepairs. Upon doing so, we observe rates varying from 1.6 ± 0.4 to 4.8 ± 1.6 s−1 (Figure 4). To our surprise, these rates do not depend monotonically on length but instead rise and fall. Looking at this in more detail, we find that the rates are reasonably well described by a sine curve with an unconstrained “best fit” period of 11.3 ± 0.7 base pairs, a value corresponding (within error) to the number of base pairs per turn in A−T-rich DNA.58 Given this, we believe that higher rates are seen when the periodicity of the helix positions the reporter on the same side of the double helix as the linker, thus allowing the reporter to approach the electrode surface more closely (Figure 4), suggesting in turn that transfer is driven by direct contact between the redox reporter and the surface and is not mediated by through-DNA transport. That is, we believe transfer requires direct contact with the surface, and this contact becomes “easier” every 11.3 base pairs, presumably because the DNA duplexes are either lying flat on or otherwise transit to the surface due to rapid structural dynamics.
Figure 4.

Studies of the length dependence of transfer from guanine-free duplexes suggest that electron transfer is influenced by the position of the redox reporter with respect to the electrode surface. Specifically, the length dependence of transfer from a series of A−T duplexes fits a sinusoidal function with a period of 11.3 ± 0.7 base pairs (this is the best-fit value using an unconstrained value for the period of the sine wave). Perhaps tellingly, this periodicity is within error of the number of base pairs per turn for A−T-rich duplexes.58 From this we hypothesized that the surface-attached DNA duplexes are either lying flat or transiently “lean over” and collide with the electrode surface, and thus, the orientation of the redox reporter relative to the face of the DNA on which the flexible surface attachment point is located defines the transfer rate. This implies that the transfer rates measured electrochemically in systems such as the ones we (and others16,21,26,36) have employed originate from an alternate mechanism in which electrons tunnel to the redox reporter only when structural dynamics in the DNA bring the reporter in close proximity to the electrode surface.59
The electron transfer we describe here from a redox-reporter-modified, double-stranded DNA appears to occur via contact-mediated exchange between the reporter and the surface and does not involve significant through-DNA charge transport. Our arguments to this end are 3-fold. First, the presence or absence of guanine, which are thought necessary to support rapid, hopping-based through-DNA transfer,1 does not significantly alter the transfer rate. Second, the presence of a mismatch, which is argued to disrupt the integrity of the π−π stacking necessary to support rapid through-DNA transfer,21,26likewise does not significantly alter the transfer rate. Finally, the transfer rate exhibits the sine-wave dependence on length expected for contact-mediated transfer rather than the power-law relationship expected for guanine-mediated through-DNA transport or the exponential distance dependence expected for through-DNA tunneling. Thus, while we obviously cannot speak to all possible experimental conditions, effcient through-DNA electron transfer does not appear to occur under the (commonly employed)10,22,23,28 conditions we have explored here.
MATERIALS AND METHODS
Materials and Instruments.
Potassium chloride, potassium phosphate dibasic, sodium chloride, sodium phosphate monobasic, and sodium hydroxide were acquired from Fischer Scientific. 6-Mercapto-1-hexanol, phosphate-buffered saline (1× PBS, pH 7.4), and tris(2-carboxyethyl)phosphine hydrochloride (TCEP) were obtained from Sigma-Aldrich. Sulfuric acid was obtained from EMD, and fritted Ag|AgCl electrodes and platinum wire were from CH Instruments. Insulated pure gold (75 μm diameter, 64 μm insulation thickness) was purchased from A-M Systems. To fabricate working electrodes, segments of gold wire were insulated using a heat-shrink polytetrafluoroethylene insulation (PTFE, HS Sub-Lite-Wall, 0.02 in, black-opaque, Lot #17747112–3) from ZEUS and soldered to gold pins purchased from CH Instruments. All of the above materials and reagents were used as received. In all electrochemical measurements, a three-electrode setup was used consisting of Ag|AgCl reference, platinum counter, and gold working electrodes, with a GAMRY Reference 600+ potentiostat/galvanostat/ZRA.
