

Abstract
Purpose:
To identify effective metabolicinhibitors to suppress the aggressive growth of pancreatic ductal adenocarcinoma (PDAC), we explored the in vivo antitumor efficacy of metabolic inhibitors, as single agents, in a panel of patient-derived PDAC xenograft models (PDX) and investigated whether genomic alterations of tumors correlate with the sensitivity to metabolic inhibitors.
Experimental Design:
Mice with established PDAC tumors from 6 to 13 individual PDXs were randomized and treated, once daily for 4 weeks, with either sterile PBS (vehicle) or the glutaminase inhibitor bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES), transaminase inhibitor aminooxyacetate (AOA), pyruvate dehydrogenase kinase inhibitor dichloroacetate (DCA), autophagy inhibitor chloroquine (CQ), and mitochondrial complex I inhibitor phenformin/metformin.
Results:
Among the agents tested, phenformin showed significant tumor growth inhibition (>30% compared with vehicle) in 5 of 12 individual PDXs. Metformin, at a fivefold higher dose, displayed significant tumor growth inhibition in 3 of 12 PDXs similar to BPTES (2/8 PDXs) and DCA (2/6 PDXs). AOA and CQ had the lowest response rates. Gene set enrichment analysis conducted using the baseline gene expression profile of pancreatic tumors identified a gene expression signature that inversely correlated with phenformin sensitivity, which is in agreement with the phenformin gene expression signature of NIH Library of Integrated Network-based Cellular Signatures (LINCS). The PDXs that were more sensitive to phenformin showed a baseline reduction in amino acids and elevation in oxidized glutathione. There was no correlation between phenformin response and genetic alterations in KRAS, TP53, SMAD4, or PTEN.
Conclusions:
Phenformin treatment showed relatively higher antitumor efficacy against established PDAC tumors, compared with the efficacy of other metabolic inhibitors and metformin. Phenformin treatment significantly diminished PDAC tumor progression and prolonged tumor doubling time. Overall, our results serve as a foundation for further evaluation of phenformin as a therapeutic agent in pancreatic cancer.
Introduction
Despite significant advances in the understanding of pancreatic cancer biology and new treatment strategies, pancreatic ductal adenocarcinoma (PDAC) remains the most deadly major cancer (1, 2). Current treatment options for advanced pancreatic cancer involve a number of cytotoxic cocktails (3), which yield only minimal survival benefit. Successful treatment of PDACs is challenged by their biological complexity. Despite the ubiquity of mutant KRAS in PDAC, tumors show variable additional mutations, extensive and heterogeneous stromal content, and a strongly immunosuppressive microenvironment (4, 5).
The energy requirements of cancer cells vary from those of quiescent, terminally differentiated cells (6). Many cancers rely on metabolic rewiring to sustain growth and survive in the tumor microenvironment (5). Metabolic rewiring, including induction of macropinocytosis, plays a role in the pathogenesis of PDAC and is a critical component of the tumorigenic program driven by KRAS (7). We sought to conduct an unbiased study to determine the antitumor efficacy of agents targeting metabolic pathways in a diverse set of pancreatic cancer patient-derived xenografts (PDX). We also aimed to determine whether genomic alterations might correlate with the specific metabolic patterns and hence sensitivity to specific metabolic inhibitors. Toward this end, we selected a glutaminase inhibitor (Bis-2-(5-phenylaceta- mido-1,3,4-thiadiazol-2-yl)ethyl sulfide, BPTES), a nonspecific transaminase inhibitor aminooxyacetate (AOA); dichloroacetate (DCA), an inhibitor of pyruvate dehydrogenase kinase (PDK), an autophagy inhibitor chloroquine (CQ), and two antidiabetic biguanides, metformin and phenformin, which are mitochondrial complex I inhibitors. These inhibitors were evaluated in PDXs varying from 6 and up to 13 individual patient tumors. We recently reported the in vivo antitumor efficacy of FX11 and hydroxy-chloroquine (HCQ; refs. 8, 9). Our results demonstrated that mutant P53 status correlated with increased glycolytic metabolism and heightened response to FX11, in a fashion that could be monitored in vivo using 13C-pyruvate hyperpolarized MRI (8).
The present preclinical study used a large number of animals (>500 athymic nude mice) with established human pancreatic tumors for the comprehensive evaluation of the antitumor efficacy of metabolic inhibitors in pancreatic cancer. Among the six metabolic inhibitors tested, AOA, BPTES, CQ, and DCA were moderately effective without any clear patterns that could be correlated with the genomic statuses of the tumors. Phenformin was the most effective single agent across the PDXs. Metformin was less potent and less effective, but showed a similar trend in responses, consistent with phenformin and metformin both targeting complex I. Baseline metabolomics performed on untreated tumors revealed patterns of metabolites in phenformin-sensitive tumors compatible with higher oxidative stress, slower glycolysis, and faster amino acid utilization. Using a phenformin transcriptomic signature from the LINCS database (http://www.lincsproject.org), wefound an inverse correlation between baseline expression of the signature with the in vivo sensitivity to phenformin. Although a recent clinical study of metformin failed to demonstrate efficacy (10), our data suggest that with appropriate patient selection markers, the more potent biguanide phenformin may have efficacy in human pancreatic cancer.
