

Abstract
Fluorination of 13-epimeric estrones and their 17-deoxy counterparts was performed with Selectfluor as the reagent. In acetonitrile or trifluoroacetic acid (TFA), 10β-fluoroestra-1,4-dien-3-ones were formed exclusively. Mechanistic investigations suggest that fluorinations occurred via SET in acetonitrile, but another mechanism was operative in TFA. Simultaneous application of N-chlorosuccinimide (NCS) and Selectfluor in TFA led to a 1.3:1 mixture of 10β-fluoroestra-1,4-dien-3-one and 10β-chloroestra-1,4-dien-3-one as the main products. The potential inhibitory action of the 10-fluoro- or 10-chloroestra-1,4-dien-3-one products on human aromatase was investigated via in vitro radiosubstrate incubation. The classical estrane conformation with trans ring anellations and a 13β-methyl group seems to be crucial for the inhibition of the enzyme, while test compounds bearing the 13β-methyl group exclusively displayed potent inhibitory action with submicromolar or micromolar IC50 values. Concerning molecular level explanation of biological activity or inactivity, computational simulations were performed. Docking studies reinforced that besides the well-known Met374 H-bond connection, the stereocenter in the 13 position has an important role in the binding affinity. The configuration inversion at C-13 results in weaker binding of 13α-estrone derivatives to the aromatase enzyme.
Keywords: 13α-estrone, Selectfluor, aromatase, docking, TEMPO, single electron transfer
1. Introduction
Aromatase is responsible for the aromatization of androgens to estrogens [1]. The overproduction of estrogens stimulates the proliferation of estrogen-sensitive cells, leading to estrogen-dependent cancers. The proliferative action of estrogens might be prevented by inhibition of the aromatase enzyme [2]. Aromatase inhibitors can be categorized by their mechanism of action. Type I inhibitors are known as steroidal, and type II as nonsteroidal inhibitors [1]. The type I compounds are usually related to substrates of the enzyme and they are either competitive inhibitors or act as suicide inhibitors [3,4]. Formestane and Atamestane belong to the type I group. Formestane is considered to be the structural analog of androstenedione and it was the first member to enter clinical trials. Despite their high inhibitory potency, both agents have unfavorable metabolism and poor oral bioavailability. These disadvantages led to the discovery of a more potent compound, named Exemestane, acting both as a competitive and irreversible inhibitor. Additionally, it is efficient in metastatic breast cancer after the failure of selective estrogen receptor modulators (SERMs) [3]. This agent has substantial advantages over the nonsteroidal aromatase inhibitors. Thanks to the mentioned benefits of Exemestane, subsequent novel therapeutics were developed using a steroidal backbone as the scaffold. Nowadays, there is a high need for new aromatase inhibitory agents, because drawbacks as androgenic effect or metabolism by other CYP enzymes could still not be avoided.
Attempts have been made in recent decades to decrease the side-effects of the known inhibitors by performing minor structural modifications on the sterane skeleton [1]. Certain modifications were based on retaining the ring A dienone and androstane structure. One of the most explored groups of aromatase inhibitors is the 19-substituted androstane class. Marcotte and Robinson published work on C-19-modified androsta-4-en-3-one derivatives [5]. The 19-monofluoro and its difluoro counterpart displayed aromatase inhibitory activity with a Ki value of 1 µM. These literature data indicate that both the presence of an enone moiety in ring A and the fluorinated β-oriented angular 10-methyl group are advantageous structural elements concerning aromatase inhibition. Halogenation was also performed on the androsta-1,4-diene-3,17-dione skeleton [6]. Chlorine was introduced onto C-19, and the resulting compound displayed potent inhibitory activity with a 1-micromolar IC50 value. Not only ring A, but also ring D modifications have been performed. Sherwin et al. published the comparison of the inhibitory data of 17-oxo and the parent 17-deoxy androsta-1,4-dien-3-one compounds [7]. They concluded that the removal of the 17-oxo function causes only minor differences in the aromatase inhibitory potential of androsta-1,4-dien-3-ones.
Incorporation of fluorine into a biomolecule may lead to beneficial biological properties [8]. The C–F bond participates in attractive interactions with hydrogen bond donors, certain polar functional groups, and hydrophobic moieties. This is due to the large C–F bond polarization, which originates from the high electronegativity of fluorine. Fluorinated molecules usually have high binding affinity to certain proteins and increased metabolic stability. Recently, a class of stable and crystalline N–F fluorinating agents has been developed. 1-Chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) (Selectfluor (2), F-TEDA-BF4) belongs to the latter group and behaves as a selective fluorinating agent with high functional group tolerance [9,10,11,12]. Selectfluor is exceptionally stable and may serve as a fluoronium cation source. It is soluble in a few polar solvents, namely, acetonitrile, N,N-dimethylformamide (DMF), nitromethane and water. Literature data reveal that the type of fluorination of aromatic molecules with Selectfluor strongly depends on both the nature and the position of the aromatic ring substituents as well as on reaction conditions. Pravst et al. performed fluorinations of p-substituted phenols (1) using Selectfluor (2) as fluorinating agent in acetonitrile or methanol under different conditions (Scheme 1) [13]. The greatest solvent-dependent difference in product distributions was observed starting from 4-methylphenol (1a), where products 3a and 4a were formed in a ratio of nearly 2:1 in acetonitrile (reflux, 2 h), but in 3a:4a = 0:1 in methanol (reflux, 2 h). Concerning the largest substituent in the starting compound (1d), no dienone (3d) was formed, and the 4d:5d = 88:12 ratio was the same, independent of the solvent used.
Scheme 1.

Fluorination of p-substituted phenols (1) with Selectfluor (2).
According to the literature, 17β-estradiol (6), estrone (7) or their certain ring D-substituted derivatives can be converted to 10β-fluoroestra-1,4-dien-3-one analogues (8 and 9) with Selectfluor in different solvents (acetonitrile or bmimBF4:MeOH = 1:1, H2O) or under solvent-free conditions (Scheme 2) [14,15,16,17]. Independent of the nature of the solvent, all reactions resulted in dienones 8 and 9 as main products. ortho-substituted derivatives were observed only in trace amounts. Bogautdinov et al. reported the stereoselective fluorination of the 8α-epimer of 17β-estradiol (10) in the 1:1 mixture of bmimBF4 and methanol (Scheme 2) [14]. They proved that the reaction leads to 10-fluoro derivative 11 as the main product with α-orientation of fluorine. It was established that the configuration of C-8 influences the chirality of the newly formed C-10 stereogenic center.
Scheme 2.

Fluorination of estrone derivatives (6, 7 or 10) with Selectfluor (2).
Natural estrone derivatives (6 and 7) exhibit a relatively rigid molecular framework with well-defined distances between the two oxygen functionalities, which might be essential in the binding of the biomolecule to its receptors or enzymes. In contrast to natural 13β-estra-1,3,5(10)-trienes, 13α derivatives possess a cis-junction of the C and D rings, a quasi-equatorial 13α-methyl group and a ring D that is directed to the β-side (Figure 1) [18,19]. 13α-Estrone derivatives exist either in a usual conformation (with chair ring C) or in an unusual steroid conformation (with a twist-boat ring C). The conformational changes lead to a complete loss of estrogenic activity in estrone (7) or 17α/β-estradiols with inverted configuration at C-13 [20]. Thus, the inversion of certain chiral carbon atoms of the estrane core may lead to completely different biological behavior. Accordingly, 13α-estrone (12, Figure 1) may serve as a suitable scaffold for the design of biologically active estrane derivatives lacking estrogenic behavior.
Figure 1.

The structure of 13-epimeric estrones (7 and 12) and their 17-deoxy counterparts (13 and 14).
