

Abstract
Background
MADS-box genes play crucial roles in plant floral organ formation and plant reproductive development. However, there is still no information on genome-wide identification and classification of MADS-box genes in some representative plant species. A comprehensive investigation of MIKC-type genes in the orchid Dendrobium officinale is still lacking.
Results
Here we conducted a genome-wide analysis of MADS-box proteins from 29 species. In total, 1689 MADS-box proteins were identified. Two types of MADS-box genes, termed type I and II, were found in land plants, but not in liverwort. The SQUA, DEF/GLO, AG and SEP subfamilies existed in all the tested flowering plants, while SQUA was absent in the gymnosperm Ginkgo biloba, and no genes of the four subfamilies were found in a charophyte, liverwort, mosses, or lycophyte. This strongly corroborates the notion that clades of floral organ identity genes led to the evolution of flower development in flowering plants. Nine subfamilies of MIKCC genes were present in two orchids, D. officinale and Phalaenopsis equestris, while the TM8, FLC, AGL15 and AGL12 subfamilies may be lost. In addition, the four clades of floral organ identity genes in both orchids displayed a conservative and divergent expression pattern. Only three MIKC-type genes were induced by cold stress in D. officinale while 15 MIKC-type genes showed different levels of expression during seed germination.
Conclusions
MIKC-type genes were identified from streptophyte lineages, revealing new insights into their evolution and development relationships. Our results show a novel role of MIKC-type genes in seed germination and provide a useful clue for future research on seed germination in orchids.
Electronic supplementary material
The online version of this article (10.1186/s12870-019-1836-5) contains supplementary material, which is available to authorized users.
Keywords: Flower formation, Phylogenetic analysis, Classification, Gene family expansion
Background
The regulation of gene expression is a complex control mechanism that is coordinated by a number of mechanisms, including by transcription factors (TFs). TFs specifically recognize cis-regulatory regions of target genes, then regulate their expression in a way that leads to a wide range of physiological and biochemical processes. They also modulate developmental processes in plants [1]. MADS-box TFs, an ancient family of TFs found in both green algae charophyceans and land plants, play an essential role in the evolution of flower architecture [2, 3]. MADS-box genes encode proteins that share a highly conservative DNA-binding domain, the MADS domain, which recognizes similar 10-bp A/T-rich DNA sequences, the CArG-box [4].
Numerous MADS-box proteins have been identified from green algae, moss, gymnosperms and angiosperms [5]. The number of MADS-box genes varies greatly from species to species in plant lineages, suggesting that they are involved in the regulation of clade-specific functions. In the charophyceans [3], one MADS-box gene in Marchantia polymorpha [6], 26 in Physcomitrella patens [7] and 19 in Selaginella moellendorffii [8] have been reported. In angiosperms, dozens or even more than 100 MADS-box genes have been identified in dicots, including Arabidopsis thaliana [9], Brassica rapa (Chinese cabbage) [10], and Vitis vinifera [11]. In monocots, dozens of MADS-box genes have been found in Zea mays (75) [12], Sorghum bicolor (65) [12], Brachypodium distachyon (57) [13] and Oryza sativa (75) [14].
In plants, MADS-box genes can be divided into two distinct groups, namely type I and type II lineages: type I MADS-box proteins have no keratin-like (K) domain and only have the MADS (M) domain, whereas type II proteins also possess an intervening (I) domain, a K domain, and a C-terminal region followed by an M domain [15]. The type II MADS-box proteins are also known as MIKC-type MADS-box proteins in plants, comprising MIKCC and MIKC* proteins [15, 16]. The MIKCC proteins can be further subdivided into at least 13 distinct subfamilies (AG, AGL6, AGL12, AGL15, AGL17, BS, DEF/GLO, FLC, SEP, SQUA, SVP, TM3/SOC1 and TM8) on the basis of phylogenetic analysis [15].
The function of type I MADS-box genes is unclear. However, type II MADS genes are well studied and play a crucial role in the control of flower development. Investigations of type II MADS genes have led to the establishment of a well know model for the development of floral organs, the ABCDE model. A + E class genes establish the identity of sepals, A + B + E class genes establish the identity of petals, B + C + E class genes establish the identity of stamens, and D + E class genes establish the identity of carpels. ABCDE homeotic genes have been comprehensively studied in angiosperms, particularly in Antirrhinum majus and A. thaliana [17]. In Arabidopsis, AP1 is an A class gene, AP3 and PI are B class genes, AG is a C class gene, STK and SHP1/2 are D class genes, and SEP1–4 are E class genes [18]. The expression of ABCDE class genes is specific to different floral organs. For example, AP3 and PI homologs are expressed in petals and stamens, AG homologs are expressed in stamens and carpels, while SEP homologs are expressed in all floral organs in angiosperms, even in basal angiosperms [18–21]. MADS-box genes serve an important function by linking evolution and development, and should aid in understanding the mechanisms underlying flower formation while providing a foundation for analyzing variations in plant floral architecture. Therefore, it is important to investigate how MADS-box genes have evolved in plants.
To obtain a comprehensive view of MADS-box genes in plants, in this study we conducted a genome-wide identification of MADS-box genes from a broad range of plants across six lineages, including chlorophytes, a charophyte, a liverwort, mosses, a lycophyte, a gymnosperm and representatives of several angiosperm plant lineages whose genomes were available. The representative species, which belong to six lineages of plants (chlorophytes, charophyte, liverwort, mosses, lycophyte, gymnosperm) were selected to analyze as much of their genomes as was available. The species from the angiosperm lineage were selected because of their importance in human life. Genomic sequencing technology and bioinformatics tools have become faster and more efficient, and thus increasingly more genomic sequences of plants are currently available. Therefore, it is necessary to reanalyze MADS-box genes in important as well as a wide range of plant species, as has been done in this study, to identify this gene family in other plant species to reveal its evolution.
Orchids belong to the Orchidaceae and have unique floral patterning, particularly floral structures and organ identity. The labellum (the ‘lip’) and gynostemium (the ‘column’, a fused structure consisting of stamens and pistils) reflect unique development of orchid flowers. Several studies have isolated and analyzed of ABCE genes in orchids [22]. For example, the ABCE function genes in Dendrobium [23–25], B function genes (AP3 and PI lineages) in Oncidium [26], B and E function genes in Cymbidium [27], and B (AP3 lineages) in Phalaenopsis [28] were identified by RT-PCR and characterized. Dendrobium officinale and Phalaenopsis equestris are two important orchids with ornamental value. Dendrobium and Phalaenopsis diverged roughly 40 million years ago (Ma) [29]. The sizes of both P. equestris and D. officinale genomes are similar and exceed 1.1 gigabytes (Gb). The size of the P. equestris genome is 1.16 Gb, which was assessed by a whole-genome shotgun sequence and assembly strategy [30]. The size of the D. officinale genome is 1.11 Gb and was assessed by a K-mer analysis [31]. In this study, the ABCE genes of two orchids were identified using whole genomes, and phylogenetic and comparative expression analyses were performed. The expression patterns of MIKC genes in D. officinale under cold stress and during seed germination were also analyzed. These results may provide important information to understand the conservation and divergence of these genes’ functions, providing clues to explore the function of this gene family.
Results
Identification of MADS-box genes in multiple plant species
In total, 1689 MADS-box genes, including 684 type I and 1001 type II MADS-box genes, as well as four MADS-box genes from chlorophytes, were identified from entire genome sequences of 29 plant species (Fig. 1; Additional file 1: Table S1). Lower plant (chlorophyte) lineages had no or few MADS-box genes in their genomes. For example, no MADS-box gene was found in Dunaliella salina, only one MADS-box gene was observed in Micromonas pusilla and in Chlamydomonas reinhardtii, and there were just two MADS-box genes in Volvox carteri (Fig. 1). All four MADS-box genes from chlorophyte lineages only contain the M domain, and have no I, K, or C domains (Additional file 2: Figure S1), which agrees with findings in other green algae belonging to the chlorophyte lineage [5]. The basal streptophyte, K. nitens has only one MADS-box gene, which is a type II MADS-box TF that contains the M, I, K, and C domains (Additional file 3: Figure S2). The type I genes were detected in the genomes of all mosses, the lycophyte, the gymnosperm and angiosperms, but not in the charophyte and liverwort (Fig. 1). However, type II genes were widely present among all streptophyte lineages (Fig. 1). In addition, the number of MADS-box genes in angiosperms were numerous, ranging from 33 (A. trichopoda) to 169 (M. domestica), but only one or two in algae (Fig. 1).
Fig. 1.

The corresponding number of type I, type II and total MADS-box genes for each plant species identified in this study. The tree topology reflects a consensus of plant phylogenies [32]. The ‘?’ indicates that the genes contained an M domain, but shared low identity with other MADS-box proteins in higher plants
Phylogenetic analysis, classification of type II (MIKC-type) genes, and analysis of the ABCDE genes in Streptophytes
To understand the classification and evolution of plant MIKC-type genes, we conducted phylogenetic analyses with all 1001 MIKC-type genes found in this study from streptophyte lineages. Based on phylogenetic reconstructions of MIKC-type members from alignments of full-length MIKC-type protein sequences, they were classified into the MIKC* and MIKCC groups (Additional file 5: Figure S3). The MIKCC group was further divided into 13 major subfamilies, namely AGL6, AGL12, AGL15, AGL17, SQUA, DEF/GLO, AG, SEP, TM3/SOC1, BS, SVP, FLC and TM8 (Fig. 2; Additional file 6: Figure S4), in agreement with previous findings [15]. Only a single MADS-box gene (GAQ89767.1) was found in the charophyte K. nitens, which falls into the SVP clade, with weak bootstrap support (34%) by phylogenetic analysis (Additional file 7: Figure S5). Moreover, only one MIKCC gene in the liverwort falls into the SVP subfamily (Additional file 8: Figure S6). This indicates that the ancestral homologs of the SVP gene evolved before the divergence of the charophyte algae lineages from land plant lineages and suggests that the ancestral charophyte emerged with one or only a few MIKCC genes, and that MIKC proteins of all modern land plants descended and radiated from these predecessors by gene duplications, as was suggested by Tanabe et al. [3].