Materials and Methods
Drugs
The following agents were evaluated for the in vivo antitumor efficacy in established human pancreatic cancer PDXs: (i) BPTES, a selective inhibitor of glutaminase; (ii) AOA, a selective inhibitor of aspartate aminotransferase; (iii) DCA, an inhibitor of PDK; (iv) chloroquine diphosphate (CQ), an inhibitor of autophagy; (v) phenformin; and (vi) metformin, both are biguanides and mitochondrial complex I inhibitors. AOA, DCA, CQ, phenformin, and metformin were purchased from Sigma-Aldrich. BPTES was kindly provided by Dr. Takashi Tsukamoto (Johns Hopkins University, Baltimore, MD).
Evaluation of antitumor efficacy of metabolic inhibitors in human pancreatic PDXs
Animal experiments were conducted following approval by the Animal Care and Use Committee guidelines of the Johns Hopkins University (Baltimore, MD). Studies were carried out in female nu/nu athymic mice (Harlan), maintained under pathogen-free conditions and a 12-hour light/12-hour dark cycle. Fresh pancreatic tumor specimens originally resected from patients at the time of surgery, with informed written patient consent, were implanted subcutaneously into the flanks of 6-week-old mice. Grafted tumors were subsequently transplanted from mouse to mouse and maintained as a live PancXenoBank according to an Institutional Review Board-approved protocol (11). Fresh tumors resected from 15 individual PDXs from the live PancXenoBank collection were subcutaneously implanted on both flanks of mice. Tumors from the same patient xenografts were allowed to grow to approximately 150 to 200 mm3. Mice with outlier tumor sizes were culled. Animals were grouped randomly (5 mice per group, with 7–10 tumors) and treated with vehicle (sterile PBS) or metabolic inhibitors. The metabolic inhibitors evaluated and doses used were BPTES (12.5 mg/kg), AOA (10 mg/kg), DCA (50 mg/kg), CQ (60 mg/kg), phenformin (50 mg/kg), and metformin (250 mg/kg). Treatment doses were selected from previously published preclinical reports (12–17). The metabolic inhibitors were prepared fresh in sterile PBS on each day and administered once daily for 4 weeks by intraperitoneal injection using 1-mL insulin syringe with a 27-gauge needle (BD). Tumor size was measured twice per week by caliper measurements, and volumes were calculated using the following formula: V = (a × b2)/2, where “a” is the largest dimension and “b” the smallest. Tumor growth in drug-treated animals was compared with vehicle-treated mice and represented as percentage tumor growth inhibition (TGI).The statistical significance of the data was evaluated by two-tailed unpaired t test using GraphPad Prism 6 software. All results are presented as mean ± SEM. P < 0.05 was considered significant. Herein, we define significant response as a 30% or greater reduction of tumor growth by an agent as compared with the tumor growth of vehicle-treated control animals.
PDX gene expression data sets
Gene expression data sets for the PDXs tested were retrieved from the NCBI’s Gene Expression Omnibus (GEO) Series with accession number GSE51798. Details regarding the sample processing have been described previously (18). Affymetrix Human Genome U133 Plus 2.0 Array were normalized using RMA (Robust Multi-Array Average). Gene expression value was collapsed by the average probe mean expression.
LINCS analysis
The Library of Integrated Network-based Cellular Signatures (LINCS, http://www.lincsproject.org) is a catalog of gene expression data associated to cell lines exposed to a variety of perturbing agents, such as small molecules (about 5,500), FDA drugs (~1,300), and shRNA silencing (22,119 genetic perturbagens). LINCS, based on L1000 high-throughput technology, is an extension of the connectivity map, which has been successfully used for drug repositioning (19).
Drug signature generation
To generate the expression signatures for each drug, the level 3 data from LINCS were used. We downloaded the codes of the instances for a given drug treatment and the DMSO controls using the InstInfo service from the LINCS: API. The differential expression analysis of the effect of the drug was carried out using the limma R package (20). Cell type and plate were considered as confounding variables, to obtain a consensus drug signature and minimize the batch effect. The top 250 probes more overexpressed and underexpressed were taken and annotated to genes using the GeneInfo service from the LINCS:API (21). The resulting genes were used to form the up- (UP) and down-regulated (DN) gene expression signatures, respectively.
Signature expression in PDX models
To measure the expression level of the consensus drug signature in each individual PDX, a single sample GSEA (ssGSEA) was applied (22). The GSVA R package was used (23). A consensus enrichment score (ES) was obtained subtracting the ES values of the DN signature from those ESs of the UP signature. A Spearman correlation coefficient was calculated using the consensus ES and PDX observed drug responses. All codes and scripts used are available at https://github.com/htejero/PhenforminPDXAnalysis.