Here, we aimed to perform fluorination of hormonally inactive 13α-estrone 12 in order to obtain novel potential steroidal aromatase inhibitors bearing the 1,4-dien-3-one structural moiety in ring A. We aimed to examine the chemo- and stereoselectivity of the fluorination with Selectfluor (2) under various conditions. The investigation of the reaction mechanism was planned by adding a radical scavenger to the reaction mixture. Another substrate, 17-deoxy-13α-estrone 14, was also subjected to these transformations with the aim of investigating the influence of the lack of the 17-oxo group on the scope of the reactions. Comparative studies in the 13β- and 13α-estrone series (from starting compounds 7 and 12–14) have also been planned. The determination of the potential inhibitory action of the 10-fluorinated estra-1,4-dien-3-one products (9, 17, 20, and 21; Scheme 3) on human aromatase enzyme was planned via in vitro radiosubstrate incubation. Finally, having obtained a molecular-level insight into the binding properties of the 10-halo-13-epimeric estrane derivatives (9, 17, and 20–22) structure-activity information was collected and computational investigations were performed. These docking simulations helped to understand the more profound consequence of our chemical modifications concerning the ligand–receptor interaction.
Scheme 3.

Fluorinations of estrone (7) and 13α-estrone (12) with Selectfluor (2).
2. Results and Discussion
2.1. Chemistry
Fluorinations of 13-epimeric estrones (7 and 12) with Selectfluor (2) were performed in different solvents (Scheme 3). Based on literature results, two solvents were selected: acetonitrile and methanol [12]. Reactions were performed at room temperature or at 80 °C. First, chemo- and regioselectivities were studied in acetonitrile (Scheme 3, Table 1, Entries 1–3 and 7–9). Stirring of the reaction mixtures at room temperature led to 10β-fluorinated derivatives (9 and 17) solely, independent of the orientation of the angular methyl group (Table 1, Entries 1 and 7). Thus, 10β-fluoroestra-1,4-dien-3-ones (9 and 17) were formed in stereo- and chemoselective manners. Aromatic electrophilic substitutions at the ortho-positions (at C-2 or at C-4) did not occur. When fluorinations were carried out at 80 °C in acetonitrile, the same 10-fluoro products (9 or 17) were formed, but the reaction was complete within 1 h (Table 1, Entries 2 and 8). Our results are not consistent with those obtained for monosubstituted phenols [13], but they show good correspondence with chemoselectivities obtained earlier for fluorinations of estrone derivatives with Selectfluor [14,15,16,17]. In methanol, however, ortho-fluorinations also occurred (15–16%: 15:16 =1:0.9 or 18:19 =1:1.5) at both reaction temperatures (Table 1, Entries 4,5,10 and 11).
Table 1.
Fluorinations of estrone (7), 13α-estrone (12), 17-deoxyestrone (13) or 17-deoxy-13α-estrone (14) with Selectfluor (2).
| Entry | Substrate | Solvent | Temperature | Reaction Time | Product | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 7 | acetonitrile | rt | 24 h | 9 | 95 |
| 2 | 7 | acetonitrile | 80 °C | 1 h | 9 | 97 |
| 3 a | 7 | acetonitrile | rt | 24 h | 9 | 3 |
| 4 | 7 | methanol | rt | 24 h | 9 + (15 + 16) b | 76 + (16) |
| 5 | 7 | methanol | reflux | 1 h | 9 + (15 + 16) | 78 + (15) |
| 6 a | 7 | methanol | rt | 24 h | 9 | 2 |
| 7 | 12 | acetonitrile | rt | 24 h | 17 | 97 |
| 8 | 12 | acetonitrile | 80 °C | 1 h | 17 | 98 |
| 9 a | 12 | acetonitrile | rt | 24 h | 17 | 4 |
| 10 | 12 | methanol | rt | 24 h | 17 + (18 + 19) c | 71 + (12) |
| 11 | 12 | methanol | reflux | 1 h | 17 + (18 + 19) | 73 + (13) |
| 12 a | 12 | methanol | rt | 24 h | 17 | 94 |
| 13 | 13 | acetonitrile | rt | 24 h | 20 | 94 |
| 14 | 14 | acetonitrile | rt | 24 h | 21 | 92 |
a 2 equiv. of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO); b Ratio: 15:16 = 1:0.9; c Ratio: 18:19 = 1:1.5.
In order to investigate the influence of minor structural modifications of the steroidal scaffold on the outcome of fluorinations, not only the 17-oxo compound, but also their 17-deoxy counterparts (13 and 14) were subjected to fluorinations in acetonitrile using 2 as the reagent (Scheme 4, Table 1, Entries 13, 14). As expected, the reactions proceeded chemo- and stereoselectively, resulting in 10β-fluoro-17-deoxy-estra-1,4-dien-3-ones (20 and 21) in both the 13β- and 13α-estrone series. It can be stated that the presence or lack of the 17-oxo function does not affect the outcome of these fluorination reactions.
Scheme 4.

Fluorinations of 17-deoxyestrone (13) and 17-deoxy-13α-estrone (14) with Selectfluor (2).
According to the literature, the reaction mechanisms of fluorination reactions with Selectfluor (2) and other N–F fluorinating agents might be highly sensitive to the applied reaction conditions [21,22,23]. Different results have been observed with the same radical probe in different solvents. However, the ability of Selectfluor for homolytic cleavage of its N–F bond has recently been proved by Zhang et al., who detected the adduct of TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) and the fluorine radical by LC-MS and F-NMR [24]. Based on these literature results, we investigated the mechanism of fluorination in acetonitrile and methanol by adding a radical scavenger to the reaction mixture (Scheme 5, Table 1, Entries 3, 6, 9, 12). Addition of 2 equiv. of TEMPO resulted in almost complete inhibition of fluorination, and only a trace of the desired 10-fluoro derivative (9) was formed. This indicates that fluorination presumably occurred via SET. Based on literature evidence [21,24] and our results, we assume that the homolytic cleavage of the N–F bond results in a fluorine radical and cationic nitrogen radical 2A. The latter is protonated and intermediate 7R is formed. Subsequent spin delocalization results in intermediate 7RD. The driving force of this delocalization is the formation of a more stable tertiary radical. The attack of the fluorine radical on C-10 results in the desired 10-fluoro derivative 9.
Scheme 5.

Proposed mechanism of fluorination of estrone (7) with Selectfluor (2) in acetonitrile or methanol.
One substrate, namely 13β-estrone 7, was chosen for further derivatization. Chlorination of compound 7 was performed using N-chlorosuccinimide (NCS) as a reagent in acetonitrile and a catalytic amount of trifluoroacetic acid (TFA) (Scheme 6). Stirring the reaction mixture at rt for 24 h resulted in 2- and 4-chloro derivatives with retained aromaticity of ring A (23 and 24; Table 2, Entry 1). Heating the reaction mixture at 80 °C afforded the same product mixture with a much shorter reaction time of only 1 h (Table 2, Entry 2). Exchanging acetonitrile for TFA, ortho-chlorination was suppressed and 10-chloro dienone 22 became the main product (Table 2, Entries 3,4). The outcome of the reaction could not be influenced by adding TEMPO to the reaction mixture (Table 2, Entry 5). Based on the above-mentioned interesting results, model compound 7 was subjected to fluorination with Selectfluor, but acetonitrile used formerly was exchanged for TFA as the solvent. Fluorination occurred solely at C-10 (Table 2, Entries 6, 7). The formation of compound 9 could not be avoided by adding TEMPO to the reaction mixture (Table 2, Entry 8). In further experiments, the two halogenating agents (Selectfluor and NCS) were used together. In acetonitrile, fluorination occurred at C-10 together with ortho-chlorinated products 23 and 24 (Table 2, Entry 9). This ratio was retained by heating the reaction mixture at 80 °C for 1 h, but the starting compound was consumed earlier (Table 2, Entry 10). The two halogenating agents were also simultaneously used in TFA. In this process, the 10-fluoro and 10-chloro compounds (9 and 22) appeared to be the main products (Table 2, Entry 11). Heating shortened the reaction time, but it did not affect the product ratio (Table 2, Entry 12). TEMPO did not affect the outcome of this reaction either (Table 2, Entry 13).