Fig. 2.

Classification of the total of 865 MIKCC proteins based on phylogenetic analysis. The MIKCC proteins were divided into 13 subfamilies: AG, AGL6, AGL12, AGL15, AGL17, BS, DEF/GLO, FLC, SEP, SQUA, SVP, TM3/SOC1 and TM8. The phylogenetic tree was constructed by MEGA 7 [33] with the NJ method based on alignments by MAFFT 7 [34]. The phylogenetic tree containing bootstrap values can be observed in Additional file 6: Figure S4
Among the 13 subfamilies, SQUA (A class), DEF/GLO (B class), AG (C/D class) and SEP (E class) subfamily genes are well known for their critical roles in flower development [35]. The evolution of the flower itself is suggested to be connected to the evolution of corresponding lineages. As expected, the plant taxonomic groups from charophytes, through to bryophytes and lycophytes, have no SQUA, DEF/GLO, AG and SEP subfamily genes, which are present in all flowering plants, while the gymnosperm Ginkgo, which has no flowers, only contains DEF/GLO, AG and SEP subfamily genes (Fig. 3).
Fig. 3.

The corresponding number of SQUA, DEF/GLO, AG and SEP subfamily MADS-box genes for each streptophyta lineage identified in this study. The tree topology reflects a consensus of streptophyta phylogenies [32]
Phylogenetic analysis of MIKC-type genes in orchids
Analysis of MIKC-type genes in orchids would help to understand the mechanisms of flower formation and provide a foundation for analyzing the diversification of flower structure. A total of 41 and 34 MIKC-type genes were identified in D. officinale and P. equestris, and could be divided into MIKC* and MIKCC groups. There were 33 and 30 MIKCC genes from D. officinale and P. equestris, respectively. Phylogenetic analysis shows that the MIKCC genes in both orchids could be divided into nine major subfamilies, SQUA, DEF/GLO, AG, BS, SEP, AGL6, AGL17, TM3/SOC1 and SVP (Fig. 4). The number of genes in each subfamily of both orchids is similar. For example, the DEF/GLO, AG, SEP and AGL6 subfamilies contained the same number of genes (5, 4, 5 and 1, respectively) in both orchids. The TM8, FLC, AGL12 and AGL15 genes were not found in D. officinale or P. equestris. Other monocots such as rice and B. distachyon had no FLC members or AG15 subfamily members but contained AGL12 subfamily genes [13, 14].
Fig. 4.

Phylogenetic analyses of MADS-box genes from two orchids, Dendrobium officinale and Phalaenopsis equestris. The AGL12, AGL15 and FLC subfamilies were lost from the two orchids. The phylogenetic tree was constructed using MEGA 7 [33] based on the alignment of MADS-box proteins by MAFFT 7 [34] with the NJ method. Numbers besides branches represent bootstrap support values from 1000 replications
Characterization of ABCE genes in two orchids
Genetic and molecular studies in core eudicots and monocots, as well as basal angiosperms, support a strong correlation between the expression pattern and function of ABCE class genes [17, 20, 36–40]. It is well known that ABCE function genes play a key role in the identity floral organs, including sepals, petals, and stamens, which in orchids differ considerably from other plants. By comparing and analyzing the expression of ABCE genes in two important orchid genera would help to understand the divergence of their gene function.
A phylogenetic reconstruction shows that the SQUA genes could be divided into dicot and monocot clades (Fig. 5A). All orchid SQUA genes did not display flower-specific expression patterns, and in some cases (PeAGL11–13, DoAGL16, DoAGL17 and DoAGL19), they even had relatively lower expression in sepals (Fig. 5A). The SQUA genes (PeAGL11–13) in P. equestris had a relatively low level of expression in all the organs and relatively strong expression in the column (Fig. 5A). DoAGL18 and DoAGL19 were detected in all organs and were strongly expressed in stems and leaves (Fig. 5A). DoAGL16 was only detected in leaves and DoAGL17 was strongly expressed in roots and stems (Fig. 5A).
Fig. 5.

Phylogenetic relationships and expression pattern of SQUA (a), DEF/GLO (b), AG (c) and SEP (d) subfamily proteins from Dendrobium officinale (Do), Phalaenopsis equestris (Pe) and Arabidopsis thaliana (At). The phylogenetic tree was constructed using MEGA 7 [33] with the NJ method and 1000 bootstrap replications based on the alignment of MADS-box proteins by MAFFT 7 [34]. R, roots; S, stems; L, leaves; FB, flower buds; Se, sepals; Pe, petals; Li, lips; Co, columns
The B class genes are required for the establishment of petal and stamen identity. AP3 and PI homologues were recognized as two subclades of B-function genes. Only one PI homologue gene each was found in D. officinale and P. equestris, while at least four AP3 homologues were present in the two orchids (Fig. 5B). The PI homologues DoAGL34 and PeAGL26 were expressed in all four floral organs (sepals, petals, lips and the column) as well as in vegetative organs (roots, stems and leaves) with a similar expression pattern (Fig. 5B). Similarly, the PI homologue OMADS3 from Oncidium Gower Ramsey was expressed in all four floral organs as well as in vegetative tissue (leaves) [26]. The AP3 subclade genes were specific to flowers and displayed similar expression patterns in both orchids. This reveals a possible positive role for AP3 genes in the regulation of sepal, petal, lip and column formation in orchids.
DoAGL21 and DoAGL22 from D. officinale, while PeAGL17 and PeAGL18 were identified as AG-lineage genes from P. equestris (Fig. 5C). DoAGL21 was expressed only in the column and floral buds but DoAGL22 and PeAGL17 were expressed in petals and predominantly in the column (Fig. 5C). Consistent with the requirement of AG lineages for normal stamen development [41], AG was expressed in the stamens in both orchids. We suggest that the C function genes are highly conserved among orchids and play an essential role in the regulation of stamen formation.
In this study, the SEP subfamily could be divided into two clades in orchids, SEP1/2 and SEP3 (Fig. 5D). SEP genes DoAGL9–13 from D. officinale were detected in roots, stems, and leaves of the reproductive stage and in flowers, with strong expression in floral buds, sepals, petals, lips and the column (Fig. 5D). DoAGL10 was detected in floral buds, sepals, petals, lips and the column, although it showed relatively lower expression in lips than in sepals, petals, and the column (Fig. 5D). The four SEP subfamily genes (PeAGL5–8) from P. equestris were expressed in all floral organs, with relatively higher expression in flower buds, sepals and petals than in the lips and stamen (Fig. 5D). The other two SEP genes (DoAGL9 and DoAGL10) belonging to the SEP1/2 clade and with a similar expression pattern, were detected in stems and floral organs (Fig. 5D). This result indicates that DoAGL9 and DoAGL10 possibly oversee similar biological functions.
Expression analysis of D. officinale MIKC-type genes under cold stress
Some clades of MADS-box genes can be induced by low temperatures in tomato (Solanum lycopersicum) [42] and rice [14]. This spurred us to characterize the expression patterns of the MIKC-type genes in D. officinale. To analyze the expression of DoAGLs under cold stress, transcriptome sequencing data of D. officinale leaves were downloaded from the SRA database and used to calculated gene expression levels. Only three MIKC-type genes from the SUQA, SOC1 and DEF/GLO clades showed a cold-responsive expression pattern (Fig. 6), DoAGL18 in the SUQA subfamily was down-regulated 0.358-fold, while its homologs TM4 in tomato [42], and LcMADS1 and LcMADS2 in sheepgrass (Leymus chinensis) [43] exhibited an opposite expression pattern, being up-regulated by low temperatures. Two genes, DoAGL24 in the SOC1 subfamily and DoAGL27 in the DEF/GLO subfamily, were up-regulated 1.51- and 1.60-fold, respectively more than the control. This result is consistent with DEF/GLO lineages LcMADS5 from sheepgrass [44] and TM6 from tomato [42] which were up-regulated in response to cold stress. In Brassica rapa, five SOC1 homologss (BrMADS36, − 38, − 39, − 40 and − 44) were induced by cold stress [45]. This result suggests that some MIKC-type genes may play a role in the response of D. officinale to low temperature stress while the genes inducible by cold stress are diverse across plant species. The expression of eight MIKC-type genes (DoAGL07, − 16, − 17, − 19, − 25, − 28, − 29 and DoAGL34) was not different with the control under cold stress (Additional file 9: Table S3). Finally, the remaining 30 genes displayed very low expression or were not detected by RNA-seq (Additional file 9: Table S3).
Fig. 6.