Gold Electrode Fabrication and Electrochemical Cleaning.
Segments of gold wire were cut (7 cm in length) and stripped of their insulating coatings at both ends (1 cm in length). The insulated body of the wires was coated using two layers of heat-shrink PTFE tubing. To facilitate connection with the potentiostat, a gold pin was soldered to one end of the electrode and this contact further coated with insulating connector paint (MG Chemicals). Finally, the uninsulated end of the electrodes was cut to a final length of 3 mm prior to electrochemical cleaning with the following protocol: (1) 300 cycles between −1 and −1.6 V in a solution of 0.5 M NaOH at 1 V s−1 toremove any residual thiol/organic contaminants on the electrode surface and (2) pulsed between 0 and 2 V for 16 000 cycles with a pulse length of 0.02 s in 0.5 M H2SO4 to increase the electrode roughness, which can be easily observed through a darkened electrode color, as previously reported.61
Functionalization of Gold Electrodes.
The stock of the thiolmodified strands (1 μL of 100 μM) was reduced using 10 mM TCEP for 1 h at room temperature. The concentration of the reduced DNA was adjusted to 200 nM (for lower packing densities) or 3 μM (for higher packing densities) based on its adsorption at 260 nm. To functionalize the electrodes, the freshly cleaned gold electrodes were immersed into such a DNA solution for 1 h at room temperature. The electrodes were then rinsed with excess 1× PBS (137 mM sodium chloride, 10 mM hydrogen phosphate, and 1.76 mM potassium chloride; conditions under which the estimated60 melting temperature of the shortest construct is ~ 34 °C) prior to their immersion in a 5 mM 6-mercapto-1-hexanethiol solution for overnight incubation.
Chronoamperometry.
The freshly modified electrodes were rinsed and moved into a 1× PBS buffer solution. O2 was removed from the cell using a constant stream of argon and added the complementary strand to a final concentration of 100 nM. To determine the potential steps to be used in the experiment, a cyclic voltammogram was recorded between 0 and −0.4 V at a 100 mV s−1scanning rate. Using this voltammogram, a reductive potential (E1) at the negative tail end of the methylene blue reduction feature and an oxidative potential (E2) at its positive tail end (Figure S3, SI) were selected. The pulse width was adjusted to 100 μs to resolve the entire current decay trace obtained after applying the reductive step. Because the resulting chronoamperogram covered a large range of currents (nanoampere to milliampere), the electrodes were serially interrogated with the same potential pulse program but with varying sensitivity limits. To improve signal-to-noise ratios, these measurements were repeated 300 times for each electrode, and signal averaging was performed. To record time courses and study equilibration times for complete hybridization, the electron transfer rates of the current decay were monitored in real time. For this, data acquisition was started, and after collecting a baseline of 100 chronoamperograms, the corresponding DNA complement was added at a concentration of 100 nM (Figure 2A). The real-time data acquisition and calculation of the electron transfer rates were performed by employing a previously published custom-made MATLAB script.50,52 This script extracts electron transfer rates via least-squares fitting of the chronoamperograms to a single-exponential decay.
The packing densities of our constructs were calculated by integrating the area under the curve of the cyclic voltammogram corresponding to the reduction reaction of methylene blue.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported partially by a grant from the National Institutes of Health (Grant R01AI107936) and by a grant from the W. M. Keck Foundation. P.D.-D. was supported in part by Fonds de Recherche du Québec - Nature et Technologies, and the Natural Sciences and Engineering Research Council of Canada with postdoctoral fellowships. N.A.C. was supported by the Otis Williams Postdoctoral Fellowship of the Santa Barbara Foundation. We would like to acknowledge Dr. Gabriel Ortega for the development of the MATLAB script used to analyze chronoamperograms in real-time.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b11341.