Measurements of metabolites in tumors by mass spectroscopy
Subcutaneously grown tumors (150 mm3) were excised from mice (two separate tumors from 10 xenografts) and were immediately frozen in liquid nitrogen. The samples were shipped overnight on dry ice to Princeton University (Princeton, NJ) and stored in liquid nitrogen until analysis. Metabolite extraction was performed in 25 mg tumor pieces (24). Samples were analyzed by two separate LC/MS systems: (i) standalone orbitrap MS (Exactive; Thermo Fisher Scientific) operating in negative full scan mode at 100,000 resolution coupled to C18 ultra performance reverse-phase ion pair LC and (ii) triple quadrupole mass spectrometer (TSQ Quantum Discovery Max; Thermo Fisher Scientific) operating in negative multiple reaction monitoring mode coupled to C18 high-performance reverse-phase ion pair LC (25, 26). Data from each instrument are presented separately and generally yielded identical conclusions.
Results
Inhibition of glutamine metabolism pathway (BPTES and AOA)
We chose BPTES and AOA as inhibitors of glutamine metabolism, which has been implicated in the maintenance and progression of PDAC (5). BPTES is a potent allosteric inhibitor of glutaminase GLS (but not GLS2) and has in vitro and in vivo antitumor activity (27, 28). We recently documented its efficacy in a genetically engineered mouse model (GEMM) of liver cancer and human lymphoma xenografts (17). We also recently documented the efficacy of AOA in human breast cancer cell line xenografts and demonstrated a link with MYC status and expression of the transaminase GOT2 (29). Here, we report the responses of selected pancreatic cancer PDXs to BPTES or AOA. As shown in Fig. 1A, BPTES treatment significantly reduced the tumor growth in 2 of 8 individual PDXs. Figure 1B documents the antitumor efficacy of AOA. Only 1 in 13 PDXs showed over 30% TGI, which did not correlate with the pattern of tumor responses to BPTES, suggesting that the mechanisms of action of these agents do not overlap (Supplementary Fig. S1). Tumor growth curves of P198 and P253 are shown in Fig. 1B and D. Neither compound yielded a response pattern that correlates with the genomic status of the tumors (Table 1). These studies underscore the complexity of interpreting results using diverse compounds that are touted as glutamine metabolic inhibitors, particularly AOA, which is a nonspecific transaminase inhibitor that may have a wide spectrum of targets. In contrast, BPTES, a potent, specific inhibitor of GLS, appears to have on-target antitumor effects, as recently demonstrated by a rescue experiment using a BPTES-resistant allele of glutaminase in lymphoma cells (17).
Figure 1.
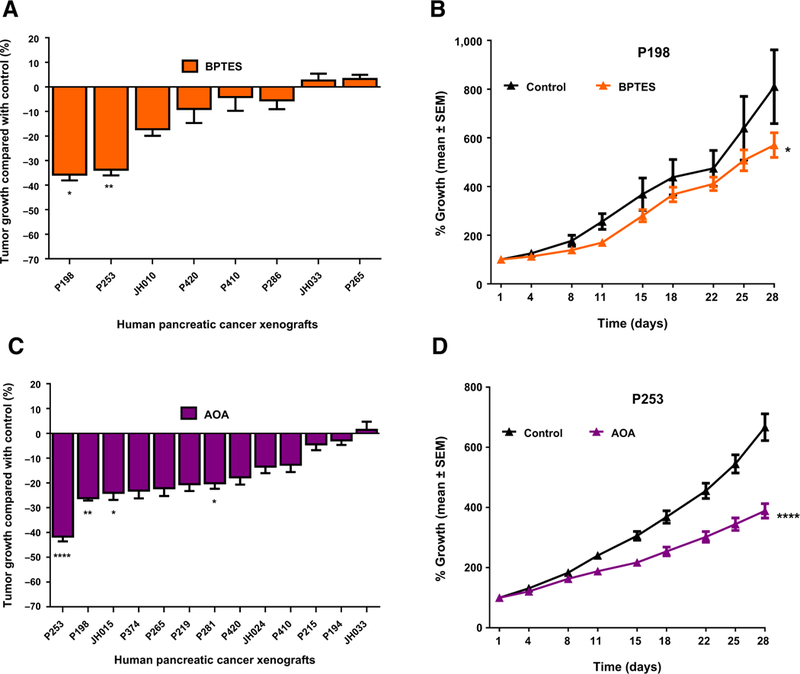
in vivo antitumor efficacy of glutaminase (BPTES) and transaminase (AOA) inhibitors in human pancreatic cancer xenografts. Mice with subcutaneously grown flank tumors (~150 mm3) were randomly assigned to two treatment arms and treated with vehicle or BPTES (12.5 mg/kg)/AOA (10 mg/kg), once daily intraperitoneal dose for 4 weeks. Tumor size was measured twice/week by using a digital caliper until the termination of experiments. A, Antitumor efficacy of BPTES in eight individual PDXs. B, Tumor growth curves of a representative xenograft (P198) treated with vehicle or BPTES, showing statistically significant tumor growth inhibition compared with vehicle treated mice. C, Antitumor efficacy of AOA in 13 PDXs. D, Tumor growth curves of representative xenograft (P253) treated with AOA showing statistically significant tumor growth inhibition compared with tumors of vehicle-treated mice. Data, mean ± SEM; n =7–10 tumors per group. **** P < 0.0001; **, P < 0.01; *, P < 0.05.
Table 1.