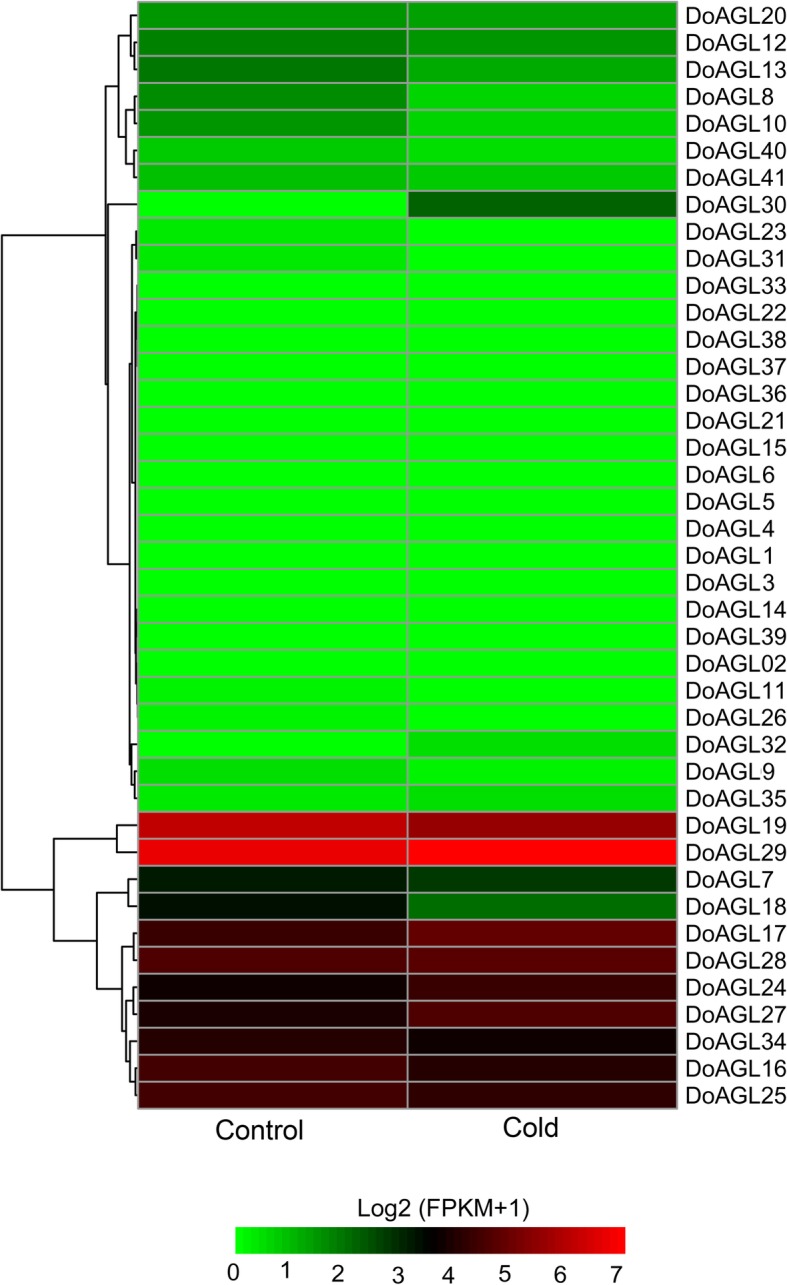
Heat map showing expression pattern of MIKC genes in leaves of D. officinale under cold (4 °C) stress
Transcript analysis of D. officinale MIKC genes during seed germination
While the involvement of MIKC genes in the regulation of floral organ development has been well established, their involvement in seed germination is largely unknown. Only two MIKC genes FLC/AGL25 and AGL21in A. thaliana were shown to be involved in seed germination by influencing the ABA catabolic pathway and regulating ABA signaling [46, 47]. However, no FLC subfamily genes have been found in orchids, such as Apostasia odorata [48], P. equestris and D. officinale (Fig. 4). This indicates that the manner in which MIKC genes regulate seed germination in orchids may differ from A. thaliana. Based on this, the expression of MIKC genes in D. officinale during four seed germination stages were analyzed. Six MIKC genes (DoAGL10, − 13, − 19, − 21, − 25, 30 and − 37) were down-regulated while four MIKC genes (DoAGL17, − 18 and − 32) were up-regulated in S2-S4 (Fig. 7; Additional file 10: Table S4). The SVP subfamily gene DoAGL27 was not detected in S1, but was detected in S2–4, with highest expression in S3 (Fig. 7; Additional file 10: Table S4). S4 is the stage in which leaves first emerge and a seedling is established. Two SQUA subfamily genes DoAGL17 and DoAGL18 showed a higher level of expression at S2-S4, while DoAGL19 expression was down-regulated in S2-S4 (Fig. 7; Additional file 10: Table S4). This suggests diverse functions of SQUA subfamily genes in D. officinale. One DEF/GLO clade gene DoAGL32 had a low level of expression (FPKM = 1) in S1, but showed a high level of expression in S2-S4 (FPKMS2 = 154; FPKMS3 = 168; FPKMS4 = 325), suggesting that DoAGL32 may play an important role in seed germination. In this study, a MIKC* gene DoAGL7 showed more than a 1.5-fold change in S3 and S4 than S1 (Fig. 7), suggesting that it may play a role during seed germination.
Fig. 7.
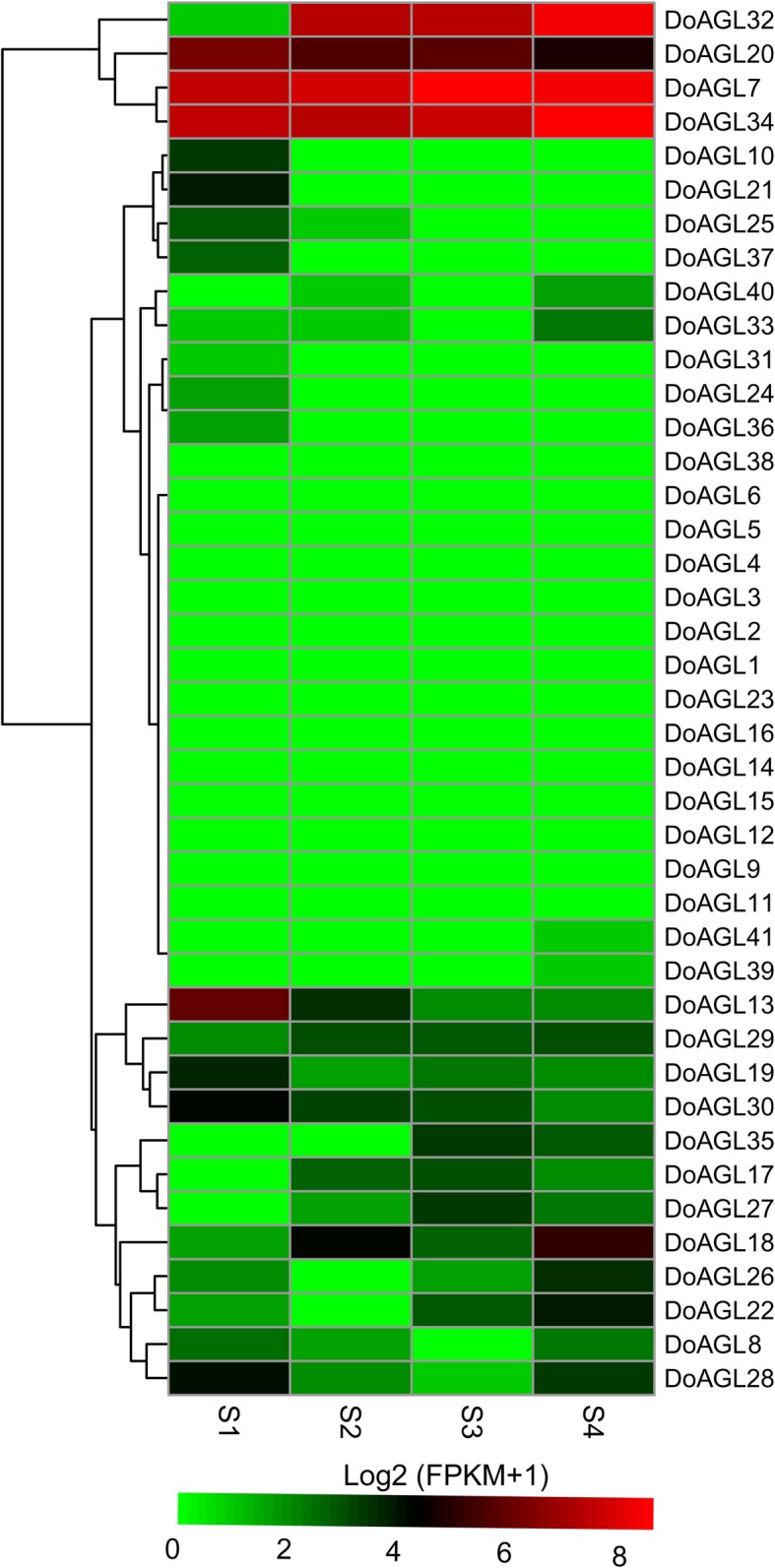
Overview of expression pattern of MIKC genes during the seed germination in D. officinale. For more detail of seed germination stages, see the materials and methods section
Discussion
Identification and classification of the MADS-box genes in plants
MADS-box genes have been reidentified in several plant species in this study (details are listed in Additional file 4: Table S2). A total of 108 MADS-box genes were identified from A. thaliana and 160 from Chinese cabbage (B. rapa), agreeing with the number of MADS-box genes in previous reports [9, 10]. Arora et al. [14] reported a genome-wide identification and annotation of rice MADS-box genes using the TIGR pseudomolecules release 4 (released on January 12, 2006) [49]. In our study, a total of 73 MADS-box genes were found in rice using the current genome assembly (MSU Release 7) [50]. Compared with the previous annotation of MADS-box genes in rice, four genes (LOC_Os02g01355, LOC_Os02g01365, LOC_Os06g01890 and LOC_Os11g12360) not previously annotated as MADS-box genes, were identified. LOC_Os02g01355 and LOC_Os02g01365, which shared the same amino acids and are located on the same chromosome, were regarded as redundant duplicate genes, so LOC_Os02g01355 was removed. Moreover, two MADS-box genes (LOC_Os12g31010 and LOC_Os08g20460) present in the previous study were absent from the latest rice peptide file and were thus excluded from our study. In addition, two genes LOC_Os01g68420 and LOC_Os02g01360 that were previously regarded as MADS-box genes, but that had low identity (less than 38%) with other MADS-box genes and have no M domain or K domain after BLAST analysis in the NCBI database, were excluded from this study. Our result provides more accurate and precise information about the MADS-box family in rice that will help to understand the role of this gene family in reproductive development. In maize, 75 MADS-box genes were found in a previous study [12] while 87 were found in our study, expanding the list by 12.