Scheme 6.

Reaction of estrone (7) with Selectfluor (2) and/or NCS.
Table 2.
Effect of the reaction conditions on the fluorination and/or chlorination of compound 7.
| Entry | Substrate | NCS and or Selectfluor (1.1 equiv.) | Solvent | Temp. | Reaction Time | Yield Products 9 + 22 + 23 + 24 (%) |
|---|---|---|---|---|---|---|
| 1 a | 7 | NCS | acetonitrile | rt | 24 h | 0 + 0 + 30 + 45 |
| 2 a | 7 | NCS | acetonitrile | 80 °C | 1 h | 0 + 0 + 30 + 45 |
| 3 | 7 | NCS | TFA | rt | 24 h | 0 + 55 + 10 + 20 |
| 4 | 7 | NCS | TFA | 80 °C | 1 h | 0 + 55 + 10 + 20 |
| 5 b | 7 | NCS | TFA | 80 °C | 1 h | 0 + 54 + 11 + 21 |
| 6 | 7 | Selectfluor | TFA | rt | 24 h | 96 + 0 + 0 + 0 |
| 7 | 7 | Selectfluor | TFA | 80 °C | 1 h | 95 + 0 + 0 + 0 |
| 8 b | 7 | Selectfluor | TFA | rt | 24 h | 95 + 0 + 0 + 0 |
| 9 a | 7 | NCS, Selectfluor | acetonitrile | rt | 24 h | 62 + 0 + 15 + 20 |
| 10 a | 7 | NCS, Selectfluor | acetonitrile | 80 °C | 1 h | 62 + 0 + 15 + 20 |
| 11 | 7 | NCS, Selectfluor | TFA | rt | 24 h | 36 + 26 + 11 + 16 |
| 12 | 7 | NCS, Selectfluor | TFA | 80 °C | 1 h | 36 + 26 + 11 + 16 |
| 13 b | 7 | NCS, Selectfluor | TFA | 80 °C | 1 h | 36 + 25 + 11 + 17 |
a catalytic amount of TFA; b 2 equiv. of TEMPO.
The structures of the newly synthesized compounds (17, 20, and 21) were established through 1H- and 13C-NMR measurements. The configuration of the newly formed chiral center at C-10 in the 13α-epimer (17) was deduced from the comparison of the 1H-NMR spectra of the two 10-fluoro-13-epimeric compounds (9 [16] and 17). The multiplets of 1-H appeared with similar shapes and coupling constants in the two spectra, suggesting the same configuration (10β-F) of the new chiral center. It can be stated that owing to the long distance of the angular methyl group from ring A, the configuration of C-13 does not influence that of C-10.
2.2. Aromatase Inhibition Studies
Literature reveals that type I inhibitors for aromatase might be designed not only based on the substrate of the enzyme, but also on its product estrone (7) [1]. It was published that 2-halogenated (with F, Cl, and Br) estrone derivatives display high binding affinity to the enzyme with Ki values in the submicromolar or micromolar range [25]. The 17-Oxo analogs seemed to be more potent than the corresponding 17β-hydroxy compounds. It was stated that the presence of the 17-carbonyl function is necessary in the binding of estrogens to the active site of the aromatase enzyme [7,26,27,28]. The 4-halogenated derivatives proved to be less potent than their 2-substituted counterparts. We reported recently that 2-, 4- or 2,4-bis-halogenated (Cl, Br, I) 13α-estrones and their 17-deoxy derivatives possess weak aromatase inhibitory action [29]. It was demonstrated that the conformational differences of 13-epimeric estrones 7 and 12 resulting from the inversion of configuration at C-13 led to different binding affinities of 13-epimers to the aromatase. The inhibitory data obtained earlier for ring A halogenated 13β- and 13α-estrones indicate that the nature of the C-17 functional group, the conformation of the sterane core, and the substitution pattern of ring A might significantly influence the inhibitory behavior.
Here, we expected that the binding affinity of estrane-based potential inhibitors might be improved by transforming the aromatic ring A into the 1,4-dien-3-one moiety. This structural element relates to that of the substrate of the enzyme. There exist numerous literature reports on the development of substrate-like aromatase inhibitors bearing the androsta-1,4-dien-3-one structure [1]. However, only a few estra-1,4-dien-3-ones have been evaluated for their inhibitory properties [30,31,32,33]. Our idea was to develop type I potential aromatase inhibitors, which possess the key structural elements, such as the 1,4-dien-3-one in ring A and the 17-oxo function, but instead of the C-19 methyl group, a promising fluorine substituent. We expected that the small, but highly electronegative fluorine will markedly improve the binding properties of the compounds. These structural modifications on the 13α-estrane core could result in compounds acting selectively without estrogenic behavior. The investigation of in vitro aromatase inhibitory action of test and reference compounds was performed by a radiosubstrate incubation method established previously [29]. Our experiments reveal that the 13β-epimer (9) was highly potent with an IC50 value in the submicromolar range (Table 3). This compound exerted similar potency to reference compounds androst-4-ene-3,17-dione and androst-1,4-diene-3,17-dione. Unfortunately, the 13α-epimer (17) displayed only a very weak inhibitory action. Concerning the 17-deoxy compounds (20 and 21), the same tendency was observed, namely that only the 13β derivative (20) inhibited the aromatase enzyme. The difference of one order of magnitude in the IC50 values of 17-oxo (9) and its 17-deoxy counterpart (20) indicates that the presence of a 17-keto function is advantageous in binding of the inhibitor. The 13α-epimer of the 17-deoxy derivative (21) displayed similar affinity to the enzyme to that of its 17-oxo (17) counterpart. The 10-chloro-13β-derivative (22) proved to be a potent inhibitor with an IC50 value in the low micromolar range, however literature reveals estrogenic activity for compound 22 comparable to that of estrone [34]. Figure 2 shows the concentration-dependent inhibitory action of the potent compounds (9, 20 and 22) and that of the reference compound androst-1,4-diene-3,17-dione. The results obtained for 10-fluoro- and 10-chloro-13β compounds (9 and 22) suggest that introduction of a more electronegative but smaller halogen onto C-10 is more advantageous.
Table 3.
In vitro inhibition of aromatase activities by the test compounds and reference agents. Relative conversions (Rel. conv., control incubation with no inhibition is 100%) measured in the presence of 10 μM concentration of the compound tested. Mean ± SD, n = 3. IC50: inhibitor concentration decreasing the enzyme activity to 50%. SD: standard deviation.
| Compound | Structure | IC50 ± SD (µM) or Rel. conv. ± SD (%) |
|---|---|---|
| 9 |
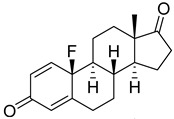
|
IC50 = 0.49 ± 0.07 |
| 20 |
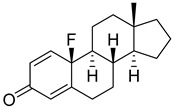
|
IC50 = 5.0 ± 2.4 |
| 17 |
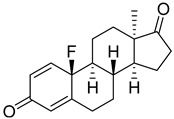
|
IC50 > 10 93 ± 11 |
| 21 |
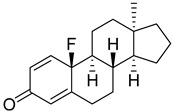
|
IC50 > 10 100 ± 6 |
| 22 |
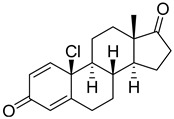
|
IC50 = 2.4 ± 0.4 |
| Androst-4-ene-3,17-dione |
|
IC50 = 0.22 ± 0.2 |
| Androst-1,4-diene-3,17-dione |
|
IC50 = 0.26 ± 0.06 |
Figure 2.