Key genetic alterations and copy-number variations in the 15 PDXs used for study
| PDXs | TP53 | KRAS | SMAD4 | PTEN |
|---|---|---|---|---|
| P410 | MUT | WT | WT | WT |
| P374 | MUT | MUT | WT | WT |
| P281 | MUT | MUT | MUT | WT |
| P265 | MUT; Amplification | MUT; Amplification | WT; Deletion | WT |
| P253 | MUT | MUT | WT | WT |
| P215 | MUT | MUT | WT | WT |
| P198 | MUT | MUT | MUT | WT; Deletion |
| P194 | MUT | MUT | Deep deletiona | WT |
| JH015 | MUT | MUT | WT; Deletion | WT |
| JH010 | MUT | MUT | MUT | WT |
| P420 | WT | WT | WT | WT |
| P286 | WT | MUT | WT | WT |
| P219 | WT | MUT | WT | WT |
| JH033 | WT | MUT | WT; Deletion | WT; Deletion |
| JH024 | WT | MUT | MUT; Deletion | WT |
Abbreviations: MUT, mutated; WT, wild type.
Deep deletion = deletion in both copies of gene.
Inhibition of PDK (DCA)
PDKs are enzymes that phosphorylate and inactivate pyruvate dehydrogenase (PDH), which converts pyruvate to acetyl CoA (30, 31). Through inhibition of PDH, PDKs divert pyruvate away from mitochondrial respiration and favor its conversion to lactate or alanine (32). We discovered that PDK1 is a target of the hypoxia-inducible factor that diminishes mitochondrial pyruvate respiration and increases its conversion to lactate (33). Knockdown of PDK1 or its inhibition by DCA increased pyruvate oxidation and increased oxidative stress as detected through an increase in reactive oxygen species (ROS; ref. 34). The increased ROS contributes to cell death, and hence, hypoxic cancer cells are expected to be sensitive to DCA treatment. In this regard, we treated a panel of pancreatic cancer PDXs and found that DCA has over 30% TGI only in 2 of 6 PDXs, without apparent correlation with the efficacy of other compounds (Fig. 2A and B; Supplementary Fig. S1).
Figure 2.
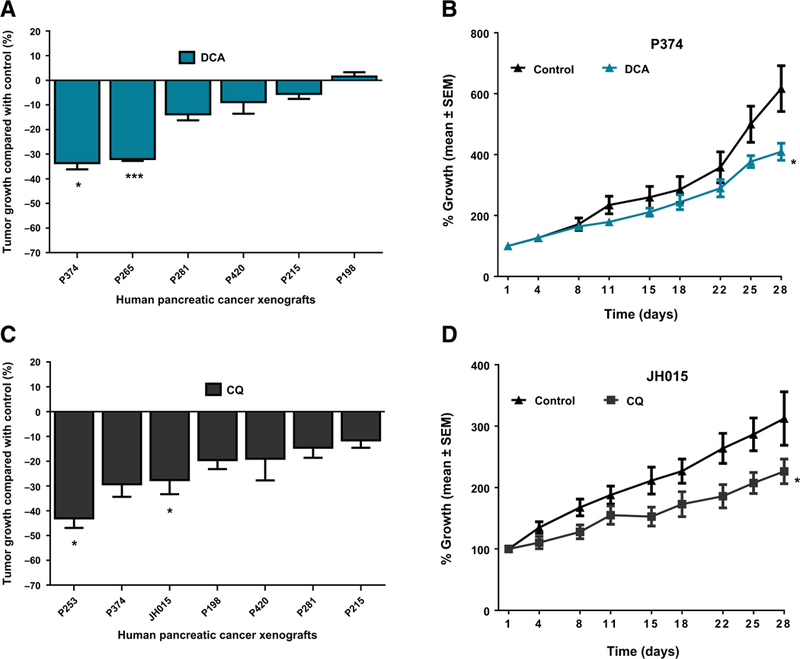
In vivo antitumor efficacy of DCA (PDK inhibitor) and CQ (autophagy inhibitor) in human pancreatic cancer xenografts. Mice with subcutaneously grown flank tumors (~150 mm3) were randomly assigned to two treatment arms and treated with vehicle or DCA (10 mg/kg) or CQ (60 mg/kg), once daily intraperitoneal dose for 4 weeks. Tumor size was monitored for 28 days. A, Antitumor efficacy of DCA in six individual PDXs. B, Tumor growth curves of P374 xenograft treated with vehicle or DCA, showing statistically significant tumor growth inhibition compared with vehicle-treated mice. C, Antitumor effect of CQ in seven individual PDXs. CQ treatment showed significant tumor growth inhibition in 2 of 7 (28.57%) PDXs. D, Tumor growth curves of representative xenograft (JH015) treated with CQ showing statistically significant tumor growth inhibition compared with vehicle-treated mice. Data, mean ± SEM; n = 7–10 tumors per group. ***, P < 0.001; *, P < 0.05.