The MADS-box genes identified from plants in this study, except for the chlorophytes and charophytes, could be divided into two distinct types, type I (SRF-like) and type II (MEF2-like), as in animals and fungi [51]. The type II genes were isolated from three charophycean green algae [3]. It was previously proposed that two main lineages (type I and type II) of MADS-box genes already existed before the divergence of plants and animals/fungi [52].
The diversification of MIKC-type genes in Streptophyte lineages
The TM8 subfamily was present in the gymnosperm G. biloba, basal angiosperms A. trichopoda and Nelumbo nucifera, and some eudicots Citrus clementina, Malus x domestica, Pyrus x bretschneideri and V. vinifera, but absent in eudicots A. thaliana and Chinese cabbage (Additional file 1: Table S1), consistent with previous findings in A. thaliana [9], Chinese cabbage [10], V. vinifera [53], A. trichopoda [54] and G. biloba [55]. In addition, the monocot Zea mays had one exclusive gene, TM8 (GRMZM2G137289_P01; Additional file 1: Table S1). This suggests that the TM8 subfamily may have been lost during the evolution of some monocots and some eudicots. The MIKC* type genes were found in all streptophyte lineages excluding charophyte algae (Additional file 1: Table S1). The liverwort M. polymorpha, regarded as the earliest diverging land plant [6], has only a single MIKC* gene. AGL15 subfamily genes were present in bryophytes, lycophytes, a gymnosperm, basal angiosperms and eudicots, but were absent in monocots (Additional file 1: Table S1). This is consistent with a report in which the AGL15 clade genes were found in gymnosperms [56, 57] and eudicots [9], but were absent in monocots [58]. The AGL15 subfamily genes may have been lost during the evolution of monocot lineages. The DEF/GLO genes were found in other gymnosperms such as the conifer Pinus radiata [59] and a gnetale Gnetum gnemon [60]. SQUA subfamily genes were not found in Ginkgo in this study, and they were also not found in previous studies [56, 61]. The SQUA subfamily genes were also absent in two other gymnosperms, Pinus taeda and Pinus sylvestris [56]. We suggest that the A-function genes were present after the divergence of angiosperms and gymnosperms and that B-, C- and E-function genes arose before the divergence of angiosperms and gymnosperms.
The functional divergence and conservation of MIKCC genes in orchids
Both AGL12 and FLC control the flowering transition and act as an enhancer and repressor, respectively in A. thaliana flowering time. XAL1 (AGL12) plays an important role in flowering transition, and the late-flowering phenotypes of the xal1 mutants are able to grow under long days [62]. FLC plays a key role in the initiation of flowering in A. thaliana, i.e., flc mutations have early flowering phenotypes [63]. The homologues of cereals (wheat and barley) VRN1, FT/VRN3, and A. thaliana AGL19, acted as key players in vernalization pathways, while those found in Dendrobium nobile were regulated by vernalization [64]. The AGL12 and FLC subfamily genes were absent in these two orchids D. officinale and P. equestris, indicating that the regulation of genes during the flowering transition in orchids is different to A. thaliana. More analyses are needed to confirm that AGL12 and FLC genes have been lost in the genomes of orchids and to determine the conservation and evolutionary importance of these genes.
In dicots such as A. thaliana and A. majus, the A class genes, which are required for the development of sepals, show highest expression in sepals but their expression is absent in roots, stems and leaves [38, 65]. However, the SQUA subfamily genes in monocots could be detected in vegetative tissues, the leaves. For example, a rice SQUA-like gene OsMADS18 is widely expressed in roots, leaves, inflorescences, and developing kernels [66]. We suggest, from our study, that SQUA-like genes underwent functional divergence in monocots and dicots, and even in orchids. The C-lineage was well demonstrated in stamen development in plants, displaying exclusive expression in stamens and carpels, such as in A. thaliana [67], Nicotiana tabacum [68] and Lilium longiflorum [69]. Only a single C-function gene was found in A. thaliana [67] and A. majus [70], which was essential for the specification of reproductive organs. However, at least two AG-lineages were found in orchids and were expressed predominantly in the column. One AG-lineage gene CeMADS1 from Cymbidium ensifolium was expressed exclusively in columns while another, CeMADS2, was detected in all floral organs with strong expression in columns, similar to two D. officinale AG-lineage genes [71]. Genetic and molecular studies have demonstrated that the SEP subfamily genes (SEP1/2/3/4) are required for the specification of identity of all four whorls of floral organs [17]. The flowers of A. thaliana sep1 sep2 sep3 triple mutant produced flowers without any petals, stamens and carpels, all of which were replaced by sepals, demonstrating that the SEP genes are required for the specification of petals, stamens, and carpels in Arabidopsis [17]. The SEP (PAP2) gene regulates spikelet meristem identity and development in rice [44]. The function E orthologous genes from D. officinale and P. equestris and other orchids such as Oncidium Gower Ramsey [72] and Dendrobium crumenatum [24] were detected in all floral organs and played a conserved role in floral organs development.
The MIKC-type genes might play a role in seed germination in D. officinale
Some MIKC genes from D. offcinale showed a different expression during the seed germination, including SVP and SQUA subfamily genes, as well as the MIKC* gene. The SVP subfamily gene PkMADS1 from Paulownia kawakamii displayed an important role in shoot morphogenesis [73], suggesting that the SVP gene in D. officinale may have a similar function with other SVP clade genes and may play a role in vegetative shoot development. The function of the MIKC* gene is not well defined. Expression analysis provided clues that MIKC* genes play a role in gametophytic generation in S. moellendorffii, Funaria hygrometrica and Ceratopteris richardii [74, 75], as well as pollen development in A. thaliana [76]. Liu et al. [77] provided evidence that the MIKC* gene in rice was involved in pollen maturation. However, a MIKC* gene showed a up-regulaed expression during seed germination and might play a role this important process in our study.
Conclusion
Genome-wide identification of 29 plant species allowed for the identification of a total of 1689 MADS-box genes. These MADS-box genes were divided into type I and type II (MIKC-type) clades. MIKCC-type genes, belonging to the MIKC-type clade, were divided into at least 13 subfamilies on the basis of their phylogenetic relationships. The SQUA (A class), AP/PI (B class), AG (C/D class) and SEP (E class) gene subfamilies were analyzed among a charophyte, a liverwort, mosses, a lycophyte, a gymnosperm and angiosperms, providing novel information that enriches our understanding of floral organ development and gene evolution. The expression patterns of ABCE genes in two orchids, as well as the expression pattern of cold stress and seed germination stage in D. officinale, were analyzed. This information will fortify our understanding of the conservation and divergence of the function of these genes, and allow floral ontogeny, stress response and seed germination in orchids to be further explained.
Methods
Plant materials and RNA isolation
D. officinale plants used in this study were grown in South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, China. Gene expression analysis was performed on D. officinale tissues: roots, stems, leaves, flower buds, lips, petals, sepals and columns. The roots, stems, leaves and floral organs were collected from 10 pots of D. officinale in the reproductive stage, then frozen in liquid nitrogen for 5 min and frozen at − 80 °C for RNA isolation as quickly as possible. Total RNA was extracted from various organs with the RNA extraction kit, Column Plant RNAout2.0 (Tiandz, Inc., Beijing, China), according to the operating manual while contaminating DNA was removed by the RNase-Free DNase I Kit (TaKaRa, Dalian, China).
Identification of MADS-box genes
All the latest peptide files of the 29 species were downloaded from the online database to generate a local protein database (details of the versions and download websites are listed in Additional file 11: Table S5). The SRF-type TF domain model (PF00319) was obtained from the Pfam database (http://pfam.xfam.org/) and was used to build a hidden Markov model (HMM) file by using the HMMER3 software package under default parameters (http://hmmer.janelia.org/). MADS-box genes were identified through the HMM search program by using the HMM file searched against the local protein databases by HMMER3. The identified sequences were confirmed to be MADS-box proteins by annotating as a MADS-box protein either in the Uniprot database (https://www.uniprot.org/) or in the NCBI database. To ensure the accuracy of the results, the chromosomal localizations of all candidate MADS-box genes were checked and redundant sequences with the same chromosome location were removed.