The concentration-dependent inhibitory action of the three potent compounds (9, 20 and 22) and that of the reference compound androst-1,4-diene-3,17-dione.
2.3. Molecular Docking Studies
Having obtained an atomic level explanation concerning biological activity, computational simulations were carried on for all 5 ligands (9, 17, 20, 21, and 22) and the original androst-1,4-diene-3,17-dione molecule. Docking studies were performed by the Glide program [35,36,37], where the concerned receptor model was based on X-ray crystal structure selected from the PDB database (pdb code:3S79, [38]). The accuracy of the chosen protocol was verified by a redocking calculation, where a ligand from the X-ray complex was taken away and redocked into the original binding pocket. The XP docking protocol could reproduce the original binding pose with 0.1893 Å RMSD accuracy, but according to the Glide-score value, it was ranked in second position. Further information about the fitted X-ray and redocked ligand structures is presented in the supporting information (see Figure S1). Considering the Emodel scoring values, we had proper binding geometry and the original crystal position was ranked in the top position. This is in line with the theoretical background of the two scoring functions [37], as the Glide score was developed for large and diverse molecules set to maximize separation of compounds with strong or weak binding affinity. On the other hand, the Emodel value was developed for comparing analogous conformers, and much less for comparing chemically distinct species. Taking into account that our ligand set consists of very similar molecules, ligand poses with the best Emodel values were selected as docking results in each case and the corresponding scoring values are summarized in Table 4.
Table 4.
The scoring values for compounds 9, 17, and 20–22.
| Compound | Emodel Score | Glide Score |
|---|---|---|
| 22 | −80.929 | −4.973 |
| 9 | −79.415 | −4.626 |
| 20 | −72.055 | −4.192 |
| 21 | −64.406 | −3.642 |
| 17 | −61.448 | −3.642 |
We can see that the Glide score could not separate characteristically active and inactive compounds, but the 13-epimer pairs (9 vs. 17 and 20 vs. 21) were always distinguished with proper binding preference order. Concerning the Emodel scores, the binding preference provided suitable separation of biologically active and inactive molecules as active molecules had better, i.e., more negative, scoring values. We would like to note that Androst-1,4-diene-3,17-done was also docked for reference reason, and it had −81.343 and −5.114 Emodel and Glide score values, respectively. This is in line with experiments, since this compound showed good biological activity (see Table 3).
Focusing now on the binding geometry of the ligands, we presented the docking pose of ligand 22 with the best Emodel score in Figure 3a. It represents clearly the possible interaction with the heme group with the Fe as well as the H-bond between ligand 22 and the Met374 residue. Other parts of the ligands fitted into the binding pocket did not provide any characteristic interactions (e.g., H-bond or π–π stacking) with the surrounding amino acids. Additionally, the poses of biologically active and inactive ligands are also presented in Figure 3b,c, respectively, where the original ligand androstenedione geometry in the X-ray structure was also represented by purple wire. It demonstrated clearly that in the non-active cases (compounds 17 and 21) the backbone had notable distortion, while biologically active molecules (9, 20 and 22) provided almost exactly the same sterane skeleton arrangement when compared to the original X-ray geometry. The distortion of the sterane skeleton in the non-active cases (compounds 17 and 21) was the consequence of inversion of C-13, which had an effect on the conformation of ring D as well. This certainly weakened the interaction between the 17-oxo function of ligand 22 and the Met374 amino acid, which was pointed out previously in the literature to be an important interaction [38]. However, we believe that this interaction is not the only important factor in the binding efficiency of the ligand, as in the case of compound 20, which was found to be biologically active with the 17-oxo function completely missing. Moreover, compound 20 performed better concerning both biological activity and chemical binding than compound 17, which bears the 17-oxo group.
Figure 3.

(a) The binding pose of ligand 22 with the best Emodel score. Distances are presented in Å. (b) The best binding pose of the biologically active ligands (9, 20, and 22) with the original X-ray ligand (molecule with purple line) pose. (c) The best binding pose of the biologically inactive ligands (17 and 21) with the original X-ray ligand (molecule with purple line) pose.
It is worth mentioning that the docking calculations of 17-deoxy compound 21 provided a geometry where the orientation of the sterane skeleton was turned around, and the oxygen on ring A tried to play the role of the ring D oxygen in the interaction with the Met379. In this case, however, neither the Glide score nor the Emodel score indicated strong interaction.
From a theoretical point of view, these results put into focus two, not necessarily exclusive, explanations. On the one hand, there should be an interaction between the heme and the methyl groups and/or halogen groups in positions 13 and 10. This interaction, however, is weakened by the inversion at C-13. On the other hand, the inversion changed the hydrophobic interaction of the ligand in the binding pocket as well, which could also lead to a weaker ligand binding. Moreover, these effects obviously overrule the loss of the interaction with the Met379 residue. This became clear when we compared the scoring of ligand 20 to the results of ligand 17.
3. Materials and Methods
3.1. Chemical Synthesis
3.1.1. General
Melting points (Mp) were determined with a Kofler hot-stage apparatus and are uncorrected. Elemental analyses were performed with a Perkin-Elmer CHN analyzer model 2400. Thin-layer chromatography: silica gel 60 F254; layer thickness 0.2 mm (Merck, Darmstadt, Germany); eluents: a: 40% ethyl acetate/60% hexanes, b: 25% ethyl acetate/75% hexanes, detection with I2 or UV (365 nm) after spraying with 5 % phosphomolybdic acid in 50 % aqueous phosphoric acid and heating at 100–120 °C for 10 min. Flash chromatography: silica gel 60, 40–63 μm (Merck). 1H-NMR spectra were recorded in DMSO-d6, CDCl3 solution with a Bruker DRX-500 instrument at 500 MHz, with Me4Si as internal standard. 13C-NMR spectra were recorded with the same instrument at 125 MHz under the same conditions. Mass spectrometry: Full scan mass spectra of the compounds were acquired in the range of 50 to 1000 m/z with a Finnigan TSQ-7000 triple quadrupole mass spectrometer (Finnigan-MAT, San Jose, CA, USA) equipped with a equipped with a Finnigan electrospray ionization source. Analyses were performed in positive ion mode using flow injection mass spectrometry with a mobile phase of 50 % aqueous acetonitrile containing 0.1 v/v % formic acid. The flow rate was 0.3 mL/min. Five µl aliquot of the samples were loaded into the flow. The ESI capillary was adjusted to 4.5 kV and N2 was used as a nebulizer gas.
3.1.2. Synthesis of 10β-Fluoroestra-1,4-dien-3-one (9) or 10β-Fluoro-13α-estra-1,4-dien-3-one (17) in acetonitrile
Estrone (7) (135 mg, 0.5 mmol) or 13α-estrone (12) (135 mg, 0.5 mmol) was dissolved in acetonitrile (5 mL) and Selectfluor (2) (195 mg, 0.55 mmol) was added. The mixture was stirred at rt for 24 h or at 80 °C for 1 h, the solvent was then evaporated off, and the crude product (9 or 17) was purified by flash chromatography with 2% ethyl acetate/98% dichloromethane as eluent.