Autophagy inhibition by chloroquine
Cancer cells, especially RAS-transformed cancer cells, upregulate autophagy to survive microenvironmental stress and to increase growth and aggressiveness (35–37). Autophagy has a critical role in PDAC pathogenesis and is elevated in PDAC cell lines and primary tumors (38). A recent report demonstrated that autophagy inhibitor, HCQ, showed promising antitumor efficacy in pancreatic PDXs as well as in a GEMM model of pancreatic cancer (9). In this study, CQ treatment showed over 30% TGI only in 1 of 7 individual PDXs with the most significant tumor growth inhibition of P253 PDX, which was sensitive to most of the tested metabolic inhibitors independent of their mode of action (Fig. 2C and D; Supplementary Fig. S1).
Inhibition of mitochondrial complex I (metformin and phenformin)
In contrast to the variable responses to other metabolic inhibitors, the biguanides phenformin and to a lesser extent metformin, displayed the best therapeutic effects across the 12 pancreatic cancer PDXs (Fig. 3A–C). The biguanides are a class of compounds that inhibit mitochondrial complex I activity and are of significant interest as cancer therapeutics, because cancer cells rely not only on aerobic glycolysis but also on oxidative phosphorylation and respiration (39). The biguanides metformin and phenformin have been studied in a number to cancer models, but to date, a comprehensive study of their activities against a relatively large panel of pancreatic cancer PDXs has not been performed. As illustrated in Fig. 3, metformin and phenformin were broadly effective against 12 pancreatic cancer PDXs. As compared with other metabolic inhibitors, the patterns of responses between the two biguanides are similar, consistent their overlapping mechanistic roles as complex I inhibitors (Supplementary Fig. S1). Phenformin displayed a higher efficacy (Fig. 3; Supplementary Fig. S1), consistent with its stronger complex I inhibition (40). Tumor growth curves and tumor doubling time of all 12 PDXs treated with vehicle and phenformin are shown in Supplementary Figs. S2 and S3, respectively. As shown in Supplementary Fig. S4A, the tumor doubling time of PDXs that were more sensitive to phenformin treatment was shorter, compared with the tumor doubling time of the less sensitive PDXs (12.77 ± 0.8833 vs. 17.42 ± 1.993 days, respectively, P = 0.0391). Aggregate tumor doubling time data of all individual tumors of control and phenformin treatment in 12 PDXs showed that phenformin treatment, although not producing tumor regression or stable disease, significantly delayed the tumor doubling time compared with vehicle treatment (P = 0.0020; Supplementary Fig. S4B).
Figure 3.
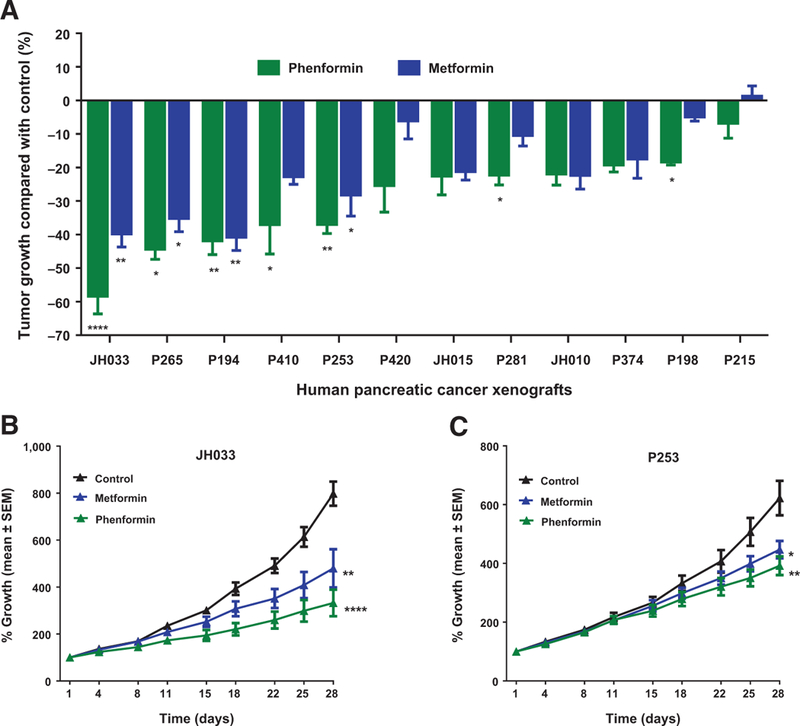
In vivo antitumor efficacy of the biguanides metformin and phenformin in human pancreatic cancer xenografts. Tumors from 12 PDXs were subcutaneously grown as flank tumors in mice. When tumors approached a size of approximately 150 mm3, animals were randomly assigned to receive vehicle or phenformin (50 mg/kg) or metformin (250 mg/kg) administered as once-a-day intraperitoneal injection for 4 weeks. Tumor size was monitored for 28 days. A, Antitumor efficacy of biguanides in 12 individual PDXs. Animals in the phenformin-treated group displayed better antitumor efficacy compared with metformin-treated animals. Phenformin treatment demonstrated tumor growth inhibition (ranging from 58.39% to 6.79%) in 12 PDXs; 7 of 12 PDXs (58.33%) showed statistically significant tumor growth inhibition when exposed to phenformin treatment compared with vehicle-treated animals. Metformin treatment demonstrated statistically significant tumor growth inhibition in 4 of 12 PDXs (33.33%). B and C, Tumor growth curves of two PDXs (JH033 and P253) sensitive to biguanides. In both xenografts, phenformin showed better tumor growth inhibition compared with metformin treatment, indicating the effectiveness of phenformin. Data, mean ± SEM; n = 7–10 tumors per group. ****, P < 0.0001; *, P < 0.01; *, P < 0.05.