Details of potentials used for chronoamperometry and chronocoulometry and cyclic voltammograms of hybridized and unhybridized electrodes (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Arnold AR; Grodick MA; Barton JK DNA Charge Transport: from Chemical Principles to the Cell. Cell Chem. Biol 2016, 23 (1), 183–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Genereux JC; Barton JK Mechanisms for DNA Charge Transport. Chem. Rev 2010, 110 (3), 1642–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Thomas JM; Chakraborty B; Sen D; Yu H-Z Analyte-Driven Switching of DNA Charge Transport: De Novo Creation of Electronic Sensors for an Early Lung Cancer Biomarker. J. Am. Chem. Soc 2012, 134 (33), 13823–13833. [DOI] [PubMed] [Google Scholar]
- (4).Ge B; Huang YC; Sen D; Yu H-Z A Robust Electronic Switch Made of Immobilized Duplex/Quadruplex DNA. Angew. Chem., Int. Ed 2010, 49 (51), 9965–9967. [DOI] [PubMed] [Google Scholar]
- (5).Lu N; Pei H; Ge Z; Simmons CR; Yan H; Fan C Charge Transport within a Three-Dimensional DNA Nanostructure Framework. J. Am. Chem. Soc 2012, 134 (32), 13148–13151. [DOI] [PubMed] [Google Scholar]
- (6).Meade TJ; Kayyem JF Electron Transfer through DNA: Site-Specific Modification of Duplex DNA with Ruthenium Donors and Acceptors. Angew. Chem., Int. Ed. Engl 1995, 34 (3), 352–354. [Google Scholar]
- (7).Boal AK; Yavin E; Lukianova OA; O’Shea VL; David SS; Barton JK DNA-Bound Redox Activity of DNA Repair Glycosylases Containing [4Fe-4S] Clusters. Biochemistry 2005, 4 (23), 8397–8407. [DOI] [PubMed] [Google Scholar]
- (8).Wong ELS; Gooding JJ Charge Transfer through DNA: A Selective Electrochemical DNA Biosensor. Anal. Chem 2006, 78 (7), 2138–2144. [DOI] [PubMed] [Google Scholar]
- (9).Buzzeo MC; Barton JK Redmond Red as a Redox Probe for the DNA-Mediated Detection of Abasic Sites. Bioconjugate Chem. 2008, 19 (11), 2110–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).O’Brien E; Holt ME; Thompson MK; Salay LE; Ehlinger AC; Chazin WJ; Barton JK The [4Fe4S] cluster of human DNA primase functions as a redox switch using DNA charge transport. Science 2017, 355 (6327), eaag1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Purugganan MD; Kumar CV; Turro NJ; Barton JK Accelerated electron transfer between metal complexes mediated by DNA. Science 1988, 241 (4873), 1645–1649. [DOI] [PubMed] [Google Scholar]
- (12).Wohlgamuth CH; McWilliams MA; Slinker JD DNA as a Molecular Wire: Distance and Sequence Dependence. Anal. Chem 2013, 85 (18), 8634–8640. [DOI] [PubMed] [Google Scholar]
- (13).Anne A; Demaille C Dynamics of Electron Transport by Elastic Bending of Short DNA Duplexes. Experimental Study and Quantitative Modeling of the Cyclic Voltammetric Behavior of 3’-Ferrocenyl DNA End-Grafted on Gold. J. Am. Chem. Soc 2006, 128(2), 542–557. [DOI] [PubMed] [Google Scholar]
- (14).Anne A; Demaille C Electron Transport by Molecular Motion of redox-DNA Strands: Unexpectedly Slow Rotational Dynamics of 20-mer ds-DNA Chains End-Grafted onto Surfaces via C6 Linkers. J. Am. Chem. Soc 2008, 130 (30), 9812–9823. [DOI] [PubMed] [Google Scholar]
- (15).Song H; Diakowski PM; Hudson RHE; Kraatz H-B Effect of Ferrocene Position on Charge Transfer in ds-DNA Films. J. Inorg. Organomet. Polym. Mater 2012, 22 (1), 178–182. [Google Scholar]