Compound 9 was obtained as a white solid (137 mg, 95% or 140 mg, 97%, Mp.: 104–102 °C, Rf = 0.42a). Compound 9 is identical with compound described in the literature [15]. 1H-NMR (DMSO-d6) δ ppm 0.86 (s, 3H, 18-H3); 2.41 and 2.53 (2 × m, 2 × 1H, 6-H2); 6.10 (s, 1H, 4-H); 6.22 (d, 1H, J = 10.2 Hz, 2-H); 7.27 (dd, 1H, J = 10.2 Hz, J = 7.4 Hz, 1-H).
Compound 17 was obtained as a white solid (140 mg, 97% or 141 mg, 98%, Mp.: 142–144 °C, Rf = 0.23b). Anal. Calcd. for C18H21FO2: C, 74.97; H, 7.34. Found: C, 74.85; H, 7.39. 1H-NMR (CDCl3) δ ppm 0.99 (s, 3H, 18-H3); 1.14–2.68 (15H); 6.04 (s, 1H, 4-H); 6.22 (d, 1H, J=10.2 Hz, 2-H); 7.06 (dd, 1H, J = 10.2 Hz, J = 7.7 Hz, 1-H). 13C-NMR (CDCl3) δ ppm 21.6; 23.6; 24.9 (C-18); 31.1; 31.5; 33.4; 34.0; 37.4; 49.1; 49.8 (C-13); 51.7 (d, J = 24.0 Hz, C-9); 88.9 (d, J = 167.9 Hz, C-10); 123.7 (d, J = 5.0 Hz, C-4); 129.7 (d, J = 8.7 Hz, C-2); 144.7 (d, J = 23.8 Hz, C-1); 159.8 (d, J = 18.9 Hz, C-5); 184.8 (C-3); 220.7 (C-17). MS m/z (%): 289 (100, [M + H]+).
3.1.3. Synthesis of 10β-Fluoroestra-1,4-dien-3-one (9) or 10β-Fluoro-13α-estra-1,4-dien-3-one (17) in methanol
Estrone (7) (135 mg, 0.5 mmol) or 13α-estrone (12) (135 mg, 0.5 mmol) was dissolved in methanol (5 mL) and Selectfluor (2) (195 mg, 0.55 mmol) was added. The mixture was stirred at rt for 24 h or at 80 °C for 1 h, the solvent was then evaporated off, and the crude product (9 or 17) was purified by flash chromatography with 2% ethyl acetate/98% dichloromethane as eluent.
Starting from compound 7, first eluted the mixture of 15:16 = 1:1.5 and was obtained as an oil (23 mg, 16% or 22 mg, 15%). Then eluted compound 9 and was obtained as a white solid (110 mg, 76% or 112 mg, 78%). Compounds 15 and 16 have not been separated. The relevant signals selected from the 1H-NMR spectrum of the mixture for compound 16 (DMSO-d6) δ ppm: 0.82 (s, 3H, 18-H3); 6.71 (t, 1H, J = 8.8 Hz, 2-H); 6.88 (d, 1H, J = 8.8 Hz, 1-H); 9.43 (s, 1H, OH). The relevant signals selected from the 1H-NMR spectrum of the mixture for compound 15 (DMSO-d6) δ ppm: 0.82 (s, 3H, 18-H3); 6.61 (d, 1H, J = 9.3 Hz, 4-H); 6.97 (d, 1H, J = 13.2 Hz, 1-H); 9.47 (s, 1H, OH). Then eluted compound 9 and was obtained as a white solid.
Starting from compound 12, first eluted the mixture of 18:19 = 1:1.5 and was obtained as an oil (17 mg, 12% or 19 mg, 13%). Compounds 18 and 19 have not been separated. The relevant signals selected from the 1H-NMR spectrum of the mixture for compound 18 (DMSO-d6) δ ppm: 0.96 (s, 3H, 18-H3); 6.58 (d, 1H, J = 9.2 Hz, 4-H); 6.97 (d, 1H, J = 13.9 Hz, 1-H); 9.47 (s, 1H, OH). The relevant signals selected from the 1H-NMR spectrum of the mixture for compound 19 (DMSO-d6) δ ppm: 0.96 (s, 3H, 18-H3); 6.69 (t, 1H, J = 8.8 Hz, 2-H); 6.88 (d, 1H, J = 8.8 Hz, 1-H); 9.42 (s, 1H, OH). Then eluted compound 17 and was obtained as a white solid (102 mg, 71% or 105 mg, 73%).
3.1.4. General Procedure for the Fluorination with Selectfluor in Acetonitrile or in Methanol by Adding TEMPO
Estrone (7) (135 mg, 0.5 mmol) or 13α-estrone (12) (135 mg, 0.5 mmol) was dissolved in acetonitrile (5 mL) or in methanol (5 mL), Selectfluor (2) (195 mg, 0.55 mmol) and TEMPO (156 mg, 1.0 mmol) were added. The mixture was stirred at rt for 24 h, and the reaction was monitored by TLCa. The crude products were purified by flash chromatography with 2% ethyl acetate/98% dichloromethane as eluent. Compound 9 or 17 was obtained as a white solid only in traces (2–4%).
3.1.5. Synthesis of 10β-Fluoro-17-deoxyestra-1,4-dien-3-one (20) or 10β-Fluoro-17-deoxy-13α-estra-1,4-dien-3-one (21)
17-Deoxy-estrone (13) (128 mg, 0.50 mmol) or 17-deoxy-13α-estrone (14) (128 mg, 0.50 mmol) was dissolved in acetonitrile (5 mL) and Selectfluor (2) (195 mg, 0.55 mmol) was added. The mixture was stirred at rt for 24 h, the solvent was then evaporated off, and the crude product was purified by flash chromatography with 5% ethyl acetate/95% hexane.
Compound 20 was obtained as an oil (129 mg, 94%, Rf = 0.72b). Anal. Calcd. for C18H23FO: C, 78.80; H, 8.45. Found: C, 78.71; H, 8.37. 1H-NMR (CDCl3) δ ppm 0.80 (s, 3H, 18-H3); 2.41 and 2.67 (2 × m, 2 × 1H, 6-H2); 6.03 (s, 1H, 4-H); 6.23 (d, 1H, J = 9.8 Hz, 2-H); 7.10 (t, 1H, J = 9.8 Hz, 1-H). 13C-NMR (CDCl3) δ ppm 17.3 (C-18); 20.3; 23.1; 25.7; 32.0; 33.5; 36.1; 38.1; 40.1; 40.9 (C-13); 53.2; 54.5 (d, J = 24.8 Hz, C-9); 88.9 (d, J = 167.8 Hz, C-10); 123.4 (d, J = 4.6 Hz); 129.2 (d, J = 8.5 Hz); 145.7 (d, J = 14.5 Hz, C-1); 161.0 (d, J = 18.6 Hz, C-5); 185.3 (C-3). MS m/z (%): 275 (100, [M + H]+).
Compound 21 was obtained as an oil (126 mg, 92%, Rf = 0.67b). Anal. Calcd. for C18H23FO: C, 78.80; H, 8.45. Found: C, 78.73; H, 8.35. 1H-NMR (CDCl3) δ ppm 0.89 (s, 3H, 18-H3); 2.39 and 2.63 (2 × m, 2 × 1H, 6-H2); 6.02 (s, 1H, 4-H); 6.22 (d, 1H, J = 10.3 Hz, 2-H); 7.11 (dd, 1H, J = 10.3 Hz, J = 7.5 Hz, 1-H). 13C-NMR (CDCl3) δ ppm 21.0; 22.2; 28.7; 29.7 (C-18); 31.9; 33.6; 34.3; 34.8; 37.3; 41.6 (C-13); 51.9; 52.6 (d, J = 24.1 Hz, C-9); 88.9 (d, J = 161.1 Hz, C-10); 123.4 (d, J = 4.7 Hz, C-4); 129.5 (d, J = 8.6 Hz, C-2); 145.2 (d, J = 14.4 Hz, C-1); 160.8 (d, J = 19.2 Hz, C-5); 185.1 (C-3). MS m/z (%): 275 (100, [M + H]+).