Biomarkers of responses to biguanides
To gain insight into the potential biomarkers of responses to biguanides, we examined the oncogenotype of the tumors (41, 42) and found no correlation of response with the mutational statuses of KRAS, TP53, SMAD4, or PTEN (Table 1). In contrast, using basal gene expression profile of the PDXs, we found that the phenformin gene expression signature from the LINCS-L1000 project (which represents the gene expression changes observed when cell lines were treated with phenformin compared with DMSO controls; see Materials and Methods) was inversely associated with phenformin sensitivity (Spearman R = –0.89, P = 0.0004; Fig. 4). Because of the exceptional dominant PALB2 mutation in the JH033 PDX, we excluded it from the biomarker analysis (43, 44). As shown in Fig. 4, the human pancreatic PDXs with higher basal phenformin-induced gene expression signature had least response to phenformin.
Figure 4.
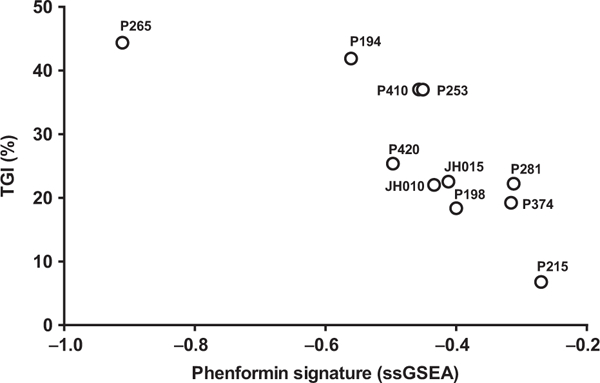
Antitumor efficacy of the biguanide phenformin in human pancreatic cancer xenografts versus the expression of LINCS-L1000 phenformin signature. Tumor responses of human pancreatic cancer xenografts demonstrated a statistically significant negative Spearman correlation ρ = –0.89 (P = 0.0004) with the LINCS phenformin signature (see Materials and Methods).
We also postulated that the basal transcriptomes of tumors might reflect the metabolomes that could be more susceptible to inhibition by biguanides. In this regard, we determined the basal metabolic profiles of PDXs using LC/MS and found general metabolic differences between tumors that are more sensitive (>30% reduction of growth) versus those that are less sensitive (Fig. 5). The levels of metabolites involved in glycolysis (such as lactate) and amino acid levels were diminished in phenformin-sensitive tumors, while oxidized glutathione was elevated. These observations suggest the possibility that sensitivity to biguanides correlates with increased use of amino acids for oxidation, resulting in lower amino acids and increased oxidized glutathione.
Figure 5.
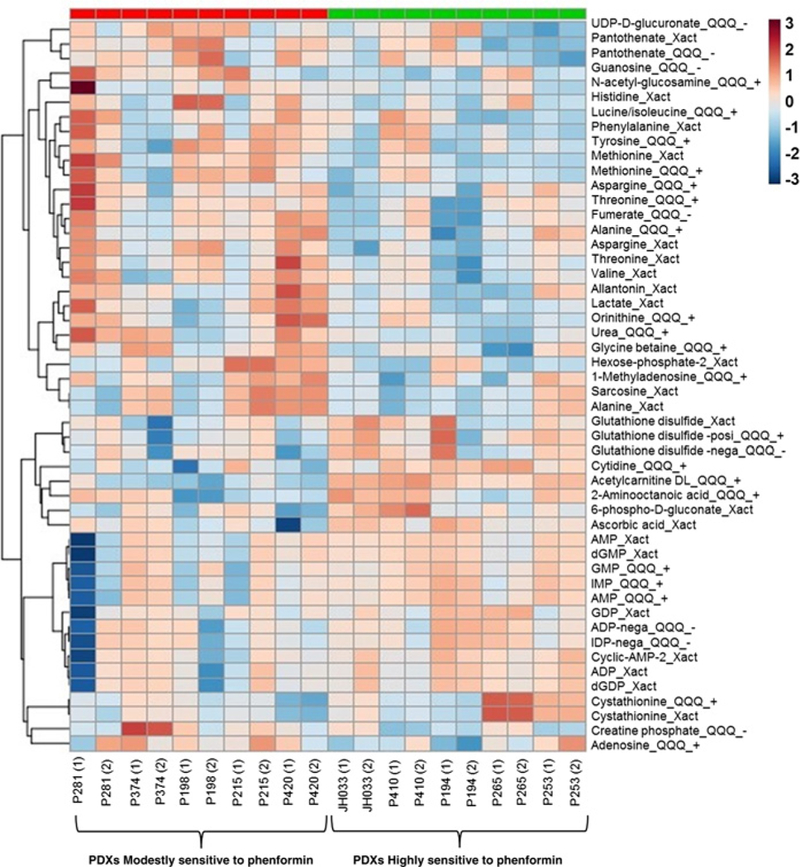
Heatmapshowing expression levelsof top 50 metabolites in PDXs that were modestly sensitive (<30% reduction of tumor growth compared with vehicle treatment) or highly sensitive (>30% reduction of tumor growth compared with vehicle treatment) to phenformin treatment. Metabolites were quantitated in the baseline tumors by LC/MS (details on LC/MS are provided in Materials and Methods). “Xact” indicates the metabolite was analyzed using an Exactive high-resolution mass spectrometer (Thermo Fisher Scientific) in negative mode, “QQQ_+” and “QQQ_−“ indicate that metabolite was analyzed using triple quad mass spectrometry analysis (both from Thermo Fisher Scientific) in positive and negative mode, respectively. Frequently, the same metabolites were measured by both methods, in which case the results from both methods are presented and generally agree.