- (16).Kelley SO; Barton JK; Jackson NM; Hill MG Electrochemistry of Methylene Blue Bound to a DNA-Modified Electrode. Bioconjugate Chem. 1997, 8 (1), 31–37. [DOI] [PubMed] [Google Scholar]
- (17).Zwang TJ; Hürlimann S; Hill MG; Barton JK Helix-Dependent Spin Filtering through the DNA Duplex. J. Am. Chem. Soc 2016, 138 (48), 15551–15554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wohlgamuth CH; McWilliams MA; Slinker JD Temperature Dependence of Electrochemical DNA Charge Transport: Influence of a Mismatch. Anal. Chem 2013, 85 (3), 1462–1467. [DOI] [PubMed] [Google Scholar]
- (19).Pheeney CG; Barton JK Intraduplex DNA-Mediated Electrochemistry of Covalently Tethered Redox-Active Reporters. J. Am. Chem. Soc 2013, 135 (40), 14944–14947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Pheeney CG; Barton JK DNA Electrochemistry with Tethered Methylene Blue. Langmuir 2012, 28 (17), 7063–7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Slinker JD; Muren NB; Renfrew SE; Barton JK DNA charge transport over 34 nm. Nat. Chem 2011, 3 (3), 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kekedy-Nagy L; Shipovskov S; Ferapontova EE Effect of a Dual Charge on the DNA-Conjugated Redox Probe on DNA Sensing by Short Hairpin Beacons Tethered to Gold Electrodes. Anal. Chem 2016, 88 (16), 7984–7990. [DOI] [PubMed] [Google Scholar]
- (23).Abi A; Ferapontova EE Unmediated by DNA Electron Transfer in Redox-Labeled DNA Duplexes End-Tethered to Gold Electrodes. J. Am. Chem. Soc 2012, 134 (35), 14499–14507. [DOI] [PubMed] [Google Scholar]
- (24).Drummond TG; Hill MG; Barton JK Electron Transfer Rates in DNA Films as a Function of Tether Length. J. Am. Chem. Soc 2004, 126 (46), 15010–15011. [DOI] [PubMed] [Google Scholar]
- (25).Jackson NM; Hill MG Electrochemistry at DNA-modified surfaces: new probes for charge transport through the double helix. Curr. Opin. Chem. Biol 2001, 5 (2), 209–215. [DOI] [PubMed] [Google Scholar]
- (26).Kelley SO; Jackson NM; Hill MG; Barton JK Long-Range Electron Transfer through DNA Films. Angew. Chem., Int. Ed 1999, 38 (7), 941–945. [DOI] [PubMed] [Google Scholar]
- (27).Gorodetsky AA; Green O; Yavin E; Barton JK Coupling into the Base Pair Stack Is Necessary for DNA-Mediated Electro-chemistry. Bioconjugate Chem. 2007, 18 (5), 1434–1441. [DOI] [PubMed] [Google Scholar]
- (28).Wohlgamuth CH; McWilliams MA; Mazaheripour A; Burke AM; Lin K-Y; Doan L; Slinker JD; Gorodetsky AA Electrochemistry of DNA Monolayers Modified With a Perylenediimide Base Surrogate. J. Phys. Chem. C 2014, 118 (50), 29084–29090. [Google Scholar]
- (29).Furst AL; Smith MJ; Lee MC; Francis MB DNA Hybridization To Interface Current-Producing Cells with Electrode Surfaces. ACS Cent. Sci 2018, 4 (7), 880–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Sontz PA; Muren NB; Barton JK DNA Charge Transport for Sensing and Signaling. Acc. Chem. Res 2012, 45 (10), 1792–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kawai K; Kodera H; Osakada Y; Majima T Sequence-independent and rapid long-range charge transfer through DNA. Nat. Chem 2009, 1, 156. [DOI] [PubMed] [Google Scholar]