Multiple sequence alignments
MAFFT version 7 software [34] was used to align multiple sequence of MADS-box proteins with default parameters to generate a multiple sequence alignment file in FASTA format. ClustalX 2.1 software [78] was used to align sequences and recheck the above results.
Classification of MADS-box genes by phylogenetic analysis
Phylogenetic and molecular evolutionary analyses of the MADS-box proteins were conducted using MEGA version 7 [33] based on the alignment of MADS-box proteins. The NJ method was used to construct phylogenetic trees in this study. To determine statistical reliability, we reconducted a bootstrap NJ tree by ClustalX 2.1 software [78] to recheck the classification of the MADS-box genes. MADS-box genes were classified according to their phylogenetic relationships with the corresponding Arabidopsis MADS-box genes.
Gene expression analysis
Total RNA reversion was carried out with the GoScript™ Reverse Transcription System (Promega, Madison, WI, USA) according to its protocols. Transcript levels were determined by semi-quantitative PCR using a Taq PCR Master Mix Kit (DreamTaq™ Green PCR Master Mix kit, TaKaRa, Dalian, China). One microliter of cDNA sample (about 400 ng/μL) from the RT reaction was used for PCR, consisting of 95 °C for 2 min, followed by 40 cycles of denaturation at 98 °C (10 s), annealing at 58 °C (30 s), and extension at 72 °C (60 s). Five microliters of PCR product in each reaction was analyzed by electrophoresis on 1% agarose gels. The primers of MADS-box genes were designed using Primer3.0 software (http://bioinfo.ut.ee/primer3-0.4.0/). To make the primers more specific, the sequences excluding highly conserved M domain sequences were used to the design of primers as much as possible. The specific primers of the MADS-box genes are listed in Additional file 12: Table S6. D. officinale actin (NCBI accession No. JX294908) was used as the internal control gene.
For expression analysis of SQUA, DEF/GLO, AG and SEP subfamily genes from P. equestris, raw sequencing reads of P. equestris samples (roots, stems, leaves, flower buds, sepals, petals, lips and columns) were obtained from the NCBI SRA (accession numbers SRR2080194, SRR2080204, SRR2080202, SRR2080200, SRR3602300, SRR3602299, SRR3602277, and SRR3600816) provided by Niu et al. [79]. Low quality reads were excluded from the raw reeds to generate clean reads, which were used to analyze gene expression. All clean reads were mapped with the nucleotide sequences of SQUA, DEF/GLO, AG and SEP subfamily genes using TopHat version 2.0.8 [80]. Gene expression level was calculated by the FPKM method using HTSeq [81].
For gene expression analysis under cold stress, the transcriptome sequencing data of D. offficinale leaves under cold stress (4 °C) and control (20 °C) (SRA, accession numbers SRR3210613, SRR3210621, SRR3210626, SRR3210630, SRR3210635 and SRR3210636) were downloaded from SRA, as provided by Wu et al. [82]. The raw reeds were cleaned and used to calculate the level of expression under cold stress as described above. Genes with a mean FPKM > 5 in control or treated leaves were used to calculate fold change (mean of FPKM treated leaves / mean of FPKM control leaves). Genes with a ≥ 1.5-fold change were regarded as up-regulated genes, and those with a ≤ 0.66-fold change were defined as down-regulated genes.
During seed germination, raw sequence reads of four stages of D. officinale seed germination (S1, SRR1951787; S2, SRR1951788; S3, SRR1951789; S4, SRR1951790) were downloaded to analyze gene expression using the gene expression methods indicated above. SRA data was provided in Chen et al. [83]. Genes with FPKM > 5 in any one of the stages were regarded as valid genes and were used to calculate fold change (FPKMS2–4 / FPKMS1). Genes with a ≥ 1.5-fold change or with a ≤ 0.66-fold change were defined as genes with different expression. S1 is the stage in which seeds do not germinate and before sowing on half-strength Murashige and Skoog (MS) [84] culture medium; in S2, the embryo enlarges and the testa ruptures; in S3, a protocorm forms; in S4, a seedling is established with the emergence of a first leaf.
Additional files
Table S1. Information of MADS-box genes in the 29 tested species. (DOCX 153 kb)
Figure S1. BLASTP graphic overview of four MADS-box proteins from chlorophytes based on the National Center for Biotechnology Information (NCBI) database. (A) MADS-box protein (Cre18.g749550.t1.1) from Chlamydomonas reinhardtii. (B) and (C) MADS-box proteins (Vocar.0002 s0667.1.p and Vocar.0014 s0224.1.p) from Volvox carteri. (D) MADS-box protein (21861) from Micromonas pusilla CCMP1545. (DOCX 155 kb)
Figure S2. Alignment of the amino acid sequences of only one MADS-box gene (GAQ89767.1) from Klebsormidium nitens and AtSVP (AT2G22540.1) and AtAGL24 (AT4G24540.1) of the SVP subfamily, as well as AtAGL65 (AT1G18750.1) and AtAGL66 (AT1G77980.1) of MIKC* genes from Arabidopsis thaliana. The GAQ89767.1 protein contains M-, I, K- and C domains, which are indicated by red, yellow, green and blue boxes, respectively. (DOCX 1228 kb)
Table S2. Information of MADS-box genes in Arabidopsis thaliana, Oryza sativa, Brassica rapa, Brachypodium distachyon, Zea mays, Sorghum bicolor, Phalaenopsis equestris, Dendrobium officinale, Physcomitrella patens, and Selaginella moellendorffii. (DOCX 17 kb)
Figure S3. Classification of the total of 983 MIKC-type proteins based on a phylogenetic analysis. MIKC-type proteins can be divided into two main groups MIKC* and MIKCC. The phylogenetic tree was constructed by MEGA 7 with the NJ method based on MAFFT 7-based alignments. (DOCX 767 kb)
Figure S4. Classification of the total 865 MIKCC proteins based on phylogenetic analysis. The phylogenetic tree was conducted using MEGA 7 based on the alignment of MADS-box proteins by MAFFT 7 with the Neighbor-Joining method. Numbers besides branches represent bootstrap support values from 1000 replications. Values lower than 40% are hidden. (DOCX 2621 kb)
Figure S5. Phylogenetic analyses of GAQ89767.1 from Klebsormidium nitens and MADS-box proteins from Arabidopsis thaliana (At). The phylogenetic tree was conducted using MEGA 7 based on the alignment of MADS-box proteins by MAFFT 7 with the NJ method. Numbers besides branches represent bootstrap support values from 1000 replications. (DOCX 428 kb)
Figure S6. Phylogenetic analyses of the only two MADS-box proteins (Mapoly0011s0161.1.p and Mapoly0174s0011.1.p) from Marchantia polymorpha and MADS-box proteins from Arabidopsis thaliana (At). Mapoly0011s0161.1.p falls into the SVP subfamily (29% similarity). Mapoly0174s0011.1.p falls into the MIKC* group. The phylogenetic tree was conducted using MEGA 7 based on the alignment of MADS-box proteins by MAFFT 7 with the NJ method. Numbers besides branches represent bootstrap support values from 1000 replications. (DOCX 413 kb)
Table S3. The mean of FPKM value and fold change of MIKC gene under control and cold stress in D. officinale leaf. (DOCX 17 kb)
Table S4. FPKM value of the MIKC gene in Dendrobium officinale during four seed germination stages. (DOCX 16 kb)
Table S5. The versin of genomic data and download websites. (DOCX 16 kb)
Table S6. The primers used for Semi-quantitative RT-PCR. (DOCX 14 kb)
Acknowledgements
We thank Dr. Hanghui Kong (South China Botanical Garden, Chinese Academy of Sciences) for valuable suggestions in the evolution analysis.
Abbreviations
- AG
AGAMOUS
- FLC
FLOWERING LOCUS C
- FPKM
Fragments per kilobase of exon per million fragments mapped
- HMM
Hidden Markov Model
- NCBI
The National Center for Biotechnology Information
- NJ
Neighbor-Joining
- RNA-seq
RNA-sequencing
- SQUA
SQUAMOSA
- STK
SEEDSTICK
- TF
Transcription factor
- SRA
Sequence read archive
- SEP1/2/3/4
SEPALLATA1/2/3/4;
- SHP1/2
SHATTERPROOF1/2
- AP1/3
APETALA1/3
Authors’ contributions
JD supervised the project. CH conceived the research and designed the experiments. CS performed Semi-quantitative RT-PCR. ML performed the bioinformatics analyses. CH and JATdS collectively interpreted the results and wrote all drafts of the manuscript. All five authors approved the final draft for submission and take full public responsibility for the content of the manuscript.
Funding
This research was supported by funding from the Natural Science Foundation of Guangdong Province Projects (Grant number 2016A030310012) and Science and Technology Program of Guangzhou (Grant number 201704020192 and 201704020010). The funding agencies were not involved in study design, data collection and analysis, or preparation of the manuscript.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files. The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Chunmei He, Email: hechunmei2012@scbg.ac.cn.
Can Si, Email: cans2013@163.com.
Jaime A. Teixeira da Silva, Email: jaimetex@yahoo.com
Mingzhi Li, Email: omics-limz@foxmail.com.
Jun Duan, Phone: +86-20-37252993, Email: duanj@scib.ac.cn.