3.1.6. Fluorination of estrone (7) with Selectfluor in TFA
Estrone (7) (135 mg, 0.5 mmol) was dissolved in TFA (3 mL) and Selectfluor (2) (195 mg, 0.55 mmol) was added. The mixture was stirred at rt for 24 h or at 80 °C for 1 h, then cooled to rt, poured onto 100 mL water and extracted with diethyl ether. The organic phase was dried over anhydrous sodium sulfate, filtered and evaporated. The crude product was purified by flash chromatography with 2% ethyl acetate/98% dichloromethane as eluent. Compound 9 was obtained as a white solid (138 mg, 96% or 137 mg, 95%).
3.1.7. Fluorination of Estrone (7) with Selectfluor in TFA by Adding TEMPO
Estrone (7) (135 mg, 0.5 mmol) was dissolved in TFA (3 mL), Selectfluor (2) (195 mg, 0.55 mmol) and TEMPO (156 mg, 1.0 mmol) were added. The mixture was stirred at rt for 24 h, then poured onto 100 mL water and extracted with diethyl ether. The organic phase was dried over anhydrous sodium sulfate, filtered and evaporated. The crude product was purified by flash chromatography with 2% ethyl acetate/98% dichloromethane as eluent. Compound 9 was obtained as a white solid (137 mg, 95%).
3.1.8. Chlorination of Estrone (7) with NCS in Acetonitrile
Estrone (7) (135 mg, 0.5 mmol) was dissolved in acetonitrile (5 mL), trifluoroacetic acid (0.005 mL) and NCS (74 mg, 0.55 mmol) were added. The mixture was stirred at rt for 24 h or at 80 °C for 1 h, the solvent was then evaporated off, and the crude product was purified by flash chromatography with 10% ethyl acetate/90% hexane. First eluted compound 24 (37 mg, 24%), which was obtained as a white solid. Mp.: 181–183 °C, [34]: 272–274 °C, Rf: 0.46a. Continued elution yielded the mixture of compounds 24 (32 mg, 21%) and 23 (26 mg, 17%). Finally eluted 23 (20 mg, 13%), which was obtained as a white solid. Mp.: 203–205 °C, [35]: 262–264 °C, Rf: 0.42a. Compounds 23 and 24 are identical with those described in [28].
3.1.9. Chlorination of Estrone (7) with NCS in TFA
Estrone (7) (135 mg, 0.5 mmol) was dissolved in TFA (3 mL) and NCS (74 mg, 0.55 mmol) was added. The mixture was stirred at rt for 24 h or at 80 °C for 1 h, and then cooled to rt, poured onto 100 mL water and extracted with diethyl ether. The organic phase was dried over anhydrous sodium sulfate, filtered and evaporated. The crude product was purified by flash chromatography with 10% ethyl acetate/90% hexane. First eluted 24 (20 mg, 13%). Continued elution yielded the mixture of 24 (11 mg, 7%) and 23 (9 mg, 5.6%). Then eluted 23 (7 mg, 4.4%). Finally eluted 22 (84 mg, 55%).
Compound 22 was obtained as a white solid. 1H-NMR (CDCl3) δ ppm 0.98 (s, 3H, 18-H3); 6.09 (s, 1H, 4-H); 6.20 (d, 1H, J = 10.1 Hz, 2-H); 7.12 (d, 1H, J = 10.1 Hz, 1-H). 13C-NMR (CDCl3) δ ppm 13.7 (C-18); 21.9; 22.4; 30.6; 31.4; 32.0; 35.5; 35.6; 47.6 (C-13); 49.7; 53.2; 67.5 (C-10); 124.1; 126.9; 147.4 (C-1); 160.4 (C-5); 184.9 (C-3); 219.7 (C-17). Compound 22 is identical with compound described in the literature [34].
3.1.10. Chlorination of estrone (7) with NCS in TFA by adding TEMPO
Estrone (7) (135 mg, 0.5 mmol) was dissolved in TFA (3 mL), NCS (74 mg, 0.55 mmol) and TEMPO (156 mg, 1.0 mmol) were added. The mixture was stirred at 80 °C for 1 h, and then cooled to rt, poured onto 100 mL water and extracted with diethyl ether. The organic phase was dried over anhydrous sodium sulfate, filtered and evaporated. The crude product was purified by flash chromatography with 10% ethyl acetate/90% hexane. First eluted 24 (21 mg, 14%). Continued elution yielded the mixture of 24 (11 mg, 7%) and 23 (10 mg, 7.0%). Then eluted 23 (6 mg, 4.0%). Finally eluted 22 (82 mg, 54%).
3.1.11. Fluorination and Chlorination of Estrone (7) with Selectfluor and NCS in Acetonitrile
Estrone (7) (135 mg, 0.5 mmol) was dissolved in acetonitrile (5 mL), Selectfluor (2) (195 mg, 0.55 mmol), NCS (74 mg, 0.55 mmol) and trifluoroacetic acid (0.005 mL) were added. The mixture was stirred at rt for 24 h or at 80 °C for 1 h, the solvent was then evaporated off, and the crude product was purified by flash chromatography with 10% ethyl acetate/90% hexane. First eluted 24 (15 mg, 9.8%). Continued elution yielded the mixture of 24 (14 mg, 9.2%) and 23 (10 mg, 6.6%). Then eluted 23 (12 mg, 8.4%). Finally eluted 9 (94 mg, 62%).
3.1.12. Fluorination and Chlorination of Estrone (7) with Selectfluor and NCS in TFA
Estrone (7) (135 mg, 0.5 mmol) was dissolved in TFA (3 mL), Selectfluor (2) (195 mg, 0.55 mmol) and NCS (74 mg, 0.55 mmol) were added. The mixture was stirred at 80 °C for 1 h, and then cooled to rt, poured onto 100 mL water and extracted with diethyl ether. The organic phase was dried over anhydrous sodium sulfate, filtered and evaporated. The crude product was purified by flash chromatography with 10% ethyl acetate/90% hexane. First eluted 24 (14 mg, 9.2%). Continued elution yielded the mixture of 24 (10 mg, 6.8%) and 23 (9 mg, 5.9%). Then eluted 23 (8 mg, 5.1%). Finally eluted separately 22 (40 mg, 26%) 9 (55 mg, 36%).
3.1.13. Fluorination and Chlorination of Estrone (7) with Selectfluor and NCS in TFA by Adding TEMPO
Estrone (7) (135 mg, 0.5 mmol) was dissolved in TFA (3 mL), Selectfluor (2) (195 mg, 0.55 mmol), NCS (74 mg, 0.55 mmol) and TEMPO (156 mg, 1.0 mmol) were added. The mixture was stirred at 80 °C for 1 h, and then cooled to rt, poured onto 100 mL water and extracted with diethyl ether. The organic phase was dried over anhydrous sodium sulfate, filtered and evaporated. The crude product was purified by flash chromatography as it is described in Section 3.1.11.
3.2. Aromatase Inhibition
3.2.1. General
[4(N)-14C]Testosterone was obtained from American Radiolabeled Chemicals, St. Louis, MO, USA). Chemicals and solvents of analytical grade purity were purchased from Sigma (St. Louis, MO, USA), from Fluka (Buchs, Switzerland) or from Merck (Darmstadt, Germany).