Discussion
PDAC is an aggressive malignancy, with less than a third of patients surviving even early-stage disease. Our comprehensive preclinical study of metabolic inhibitors using patient-derived pancreatic cancer xenografts, which preserve the heterogeneity and histologic characteristics of pancreatic cancer, demonstrated diverse antitumor responses in a large set of human pancreatic cancerxenografts (Figs. 1–3; Supplementary Fig. S1). Inhibitors of glutaminase (GLS), transaminases, and PDK showed significant antitumor activity only in a small subset of pancreatic cancer PDXs. Only the LDHA inhibitor FX11, as previously reported, seems to have antitumor activity that correlates with mutant P53 status of tumors (7). Importantly, as compared with inhibitors of autophagy, glutaminase, transaminases, or PDK, we found that the biguanides have significant tumor growth-inhibitory activity, albeit not leading to tumor growth regressions across a wide spectrum of PDXs as single agents, with phenformin being notably most active. A recent report showed that phenformin, but not metformin, strongly reduces melanoma cell viability, growth, and abrogates melanoma cell invasion (45). Furthermore, phenformin has been reported to enhance the therapeutic efficacy ofBRAF (V600E) and ERK inhibitors in melanoma (46, 47). The encouraging antitumor efficacy of phenformin in our study, in the context of the disappointing efficacy of metformin in a recent pancreatic cancer clinical trial, suggests that phenformin merits clinical investigation as an anticancer agent for the treatment of pancreatic cancer. Although phenformin was withdrawn from the U.S. markets by the FDA in the late 1978s because of its predisposition to cause lactic acidosis in diabetic patients (48), with proper clinical vigilance, this side effect may be manageable in the context of PDAC therapy (49). Our study demonstrates that phenformin as a single agent reduces tumor progression in human pancreatic cancer PDXs and provides rationale for further evaluation of phenformin in pancreatic cancer.
Supplementary Material
Translational Relevance.
Pancreatic ductal adenocarcinoma (PDAC) remains mystifyingly difficult to treat, and outcomes are persistently poor, demanding the exploration of new therapeutic options. Here, we used a clinically relevant and genetically characterized platform of human PDAC PDXs to explore the in vivo antitumor efficacy of cancer metabolism-targeted agents. Among the six metabolic inhibitors tested, phenformin was the most effective single agent across the PDXs. Phenformin significantly diminished PDAC tumor progression and prolonged tumor doubling time. Efficacy of metformin, at a fivefold higher dose of phenformin, was less compared with phenformin. Our results raise the possibility that phenformin might represent a better biguanide to inhibit PDAC progression and provide the foundation for further evaluation of phenformin in pancreatic cancer.
Acknowledgments
Grant Support
This study was supported by funding from a Stand Up To Cancer Dream Team Translational Cancer Research grant (grant number: SU2C-AACR-DT0509; to N.V. Rajeshkumar, M. Hidalgo, A. Maitra, and C.V. Dang). Research grants were administered by the American Association for Cancer Research, the Scientific Partner of SU2C. This research was also supported by the following NIH grants: P30CA016520 (to C.V. Dang), R01CA051497 (to C.V. Dang), R01CA057341 (to C.V. Dang), R01CA163591 (to J.D. Rabinowitz), and R01CA113669 (to A. Maitra).