References
- 1.Franco-Zorrilla JM, López-Vidriero I, Carrasco JL, Godoy M, Vera P, Solano R. DNA-binding specificities of plant transcription factors and their potential to define target genes. Proc Natl Acad Sci U S A. 2014;111(6):2367–2372. doi: 10.1073/pnas.1316278111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kater M. M., Dreni L., Colombo L. Functional conservation of MADS-box factors controlling floral organ identity in rice and Arabidopsis. Journal of Experimental Botany. 2006;57(13):3433–3444. doi: 10.1093/jxb/erl097. [DOI] [PubMed] [Google Scholar]
- 3.Tanabe Y, Hasebe M, Sekimoto H, Nishiyama T, Kitani M, Henschel K, Münster T, Theissen G, Nozaki H, Ito M. Characterization of MADS-box genes in charophycean green algae and its implication for the evolution of MADS-box genes. Proc Natl Acad Sci U S A. 2005;102(7):2436–2441. doi: 10.1073/pnas.0409860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Folter SD, Angenent GC. Trans meets cis in MADS science. Trends Plant Sci. 2006;11(5):224–31. [DOI] [PubMed]
- 5.Gramzow L, Theissen G. A hitchhiker’s guide to the MADS world of plants. Genome Biol. 2010;11(6):214. doi: 10.1186/gb-2010-11-6-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowman JL, Kohchi T, Yamato KT, Jenkins J, Shu S, Ishizaki K, Yamaoka S, Nishihama R, Nakamura Y, Berger F: Insights into land plant evolution garnered from the Marchantia polymorpha genome. Cell 2017, 171(2):287–304. [DOI] [PubMed]
- 7.Barker EI, Ashton NW. A parsimonious model of lineage-specific expansion of MADS-box genes in Physcomitrella patens. Plant Cell Rep. 2013;32(8):1161–1177. doi: 10.1007/s00299-013-1411-8. [DOI] [PubMed] [Google Scholar]
- 8.Gramzow L, Barker E, Schulz C, Ambrose B, Ashton N, Theissen G, Litt A. Selaginella genome analysis – entering the “homoplasy heaven” of the MADS world. Front Plant Sci. 2012;3:214. doi: 10.3389/fpls.2012.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parenicová L, De SF, Kieffer M, Horner DS, Favalli C, Busscher J, Cook HE, Ingram RM, Kater MM, Davies B. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell. 2003;15(7):1538–1551. doi: 10.1105/tpc.011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duan W, Song X, Liu T, Huang Z, Ren J, Hou X, Li Y. Genome-wide analysis of the MADS-box gene family in Brassica rapa (Chinese cabbage) Mol Gen Genomics. 2015;290(1):239–255. doi: 10.1007/s00438-014-0912-7. [DOI] [PubMed] [Google Scholar]
- 11.Grimplet J, Martínez-Zapater JM, Carmona MJ. Structural and functional annotation of the MADS-box transcription factor family in grapevine. BMC Genomics. 2016;17(1):80. doi: 10.1186/s12864-016-2398-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y, Li X, Chen W, Peng X, Cheng X, Zhu S, Cheng B. Whole-genome survey and characterization of MADS-box gene family in maize and sorghum. Plant Cell Tiss Organ Culture. 2011;105(2):159–173. doi: 10.1007/s11240-010-9848-8. [DOI] [Google Scholar]
- 13.Wei B, Zhang RZ, Guo JJ, Liu DM, Li AL, Fan RC, Mao L, Zhang XQ. Genome-wide analysis of the MADS-box gene family in Brachypodium distachyon. Genome. 2014;9(1):e84781. doi: 10.1371/journal.pone.0084781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arora R, Agarwal P, Ray S, Singh AK, Singh VP, Tyagi AK, Kapoor S. MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics. 2007;8(1):242. doi: 10.1186/1471-2164-8-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smaczniak C, Immink RG, Angenent GC, Kaufmann K. Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development. 2012;139(17):3081. doi: 10.1242/dev.074674. [DOI] [PubMed] [Google Scholar]
- 16.Henschel K, Kofuji R, Hasebe M, Saedler H, Münster T, Theissen G. Two ancient classes of MIKC-type MADS-box genes are present in the moss Physcomitrella patens. Mol Biol Evol. 2002;19(6):801–814. doi: 10.1093/oxfordjournals.molbev.a004137. [DOI] [PubMed] [Google Scholar]
- 17.Pelaz S, Ditta GS, Baumann E, Wisman E, Yanofsky MF. B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature. 2000;405(6783):200–203. doi: 10.1038/35012103. [DOI] [PubMed] [Google Scholar]
- 18.Ng M, Yanofsky MF. Function and evolution of the plant MADS-box gene family. Nat Rev Genet. 2001;2(3):186–195. doi: 10.1038/35056041. [DOI] [PubMed] [Google Scholar]
- 19.Jing D, Liu Z, Zhang B, Ma J, Han Y, Chen F. Two ancestral APETALA3 homologs from the basal angiosperm Magnolia wufengensis (Magnoliaceae) can affect flower development of Arabidopsis. Gene. 2014;537(1):100–107. doi: 10.1016/j.gene.2013.11.076. [DOI] [PubMed] [Google Scholar]
- 20.Kim S, Jin K, Yoo MJ, Kong H, Hu Y, Ma H, Soltis PS, Soltis DE. Expression of floral MADS-box genes in basal angiosperms: implications for the evolution of floral regulators. Plant J. 2005;43(5):724–744. doi: 10.1111/j.1365-313X.2005.02487.x. [DOI] [PubMed] [Google Scholar]
- 21.Zhang B, Liu ZX, Ma J, Song Y, Chen FJ. Alternative splicing of the AGAMOUS orthologous gene in double flower of Magnolia stellata (Magnoliaceae) Plant Sci. 2015;241:277–285. doi: 10.1016/j.plantsci.2015.10.017. [DOI] [PubMed] [Google Scholar]
- 22.Teixeira da Silva JA, Aceto S, Liu W, Yu H, Kanno A. Genetic control of flower development, color and senescence of Dendrobium orchids. Sci Hortic. 2014;175(1):74–86. doi: 10.1016/j.scienta.2014.05.008. [DOI] [Google Scholar]
- 23.Sawettalake N, Bunnag S, Wang Y, Shen L, Yu H. DOAP1 promotes flowering in the orchid Dendrobium Chao Praya smile. Front Plant Sci. 2017;8:400. doi: 10.3389/fpls.2017.00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Y, Teo LL, Zhou J, Kumar PP, Yu H. Floral organ identity genes in the orchid Dendrobium crumenatum. Plant J. 2006;46(1):54. doi: 10.1111/j.1365-313X.2006.02669.x. [DOI] [PubMed] [Google Scholar]
- 25.Yu H, Goh CJ. Identification and characterization of three orchid MADS-box genes of the AP1/AGL9 subfamily during floral transition. Plant Physiol. 2000;123(4):1325–1336. doi: 10.1104/pp.123.4.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang YY, Kao NH, Li JY, Hsu WH, Liang YL, Wu JW, Yang CH. Characterization of the possible roles for B class MADS box genes in regulation of perianth formation in orchid. Plant Physiol. 2010;152(2):837–853. doi: 10.1104/pp.109.147116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiang Lin, Chen Yue, Chen Liping, Fu Xiaopeng, Zhao Kaige, Zhang Jie, Sun Chongbo. B and E MADS-box genes determine the perianth formation in Cymbidium goeringii Rchb.f. Physiologia Plantarum. 2017;162(3):353–369. doi: 10.1111/ppl.12647. [DOI] [PubMed] [Google Scholar]
- 28.Tsai W-C, Hsiao Y-Y, Lee S-H, Tung C-W, Wang D-P, Wang H-C, Chen W-H, Chen H-H. Expression analysis of the ESTs derived from the flower buds of Phalaenopsis equestris. Plant Sci. 2006;170(3):426–432. doi: 10.1016/j.plantsci.2005.08.029. [DOI] [Google Scholar]
- 29.Givnish TJ, Spalink D, Ames M, Lyon SP, Hunter SJ, Zuluaga A, Doucette A, Caro GG, McDaniel J, Clements MA. Orchid historical biogeography, diversification, Antarctica and the paradox of orchid dispersal. J Biogeogr. 2016;43(10):1905–1916. doi: 10.1111/jbi.12854. [DOI] [Google Scholar]
- 30.Cai J, Liu X, Vanneste K, Proost S, Tsai W-C, Liu K-W, Chen L-J, He Y, Xu Q, Bian C. The genome sequence of the orchid Phalaenopsis equestris. Nat Genet. 2015;47(1):65. doi: 10.1038/ng.3149. [DOI] [PubMed] [Google Scholar]
- 31.Zhang G-Q, Xu Q, Bian C, Tsai W-C, Yeh C-M, Liu K-W, Yoshida K, Zhang L-S, Chang S-B, Chen F. The Dendrobium catenatum Lindl. Genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Sci Rep. 2016;6:19029. doi: 10.1038/srep19029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bowman JL, Floyd SK, Sakakibara K. Green genes—comparative genomics of the green branch of life. Cell. 2007;129(2):229–234. doi: 10.1016/j.cell.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 33.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kazutaka K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Theißen G, Melzer R, Rümpler F. MADS-domain transcription factors and the floral quartet model of flower development: linking plant development and evolution. Development. 2016;143(18):3259. doi: 10.1242/dev.134080. [DOI] [PubMed] [Google Scholar]