3.2.2. Preparation of Enzyme Sources
Human term placentae were collected immediately after delivery and stored frozen at −80 °C. Tissue specimens were homogenized with an Ultra-Turrax in 0.1 M HEPES buffer solution (pH = 7.3) containing 1 mM EDTA and 1 mM dithiotreitol, and microsomas were obtained with fractionated centrifugation. Application of the human tissue was approved by the institutional Human Investigation Review Board.
3.2.3. Incubation Procedures
The microsoma suspension was incubated with 1.0 μM [4(N)-14C]testosterone substrate in the presence of 0.1 mM NADPH cofactor excess. Enzymatic incubations were carried out in the HEPES buffer medium at a final volume of 200 µL. The substrate was added to the incubate in 20 μL of a 25 v/v% propylene glycol in HEPES buffer solution, whereas test compounds were applied in 10 μL of dimethyl sulfoxide solution. Incubations were performed at 37 °C and lasted for 40 min. Enzymatic reaction was terminated by cooling and the addition of organic solvents of the subsequent extraction procedure. Control samples with no inhibitor and blank samples were incubated simultaneously. The incubation mixture was extracted with toluene, then the toluene phase was drained and washed with HEPES buffer. Aromatase products containing phenolic hydroxy group were extracted with 1.2 M sodium hydroxide solution from the toluene extract and radioactivity of the alkaline phase was measured in liquid scintillation counting.
3.2.4. Inhibition Studies
In the general procedure, test compounds were applied at 10 μM final concentration in the incubate. Relative conversions compared to non-inhibited controls (100%) were determined. The assays were performed in triplicate, and the mean value and the standard deviation (SD) were calculated. IC50 values (the inhibitor concentration that decreases the enzyme activity to 50%) were determined for the most effective test compounds and for reference compounds. In these cases, conversions were measured at 10–15 different concentrations in the appropriate interval between 0.001–50 μM. IC50 results were calculated by using unweighted iterative least squares logistic curve fitting by means of the “absolute IC50 calculation“ function of the GraphPad Prism 4.0 software (GraphPad Software, Inc., San Diego, CA, USA).
3.3. Computational Simulations
Docking studies were performed with the Glide [33,34,35] program from the Schrodinger suits using the XP protocol. Docking grid generation was based on X-ray crystal structure as it was deposited into the Protein Database (pdb code: 3S79) and refined by the Protein Preparation Wizard. During the XP docking calculations, enhanced sampling was selected and the energy window for ring sampling was also increased to 100 kcal/mol and the number of final outputs per ligand was enlarged to 10. All figures were prepared with the Maestro program [35] which is the GUI part of the Schrödinger program package.
4. Conclusions
In conclusion, fluorinations of 13-epimeric estrones (7 and 12) and their 17-deoxy counterparts (13 and 14) with Selectfluor (2) in acetonitrile furnished exclusively the corresponding 10-fluoro derivatives (9, 17, 20, and 21). Mechanistic studies suggest that reactions in acetonitrile occur via SET, while halogenations in TFA follow a different mechanism. The simultaneous application of the two halogenating agents (Selectfluor and NCS) in TFA results in 10-halogenated compounds 9 and 22 as the main products at a ratio of about 1.3:1.0. The results obtained from the aromatase assay suggest that in estrane-based aromatase inhibitors, the presence of the small, β-oriented halogen at C-10 and the 1,4-diene-3,17-dione moiety are advantageous and might lead to potent derivatives with submicromolar inhibitory potential. Docking calculations reinforced that besides the well-known Met374 H-bond connection, the stereocenter in the 13 position has an important role in the binding affinity. Our results might contribute not only to the research field of type I steroidal inhibitors of aromatase, but also to the development of biologically active compounds bearing substituted phenol moieties, by improving their biological potency via halogenations.
Acknowledgments
The work of Erzsébet Mernyák and Csilla Özvegy-Laczka was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences. The work of Erzsébet Mernyák in this project was supported by the ÚNKP-18-4-SZTE-45 „NEW NATIONAL EXCELLENCE PROGRAM OF THE MINISTRY OF HUMAN CAPACITIES”.
Supplementary Materials
The following are available online at https://www.mdpi.com/1420-3049/24/9/1783/s1: Synthetic procedures, characterization data for the synthesized compounds, aromatase inhibition method and computational simulation methods.
Author Contributions
R.J., P.T., É.K. and Á.H. peformed the experiments; E.M. and M.S. contributed reagents, materials and analysis tools; E.M. and M.S. conceived and designed the experiments; G.P. designed and performed the docking calculations; E.M., G.P., G.S., M.S. and P.T. analyzed the data; A.P. performed the MS experiments; E.M., R.J. and G.P. wrote the paper.
Funding
This research was funded by Hungarian Scientific Research Fund OTKA SNN124329.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds are not available from the authors.
References
- 1.Yadav M.R., Barmade M.A., Tamboli R.S., Murumkar P.R. Developing steroidal aromatase inhibitors-an effective armament to win the battle against breast cancer. Eur. J. Med. Chem. 2015;105:1–38. doi: 10.1016/j.ejmech.2015.09.038. [DOI] [PubMed] [Google Scholar]
- 2.Miller W.L., Auchus R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011;32:81–151. doi: 10.1210/er.2010-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brueggemeier R.W. Update on the use of aromatase inhibitors in breast cancer. Expert Opin. Pharmacother. 2006;7:1919–1930. doi: 10.1517/14656566.7.14.1919. [DOI] [PubMed] [Google Scholar]
- 4.Lonning P.E. Clinico-pharmacological aspects of different hormone treatments. Eur. J. Cancer. 2000;36:81–82. doi: 10.1016/S0959-8049(00)00237-9. [DOI] [PubMed] [Google Scholar]
- 5.Marcotte P.A., Robinson C.H. Synthesis and evaluation of 10β-substituted 4-estrene-3,17-diones as inhibitors of human placental microsomal aromatase. Steroids. 1982;39:325–344. doi: 10.1016/0039-128X(82)90151-9. [DOI] [PubMed] [Google Scholar]
- 6.Numazawa M., Nagaoka M., Handa W., Ogawa Y., Matsuoka S. Studies directed towards a mechanistic evaluation of inactivation of aromatase by the suicide substrates androsta-1,4-diene-3,17-diones and its 6-ene derivatives Aromatase inactivation by the 19-substituted derivatives and their enzymic aromatization. J. Steroid Biochem. Mol. Biol. 2007;107:211–219. doi: 10.1016/j.jsbmb.2007.03.042. [DOI] [PubMed] [Google Scholar]
- 7.Sherwin P.F., McMullan P.C., Covey D.F. Effects of steroid d-ring modification on suicide inactivation and competitive inhibition of aromatase by analogues of androsta-1,4-diene-3,17-dione. J. Med. Chem. 1989;32:651–658. doi: 10.1021/jm00123a026. [DOI] [PubMed] [Google Scholar]