Footnotes
Disclosure of Potential Conflicts of Interest
J.D. Rabinowitz is a consultant/advisory board member for Kadmon Pharmaceuticals. No potential conflicts of interest were disclosed by the other authors.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Hidalgo M Pancreatic cancer. N Engl J Med 2010;362:1605–17. [DOI] [PubMed] [Google Scholar]
- 2.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014;371:1039–49. [DOI] [PubMed] [Google Scholar]
- 3.Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet 2016; 388:73–85. [DOI] [PubMed] [Google Scholar]
- 4.Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol 2008;3: 157–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ying H, Dey P,Yao W, Kimmelman AC, Draetta GF, Maitra A, et al. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 2016;30: 355–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirschey MD, DeBerardinis RJ, Diehl AM, Drew JE, Frezza C, Green MF, et al. Dysregulated metabolism contributes to oncogenesis. Semin Cancer Biol 2015;35:S129–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perera RM, Bardeesy N. Pancreatic cancer metabolism: breaking it down to build it back up. Cancer Discov 2015;5:1247–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajeshkumar NV, Dutta P, Yabuuchi S, de Wilde RF, Martinez GV, Le A, et al. Therapeutic targeting of the warburg effect in pancreatic cancer relies on an absence of p53 function. Cancer Res 2015;75: 3355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov 2014;4:905–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kordes S, Pollak MN, Zwinderman AH, Mathot RA, Weterman MJ, Beeker A, et al. Metformin in patients with advanced pancreatic cancer: a doubleblind, randomised, placebo-controlled phase 2 trial. Lancet Oncol 2015; 16:839–47. [DOI] [PubMed] [Google Scholar]
- 11.Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, et al. MK-1775, a potentWee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res 2011;17:2799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hao Y, Samuels Y, Li Q, Krokowski D, Guan BJ, Wang C, et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat Commun 2016;7:11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang W, Finniss S, Cazacu S, Xiang C, Brodie Z, Mikkelsen T, et al. Repurposing phenformin for the targeting of glioma stem cells and the treatment of glioblastoma. Oncotarget 2016;7:56456–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar K, Wigfield S, Gee HE, Devlin CM, Singleton D, Li JL, et al. Dichloroacetate reverses the hypoxic adaptation to bevacizumab and enhances its antitumor effects in mouse xenografts. J Mol Med 2013;91: 749–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu F, Shang Y, Chen SZ. Chloroquine potentiates the anti-cancer effect of lidamycin on non-small cell lung cancer cells in vitro. Acta Pharmacol Sin 2014;35:645–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen-induced lung tumorigenesis. Cancer Prev Res 2010;3:1066–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiang Y, Stine ZE, Xia J, Lu Y, O’Connor RS, Altman BJ, et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest 2015;125:2293–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez-Garcia R, Juan D, Rausell A, Munoz M, Banos N, Menendez C, et al. Transcriptional dissection of pancreatic tumors engrafted in mice. Genome Med 2014;6:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Musa A, Ghoraie LS, Zhang SD, Galzko G, Yli-Harja O, Dehmer M, et al. A review of connectivity map and computational approaches in pharmacogenomics. Brief Bioinform 2017. January 9 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iorio F, Bosotti R, Scacheri E, Belcastro V, Mithbaokar P, Ferriero R, et al. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc Natl Acad Sci U S A 2010;107:14621–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009;462:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 2015;75:544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bajad SU, Lu W, Kimball EH, Yuan J, Peterson C, Rabinowitz JD. Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography-tandem mass spectrometry. J Chromatogr A 2006;1125:76–88. [DOI] [PubMed] [Google Scholar]
- 26.Lu W, Clasquin MF, Melamud E, Amador-Noguez D, Caudy AA, Rabinowitz JD. Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Bioanal Chem 2010;82:3212–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 2012;15:110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res 2010;70:8981–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korangath P, Teo WW, Sadik H, Han L, Mori N, Huijts CM, et al. Targeting Glutamine Metabolism in Breast Cancer with Aminooxyacetate. Clin Cancer Res 2015;21:3263–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res 2006;66:8927–30. [DOI] [PubMed] [Google Scholar]
- 31.Zhang S, Hulver MW, McMillan RP, Cline MA, Gilbert ER. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr Metab 2014;11:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gray LR, Tompkins SC, Taylor EB. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014;71:2577–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006;3:177–85. [DOI] [PubMed] [Google Scholar]
- 34.Lu J, Tan M, Cai Q. The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett 2015;356:156–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White E The role for autophagy in cancer. J Clin Invest 2015;125:42–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.White E, Mehnert JM, Chan CS. Autophagy, metabolism, and cancer. Clin Cancer Res 2015;21:5037–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov 2014;4:466–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011;25: 717–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol 2015;11:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bridges HR, Jones AJ, Pollak MN, Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J 2014;462:475–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rubio-Camarillo M, Gomez-Lopez G, Fernandez JM, Valencia A, Pisano DG. RUbioSeq: a suite of parallelized pipelines to automate exome variation and bisulfite-seq analyses. Bioinformatics 2013;29: 1687–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321:1801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 2009;324:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Villarroel MC, Rajeshkumar NV, Garrido-Laguna I, De Jesus-Acosta A, Jones S, Maitra A, et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol Cancer Ther 2011;10:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petrachi T, Romagnani A, Albini A, Longo C, Argenziano G, Grisendi G, et al. Therapeutic potential of the metabolic modulator phenformin in targeting the stem cell compartment in melanoma. Oncotarget 2017;8: 6914–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trousil S, Chen S, Mu C, Shaw FM, Yao Z, Ran Y, et al. Phenformin enhances the efficacy of ERK inhibition in NF1-mutant melanoma. J Invest Dermatol 2017;137:1135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan P, Ito K, Perez-Lorenzo R, Del Guzzo C, Lee JH, Shen CH, et al. Phenformin enhances the therapeutic benefit of BRAF(V600E) inhibition in melanoma. Proc Natl Acad Sci U S A 2013;110:18226–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bailey CJ, Turner RC. Metformin. N Engl J Med 1996;334:574–9. [DOI] [PubMed] [Google Scholar]
- 49.Pollak M Potential applications for biguanides in oncology. J Clin Invest 2013;123:3693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.