- 36.Cheng Z, Ge W, Li L, Hou D, Ma Y, Liu J, Bai Q, Li X, Mu S, Gao J. Analysis of MADS-box gene family reveals conservation in floral organ ABCDE model of Moso bamboo (Phyllostachys edulis) Front Plant Sci. 2017;8:656. doi: 10.3389/fpls.2017.00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fornara F, Pařenicová L, Falasca G, Pelucchi N, Masiero S, Ciannamea S, Lopezdee Z, Altamura MM, Colombo L, Kater MM. Functional characterization of OsMADS18, a member of the AP1/SQUA subfamily of MADS box genes. Plant Physiol. 2004;135(4):2207–2219. doi: 10.1104/pp.104.045039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huijser P, Klein J, Lönnig WE, Meijer H, Saedler H, Sommer H. Bracteomania, an inflorescence anomaly, is caused by the loss of function of the MADS-box gene squamosa in Antirrhinum majus. EMBO J. 1992;11(4):1239–1249. doi: 10.1002/j.1460-2075.1992.tb05168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moon YH, Jung JY, Kang HG, An G. Identification of a rice APETALA3 homologue by yeast two-hybrid screening. Plant Mol Biol. 1999;40(1):167–177. doi: 10.1023/A:1026429922616. [DOI] [PubMed] [Google Scholar]
- 40.Pan Z-J, Chen Y-Y, Du J-S, Chen Y-Y, Chung M-C, Tsai W-C, Wang C-N, Chen H-H. Flower development of Phalaenopsis orchid involves functionally divergent SEPALLATA-like genes. New Phytologist. 2014;202(3):1024–1042. doi: 10.1111/nph.12723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ito T, Ng K-H, Lim T-S, Yu H, Meyerowitz EM. The homeotic protein AGAMOUS controls late stamen development by regulating a Jasmonate biosynthetic gene in Arabidopsis. Plant Cell. 2007;19(11):3516–3529. doi: 10.1105/tpc.107.055467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lozano R, Angosto T, Gomez P, Payan C, Capel J, Huijser P, Salinas J, Martinezzapater JM. Tomato flower abnormalities induced by low temperatures are associated with changes of expression of MADS-box genes. Plant Physiol. 1998;117(1):91–100. doi: 10.1104/pp.117.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jia J, Zhao P, Cheng L, Yuan G, Yang W, Liu S, Chen S, Qi D, Liu G, Li X. MADS-box family genes in sheepgrass and their involvement in abiotic stress responses. BMC Plant Biol. 2018;18(1):42. doi: 10.1186/s12870-018-1259-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kobayashi Kaoru, Maekawa Masahiko, Miyao Akio, Hirochika Hirohiko, Kyozuka Junko. PANICLE PHYTOMER2 (PAP2), encoding a SEPALLATA subfamily MADS-box protein, positively controls spikelet meristem identity in rice. Plant and Cell Physiology. 2009;51(1):47–57. doi: 10.1093/pcp/pcp166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saha G, Park JI, Jung HJ, Ahmed NU, Kayum MA, Chung MY, Hur Y, Cho YG, Watanabe M, Nou IS. Genome-wide identification and characterization of MADS-box family genes related to organ development and stress resistance in Brassica rapa. BMC Genomics. 2015;16(1):178. doi: 10.1186/s12864-015-1349-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chiang GCK, Barua D, Kramer EM, Amasino RM, Donohue K. Major FLOWERING time gene, FLOWERING LOCUS C, regulates seed germination in Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2009;106(28):11661–11666. doi: 10.1073/pnas.0901367106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu LH, Wu J, Zhang ZS, Miao ZQ, Zhao PX, Wang Z, Xiang CB. Arabidopsis MADS-box transcription factor AGL21 acts as environmental surveillance for seed germination by regulating ABI5 expression. Mol Plant. 2017;10(6):834–845. doi: 10.1016/j.molp.2017.04.004. [DOI] [PubMed] [Google Scholar]
- 48.Zhang GQ, Liu KW, Li Z, Lohaus R, Hsiao YY, Niu SC, Wang JY, Lin YC, Xu Q, Chen LJ. The Apostasia genome and the evolution of orchids. Nature. 2017;549(7672):379. doi: 10.1038/nature23897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ouyang S, Zhu W, Hamilton J, Lin H, Campbell M, Childs K, Thibaudnissen F, Malek RL, Lee Y, Zheng L. The TIGR Rice genome annotation resource: improvements and new features. Nucleic Acids Res. 2007;35(Database issue):883–887. doi: 10.1093/nar/gkl976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawahara Yoshihiro, de la Bastide Melissa, Hamilton John P, Kanamori Hiroyuki, McCombie W Richard, Ouyang Shu, Schwartz David C, Tanaka Tsuyoshi, Wu Jianzhong, Zhou Shiguo, Childs Kevin L, Davidson Rebecca M, Lin Haining, Quesada-Ocampo Lina, Vaillancourt Brieanne, Sakai Hiroaki, Lee Sung Shin, Kim Jungsok, Numa Hisataka, Itoh Takeshi, Buell C Robin, Matsumoto Takashi. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice. 2013;6(1):4. doi: 10.1186/1939-8433-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu W, Huang X, Cheng J, Li Z, Folter SD, Huang Z, Jiang X, Pang H, Tao S. Conservation and evolution in and among SRF- and MEF2-type MADS domains and their binding sites. Mol Biol Evol. 2011;28(1):501–511. doi: 10.1093/molbev/msq214. [DOI] [PubMed] [Google Scholar]
- 52.Alvarez-Buylla ER, Pelaz S, Liljegren SJ, Gold SE, Burgeff C, Ditta GS, Ribas dPL, Martínez-Castilla L, Yanofsky MF. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc Natl Acad Sci U S A. 2000;97(10):5328. doi: 10.1073/pnas.97.10.5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Díazriquelme J, Lijavetzky D, Martínezzapater JM, Carmona MJ. Genome-wide analysis of MIKCC-type MADS box genes in grapevine. Plant Physiol. 2009;149(1):354–369. doi: 10.1104/pp.108.131052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Project AG The Amborella genome and the evolution of flowering plants. Science. 2013;342(6165):1467. doi: 10.1126/science.1241089. [DOI] [PubMed] [Google Scholar]
- 55.Lovisetto A, Guzzo F, Tadiello A, Toffali K, Favretto A, Casadoro G. Molecular analyses of MADS-box genes trace back to gymnosperms the invention of fleshy fruits. Mol Biol Evol. 2012;29(1):409–419. doi: 10.1093/molbev/msr244. [DOI] [PubMed] [Google Scholar]
- 56.Chen F, Zhang X, Liu X, Zhang L. Evolutionary analysis of MIKCC-type MADS-box genes in gymnosperms and angiosperms. Front Plant Sci. 2017;8:895. doi: 10.3389/fpls.2017.00895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li X, Han J-D, Fang Y-H, Bai S-N, Rao G-Y. Expression analyses of embryogenesis-associated genes during somatic embryogenesis of Adiantum capillus-veneris L. in vitro: new insights into the evolution of reproductive organs in land plants. Front Plant Sci. 2017;8:658. [DOI] [PMC free article] [PubMed]
- 58.Zhao T, Ni Z, Dai Y, Yao Y, Nie X, Sun Q. Characterization and expression of 42 MADS-box genes in wheat (Triticum aestivum L.) Mol Genet Genomics. 2006;276(4):334. doi: 10.1007/s00438-006-0147-3. [DOI] [PubMed] [Google Scholar]
- 59.Mouradov A, Hamdorf B, Teasdale RD, Kim JT, Winter KU, Theissen G. A DEF/GLO-like MADS-box gene from a gymnosperm: Pinus radiata contains an ortholog of angiosperm B class floral homeotic genes. Dev Genet. 1999;25(3):245–252. doi: 10.1002/(SICI)1520-6408(1999)25:3<245::AID-DVG7>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 60.Winter KU, Becker A, Münster T, Kim JT, Saedler H, Theissen G. MADS-box genes reveal that gnetophytes are more closely related to conifers than to flowering plants. Proc Natl Acad Sci U S A. 1999;96(13):7342–7347. doi: 10.1073/pnas.96.13.7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jager M, Hassanin A, Manuel M, Le HG, Deutsch J. MADS-box genes in Ginkgo biloba and the evolution of the AGAMOUS family. Mol Biol Evol. 2003;20(5):842–854. doi: 10.1093/molbev/msg089. [DOI] [PubMed] [Google Scholar]
- 62.Tapia-López R, Alvarez-Buylla ER. An AGAMOUS-related MADS-box gene, XAL1 (AGL12), regulates root meristem cell proliferation and flowering transition in Arabidopsis. Plant Physiol. 2008;146(3):1182–1192. doi: 10.1104/pp.107.108647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Michaels SD, Amasino RM. FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell. 1999;11(5):949–956. doi: 10.1105/tpc.11.5.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liang S, Ye Q-S, Li R-H, Leng J-Y, Li M-R, Wang X-J, Li H-Q. Transcriptional regulations on the low-temperature-induced floral transition in an Orchidaceae species, Dendrobium nobile: an expressed sequence tags analysis. Comp Funct Genom. 2012;2012:757801. doi: 10.1155/2012/757801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mandel MA, Gustafsonbrown C, Savidge B, Yanofsky MF. Molecular characterization of the Arabidopsis floral homeotic gene APETALA1. Nature. 1992;360(6401):273–277. doi: 10.1038/360273a0. [DOI] [PubMed] [Google Scholar]