- (32).Endres RG; Cox DL; Singh RRP Colloquium: The quest for high-conductance DNA. Rev. Mod. Phys 2004, 76 (1), 195–214. [Google Scholar]
- (33).Hihath J; Xu B; Zhang P; Tao N Study of single-nucleotide polymorphisms by means of electrical conductance measurements. Proc. Natl. Acad. Sci. U. S. A 2005, 102 (47), 16979–16983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Xu B; Zhang P; Li X; Tao N Direct Conductance Measurement of Single DNA Molecules in Aqueous Solution. Nano Lett. 2004, 4 (6), 1105–1108. [Google Scholar]
- (35).Boon EM; Barton JK; Bhagat V; Nersissian M; Wang W; Hill MG Reduction of Ferricyanide by Methylene Blue at a DNA-Modified Rotating-Disk Electrode. Langmuir 2003, 19 (22), 9255–9259. [Google Scholar]
- (36).Boon EM; Jackson NM; Wightman MD; Kelley SO; Hill MG; Barton JK Intercalative Stacking: A Critical Feature of DNA Charge-Transport Electrochemistry. J. Phys. Chem. B 2003, 107(42), 11805–11812. [Google Scholar]
- (37).Pittman TL; Miao W Examination of Electron Transfer Through DNA Using Electrogenerated Chemiluminescence. J. Phys. Chem. C 2008, 112 (43), 16999–17004. [Google Scholar]
- (38).Inouye M; Ikeda R; Takase M; Tsuri T; Chiba J Single-nucleotide polymorphism detection with “wire-like” DNA probes that display quasi “on−off” digital action. Proc. Natl. Acad. Sci. U. S. A 2005, 102 (33), 11606–11610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Chiba J; Akaishi A; Ikeda R; Inouye M Electrochemical detection of insertion/deletion mutations based on enhanced flexibility of bulge-containing duplexes on electrodes. Chem. Commun 2010, 46 (40), 7563–7565. [DOI] [PubMed] [Google Scholar]
- (40).Mehrgardi MA; Ahangar LE Silver nanoparticles as redox reporters for the amplified electrochemical detection of the single base mismatches. Biosens. Bioelectron 2011, 26 (11), 4308–4313. [DOI] [PubMed] [Google Scholar]
- (41).McWilliams MA; Bhui R; Taylor DW; Slinker JD The Electronic Influence of Abasic Sites in DNA. J. Am. Chem. Soc 2015, 137 (34), 11150–11155. [DOI] [PubMed] [Google Scholar]
- (42).Moradi N; Noori A; Mehrgardi MA; Mousavi MF Scanning Electrochemical Microscopy for Electrochemical Detection of Single-base Mismatches by Tagging Ferrocenecarboxylic Acid as a Redox Probe to DNA. Electroanalysis 2016, 28 (4), 823–832. [Google Scholar]
- (43).Campos R; Kekedy-Nagy L; She Z; Sodhi R; Kraatz H-B;Ferapontova EE Electron Transfer in Spacer-Free DNA Duplexes Tethered to Gold via dA10 Tags. Langmuir 2018, 34 (29), 8472–8479. [DOI] [PubMed] [Google Scholar]
- (44).Anne A; Bouchardon A; Moiroux J 3’-Ferrocene-Labeled Oligonucleotide Chains End-Tethered to Gold Electrode Surfaces: Novel Model Systems for Exploring Flexibility of Short DNA Using Cyclic Voltammetry. J. Am. Chem. Soc 2003, 125 (5), 1112–1113. [DOI] [PubMed] [Google Scholar]
- (45).Campos R; Kotlyar A; Ferapontova EE DNA-Mediated Electron Transfer in DNA Duplexes Tethered to Gold Electrodes via Phosphorothioated dA Tags. Langmuir 2014, 30 (40), 11853–11857. [DOI] [PubMed] [Google Scholar]
- (46).Farjami E; Clima L; Gothelf K; Ferapontova EE Off−On” Electrochemical Hairpin-DNA-Based Genosensor for Cancer Diagnostics. Anal. Chem 2011, 83 (5), 1594–1602. [DOI] [PubMed] [Google Scholar]