- 8.Liang T., Neumann C.N., Ritter T. Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed. 2013;52:8214–8264. doi: 10.1002/anie.201206566. [DOI] [PubMed] [Google Scholar]
- 9.Smart B.E. Fluorine substituent effects (on bioactivity) J. Fluorine Chem. 2001;109:3–11. doi: 10.1016/S0022-1139(01)00375-X. [DOI] [Google Scholar]
- 10.Leo A., Hansch C., Elkins D. Partition coefficients and their uses. Chem. Rev. 1971;71:525–616. doi: 10.1021/cr60274a001. [DOI] [Google Scholar]
- 11.Hansch C., Leo A., Unger S.H., Kim K.H., Nikaitan D., Lien E.J. “Aromatic” substituent constants for structure-activity correlations. J. Med. Chem. 1973;16:1207–1216. doi: 10.1021/jm00269a003. [DOI] [PubMed] [Google Scholar]
- 12.Hansch C., Leo A., Taft R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991;91:165–195. doi: 10.1021/cr00002a004. [DOI] [Google Scholar]
- 13.Pravst I., Papez Iskra M., Jereb M., Zupan M., Stavber S. The role of F–N reagent and reaction conditions on fluorofunctionalisation of substituted phenols. Tetrahedron. 2006;62:4474–4481. doi: 10.1016/j.tet.2006.02.051. [DOI] [Google Scholar]
- 14.Bogautdinov R.P., Fidarov A.F., Morozkina S.N., Zolotarev A.A., Starova G.L., Selivanov S.I., Shavva A.G. Fluorination of steroid estrogens with Selectfluor: Elucidation of regio- and stereoselectivity. J. Fluorine Chem. 2014;168:218–222. doi: 10.1016/j.jfluchem.2014.09.030. [DOI] [Google Scholar]
- 15.Zaikin P.A., Dyan O.T., Evtushok D.V., Usoltsev A.N., Borodkin G.I., Karpova E.V., Shubin V.G. Solvent-Free Fluorination of Electron-Rich Aromatic Compounds with F-TEDA-BF4: Toward “Dry” Processes. Eur. J. Org. Chem. 2017;17:2469–2474. doi: 10.1002/ejoc.201700179. [DOI] [Google Scholar]
- 16.Stavber G., Zupan M., Jereb M., Stavber S. Selective and Effective Fluorination of Organic Compounds in Water using SelectfluorTM F-TEDA-BF4. Org. Lett. 2004;6:4973–4976. doi: 10.1021/ol047867c. [DOI] [PubMed] [Google Scholar]
- 17.Stavber S., Jereb M., Zupan M. Efficient Synthesis of 4-Fluorocyclohexa-2,5-dienone Derivatives Using N-Fluoro-1,4-diazoniabicyclo[2.2.2]octane Salt Analogues. Synlett. 1999;9:1375–1378. doi: 10.1055/s-1999-2840. [DOI] [Google Scholar]
- 18.Butenandt A., Wolff A., Karlson P. Über Lumi-oestron. Chem. Ber. 1941;74:1308–1312. doi: 10.1002/cber.19410740724. [DOI] [Google Scholar]
- 19.Schönecker B., Lange C., Kötteritzsch M., Günther W., Weston J., Anders E., Görls H. Conformational design for 13α-steroids. J. Org. Chem. 2000;65:5487–5497. doi: 10.1021/jo000108x. [DOI] [PubMed] [Google Scholar]
- 20.Roy A.J., Maltais R., Poirier D. Impact of estradiol structural modifications (18-methyl and/or 17-hydroxy inversion of configuration) on the in vitro and in vivo estrogenic activity. J. Steroid Biochem. Mol. Biol. 2011;127:324–330. doi: 10.1016/j.jsbmb.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 21.Vincent S.P., Burkart M.D., Tsai C.Y., Zhang Z., Wong C.H. Electrophilic Fluorination−Nucleophilic Addition Reaction Mediated by Selectfluor: Mechanistic Studies and New Applications. J. Org. Chem. 1999;64:5264–5279. doi: 10.1021/jo990686h. [DOI] [PubMed] [Google Scholar]
- 22.Differding E., Rüegg G. Nucleophilic substitution versus electron transfer: 1. On the mechanism of electrophilic fluorinations. Tetrahedron Lett. 1991;32:3815–3818. doi: 10.1016/S0040-4039(00)79383-X. [DOI] [Google Scholar]
- 23.Ashby E.C., Pham T.N. The question of the validity of using radical probes for determining SET. The reaction of alkyl halides with LiAlH4. Tetrahedron Lett. 1987;28:3197–3200. doi: 10.1016/S0040-4039(00)95470-4. [DOI] [Google Scholar]
- 24.Zhang Y., Wen C., Li J. C5-Regioselective C–H fluorination of 8-aminoquinoline amides and sulfonamides with Selectfluor under metal-free conditions. Org. Biomol. Chem. 2018;16:1912–1920. doi: 10.1039/C7OB03059B. [DOI] [PubMed] [Google Scholar]
- 25.Numazawa M., Ando M., Watari Y., Tominaga T., Hayata Y., Yoshimura A. Structure-activity relationships of 2-, 4-, or 6-substituted estrogens as aromatase inhibitors. J. Steroid Biochem. Mol. Biol. 2005;96:51–58. doi: 10.1016/j.jsbmb.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 26.Kellis J.T., Jr., Vickery L.E. Purification and characterization of human placental aromatase cytochrome P-450. J. Biol. Chem. 1987;262:4413–4420. [PubMed] [Google Scholar]
- 27.Yoshida N., Osawa Y. Purification of human placental aromatase cytochrome P-450 with monoclonal antibody and its characterization. Biochemistry. 1991;30:3003–3010. doi: 10.1021/bi00226a004. [DOI] [PubMed] [Google Scholar]
- 28.Numazawa M., Kamiyama T., Tachibana M., Oshibe M. Synthesis and structure−activity relationships of 6-substituted androst-4-ene analogs as aromatase inhibitors. J. Med. Chem. 1996;39:2245–2252. doi: 10.1021/jm960047o. [DOI] [PubMed] [Google Scholar]
- 29.Bacsa I., Herman B.E., Jójárt R., Herman K.S., Wölfling J., Schneider G., Varga M., Tömböly C., Lanišnik Rižner T., Szécsi M., et al. Synthesis and structure–activity relationships of 2- and/or 4-halogenated 13β- and 13α-estrone derivatives as enzyme inhibitors of estrogen biosynthesis. J. Enzyme Inhib. Med. Chem. 2018;33:1271–1282. doi: 10.1080/14756366.2018.1490731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flynn G.A., Johnston J.O., Wright C.L., Metcalf B.W. The time-dependent inactivation of aromatase by 17beta-hydroxy-10-methylthioestra-1,4-dien-3-one. Biochem. Biophys. Res. Commun. 1998;103:913–918. doi: 10.1016/0006-291X(81)90897-4. [DOI] [PubMed] [Google Scholar]
- 31.Lovett J.A., Darby M.V., Counsell R.E. Synthesis and evaluation of 19-aza- and 19-aminoandrostenedione analogues as potential aromatase inhibitors. J. Med. Chem. 1984;27:734–740. doi: 10.1021/jm00372a005. [DOI] [PubMed] [Google Scholar]
- 32.Bednarski P.J., Porubek D.J., Nelson S.D. Thiol-containing androgens as suicide substrates of aromatase. J. Med. Chem. 1985;28:775–779. doi: 10.1021/jm00383a014. [DOI] [PubMed] [Google Scholar]
- 33.Bednarski P.J., Nelson S.D. Interactions of thiol-containing androgens with human placental aromatase. J. Med. Chem. 1989;32:203–213. doi: 10.1021/jm00121a037. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura H., Shiozawa T., Terao Y., Shiraishi F., Fukazawa H. By-products Produced by the Reaction of Estrogens with Hypochlorous Acid and their Estrogen Activities. J. Health. Sci. 2006;52:124–131. doi: 10.1248/jhs.52.124. [DOI] [Google Scholar]
- 35.Schrödinger Release 2018-4: Glide. Schrödinger, LLC; New York, NY, USA: 2018. [Google Scholar]
- 36.Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A., Sanschagrin P.C., Mainz D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 37.Halgren T.A., Murphy R.B., Friesner R.A., Beard H.S., Frye L.L., Pollard W.T., Banks J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 38.Ghosh D., Lo J., Morton D., Valette D., Xi J., Griswold J., Hubbell S., Egbuta C., Jiang W., An J., et al. Novel Aromatase Inhibitors by Structure-Guided Design. J. Med. Chem. 2012;55:8464–8476. doi: 10.1021/jm300930n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.