- 66.Masiero S, Imbriano C, Ravasio F, Favaro R, Pelucchi N, Gorla MS, Mantovani R, Colombo L, Kater MM. Ternary complex formation between MADS-box transcription factors and the histone fold protein NF-YB. J Biol Chem. 2002;277(29):26429–26435. doi: 10.1074/jbc.M202546200. [DOI] [PubMed] [Google Scholar]
- 67.Yanofsky MF, Ma H, Bowman JL, Drews GN, Feldmann KA, Meyerowitz EM. The protein encoded by the Arabidopsis homeotic gene agamous resembles transcription factors. Nature. 1990;346(6279):35–39. doi: 10.1038/346035a0. [DOI] [PubMed] [Google Scholar]
- 68.Kempin SA, Mandel MA, Yanofsky MF. Conversion of perianth into reproductive organs by ectopic expression of the tobacco floral homeotic gene NAG1. Plant Physiol. 1993;103(4):1041–1046. doi: 10.1104/pp.103.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Benedito V. A., Visser P. B., van Tuyl J. M., Angenent G. C., de Vries S. C., Krens F. A. Ectopic expression of LLAG1, an AGAMOUS homologue from lily (Lilium longiflorum Thunb.) causes floral homeotic modifications in Arabidopsis. Journal of Experimental Botany. 2004;55(401):1391–1399. doi: 10.1093/jxb/erh156. [DOI] [PubMed] [Google Scholar]
- 70.Bradley Desmond, Carpenter Rosemary, Sommer Hans, Hartley Nigel, Coen Enrico. Complementary floral homeotic phenotypes result from opposite orientations of a transposon at the plena locus of antirrhinum. Cell. 1993;72(1):85–95. doi: 10.1016/0092-8674(93)90052-R. [DOI] [PubMed] [Google Scholar]
- 71.Wang SY, Lee PF, Lee YI, Hsiao YY, Chen YY, Pan ZJ, Liu ZJ, Tsai WC. Duplicated C-class MADS-box genes reveal distinct roles in gynostemium development in Cymbidium ensifolium (Orchidaceae) Plant Cell Physiol. 2011;52(3):563–577. doi: 10.1093/pcp/pcr015. [DOI] [PubMed] [Google Scholar]
- 72.Chang YY, Chiu YF, Wu JW, Yang CH. Four orchid (Oncidium Gower Ramsey) AP1/AGL9-like MADS box genes show novel expression patterns and cause different effects on floral transition and formation in Arabidopsis thaliana. Plant Cell Physiol. 2009;50(8):1425–1438. doi: 10.1093/pcp/pcp087. [DOI] [PubMed] [Google Scholar]
- 73.Prakash AP, Kumar PP. PkMADS1 is a novel MADS box gene regulating adventitious shoot induction and vegetative shoot development in Paulownia kawakamii. Plant J. 2010;29(2):141–151. doi: 10.1046/j.0960-7412.2001.01206.x. [DOI] [PubMed] [Google Scholar]
- 74.Kofuji R, Sumikawa N, Yamasaki M, Kondo K, Ueda K, Ito M, Hasebe M. Evolution and divergence of the MADS-box gene family based on genome-wide expression analyses. Mol Biol Evol. 2003;20(12):1963–1977. doi: 10.1093/molbev/msg216. [DOI] [PubMed] [Google Scholar]
- 75.Kwantes M, Liebsch D, Verelst W. How MIKC* MADS-box genes originated and evidence for their conserved function throughout the evolution of vascular plant gametophytes. Mol Biol Evol. 2012;29(1):293–302. doi: 10.1093/molbev/msr200. [DOI] [PubMed] [Google Scholar]
- 76.Zobell O, Faigl W, Saedler H, Münster T. MIKC* MADS-box proteins: conserved regulators of the gametophytic generation of land plants. Mol Biol Evol. 2010;27(5):1201–1211. doi: 10.1093/molbev/msq005. [DOI] [PubMed] [Google Scholar]
- 77.Liu Y., Cui S., Wu F., Yan S., Lin X., Du X., Chong K., Schilling S., Theissen G., Meng Z. Functional Conservation of MIKC*-Type MADS Box Genes in Arabidopsis and Rice Pollen Maturation. The Plant Cell. 2013;25(4):1288–1303. doi: 10.1105/tpc.113.110049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Larkin M, Blackshields G, Brown N, Chenna R, McGettigan P, McWilliam H, Valentin F, Wallace I, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 79.Niu SC, Xu Q, Zhang GQ, Zhang YQ, Tsai WC, Hsu JL, Liang CK, Luo YB, Liu ZJ. De novo transcriptome assembly databases for the butterfly orchid Phalaenopsis equestris. Scientific Data. 2016;3:160083. [DOI] [PMC free article] [PubMed]
- 80.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu Z-G, Jiang W, Chen S-L, Mantri N, Tao Z-M, Jiang C-X. Insights from the cold transcriptome and metabolome of Dendrobium officinale: global reprogramming of metabolic and gene regulation networks during cold acclimation. Front Plant Sci. 2016;7:1653. doi: 10.3389/fpls.2016.01653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen Juan, Liu Si Si, Kohler Annegret, Yan Bo, Luo Hong Mei, Chen Xiao Mei, Guo Shun Xing. iTRAQ and RNA-Seq Analyses Provide New Insights into Regulation Mechanism of Symbiotic Germination of Dendrobium officinale Seeds (Orchidaceae) Journal of Proteome Research. 2017;16(6):2174–2187. doi: 10.1021/acs.jproteome.6b00999. [DOI] [PubMed] [Google Scholar]
- 84.Murashige T, Skoog F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant. 1962;15(3):473–497. doi: 10.1111/j.1399-3054.1962.tb08052.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Information of MADS-box genes in the 29 tested species. (DOCX 153 kb)
Figure S1. BLASTP graphic overview of four MADS-box proteins from chlorophytes based on the National Center for Biotechnology Information (NCBI) database. (A) MADS-box protein (Cre18.g749550.t1.1) from Chlamydomonas reinhardtii. (B) and (C) MADS-box proteins (Vocar.0002 s0667.1.p and Vocar.0014 s0224.1.p) from Volvox carteri. (D) MADS-box protein (21861) from Micromonas pusilla CCMP1545. (DOCX 155 kb)
Figure S2. Alignment of the amino acid sequences of only one MADS-box gene (GAQ89767.1) from Klebsormidium nitens and AtSVP (AT2G22540.1) and AtAGL24 (AT4G24540.1) of the SVP subfamily, as well as AtAGL65 (AT1G18750.1) and AtAGL66 (AT1G77980.1) of MIKC* genes from Arabidopsis thaliana. The GAQ89767.1 protein contains M-, I, K- and C domains, which are indicated by red, yellow, green and blue boxes, respectively. (DOCX 1228 kb)
Table S2. Information of MADS-box genes in Arabidopsis thaliana, Oryza sativa, Brassica rapa, Brachypodium distachyon, Zea mays, Sorghum bicolor, Phalaenopsis equestris, Dendrobium officinale, Physcomitrella patens, and Selaginella moellendorffii. (DOCX 17 kb)
Figure S3. Classification of the total of 983 MIKC-type proteins based on a phylogenetic analysis. MIKC-type proteins can be divided into two main groups MIKC* and MIKCC. The phylogenetic tree was constructed by MEGA 7 with the NJ method based on MAFFT 7-based alignments. (DOCX 767 kb)
Figure S4. Classification of the total 865 MIKCC proteins based on phylogenetic analysis. The phylogenetic tree was conducted using MEGA 7 based on the alignment of MADS-box proteins by MAFFT 7 with the Neighbor-Joining method. Numbers besides branches represent bootstrap support values from 1000 replications. Values lower than 40% are hidden. (DOCX 2621 kb)
Figure S5. Phylogenetic analyses of GAQ89767.1 from Klebsormidium nitens and MADS-box proteins from Arabidopsis thaliana (At). The phylogenetic tree was conducted using MEGA 7 based on the alignment of MADS-box proteins by MAFFT 7 with the NJ method. Numbers besides branches represent bootstrap support values from 1000 replications. (DOCX 428 kb)
Figure S6. Phylogenetic analyses of the only two MADS-box proteins (Mapoly0011s0161.1.p and Mapoly0174s0011.1.p) from Marchantia polymorpha and MADS-box proteins from Arabidopsis thaliana (At). Mapoly0011s0161.1.p falls into the SVP subfamily (29% similarity). Mapoly0174s0011.1.p falls into the MIKC* group. The phylogenetic tree was conducted using MEGA 7 based on the alignment of MADS-box proteins by MAFFT 7 with the NJ method. Numbers besides branches represent bootstrap support values from 1000 replications. (DOCX 413 kb)
Table S3. The mean of FPKM value and fold change of MIKC gene under control and cold stress in D. officinale leaf. (DOCX 17 kb)
Table S4. FPKM value of the MIKC gene in Dendrobium officinale during four seed germination stages. (DOCX 16 kb)
Table S5. The versin of genomic data and download websites. (DOCX 16 kb)
Table S6. The primers used for Semi-quantitative RT-PCR. (DOCX 14 kb)
Data Availability Statement
All data generated or analysed during this study are included in this published article and its supplementary information files. The datasets used and analysed during the current study are available from the corresponding author on reasonable request.