- (47).Wang K; Goyer C; Anne A; Demaille C Exploring the Motional Dynamics of End-Grafted DNA Oligonucleotides by in Situ Electrochemical Atomic Force Microscopy. J. Phys. Chem. B 2007, 111 (21), 6051–6058. [DOI] [PubMed] [Google Scholar]
- (48).Fukuzumi S; Miyao H; Ohkubo K; Suenobu T Electron-Transfer Oxidation Properties of DNA Bases and DNA Oligomers. J. Phys. Chem. A 2005, 109 (15), 3285–3294. [DOI] [PubMed] [Google Scholar]
- (49).Dauphin-Ducharme P; Arroyo-Currás N; Kurnik M; Ortega G; Li H; Plaxco KW Simulation-Based Approach to Determining Electron Transfer Rates Using Square-Wave Voltammetry. Langmuir 2017, 33 (18), 4407–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Dauphin-Ducharme P; Arroyo-Currás N;Adhikari R; Somerson J; Ortega G; Makarov DE; Plaxco KW Chain Dynamics Limit Electron Transfer from Electrode-Bound, Single-Stranded Oligonucleotides. J. Phys. Chem. C 2018, 122 (37), 21441–21448. [Google Scholar]
- (51).Boon EM; Ceres DM; Drummond TG; Hill MG; Barton JK Mutation detection by electrocatalysis at DNA-modified electrodes. Nat. Biotechnol 2000, 18 (10), 1096–1100. [DOI] [PubMed] [Google Scholar]
- (52).Arroyo-Currás N; Dauphin-Ducharme P; Ortega G; Ploense KL; Kippin TE; Plaxco KW Subsecond-Resolved Molecular Measurements in the Living Body Using Chronoamperometrically Interrogated Aptamer-Based Sensors. ACS Sensors 2018, 3(2), 360–366. [DOI] [PubMed] [Google Scholar]
- (53).Laviron E General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J. Electroanal. Chem. Interfacial Electrochem 1979, 101 (1), 19–28. [Google Scholar]
- (54).Drummond TG; Hill MG; Barton JK Electrochemical DNA sensors. Nat. Biotechnol 2003, 21 (10), 1192–1199. [DOI] [PubMed] [Google Scholar]
- (55).Johnson RP; Gale N; Richardson JA; Brown T; Bartlett PN Denaturation of dsDNA immobilised at a negatively charged gold electrode is not caused by electrostatic repulsion. Chemical Science 2013, 4 (4), 1625–1632. [Google Scholar]
- (56).Kékedy-Nagy L; Ferapontova EE; Brand I Submolecular Structure and Orientation of Oligonucleotide Duplexes Tethered to Gold Electrodes Probed by Infrared Reflection Absorption Spectroscopy: Effect of the Electrode Potentials. J. Phys. Chem. B 2017, 121(7), 1552–1565. [DOI] [PubMed] [Google Scholar]
- (57).Berlin YA; Burin AL; Ratner MA Charge Hopping in DNA. J. Am. Chem. Soc 2001, 123 (2), 260–268. [DOI] [PubMed] [Google Scholar]
- (58).Bram S Secondary Structure of DNA depends on Base Composition. Nature New Biology 1971, 232, 174. [DOI] [PubMed] [Google Scholar]
- (59).Ricci F; Lai RY; Heeger AJ; Plaxco KW; Sumner JJ Effect of Molecular Crowding on the Response of an Electrochemical DNA Sensor. Langmuir 2007, 23 (12), 6827–6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Kibbe WA OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 35, W43–W46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Arroyo-Currás N; Scida K; Ploense KL; Kippin TE; Plaxco KW High Surface Area Electrodes Generated via Electrochemical Roughening Improve the Signaling of Electro-chemical Aptamer-Based Biosensors. Anal. Chem 2017, 89 (22), 12185–12191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.