

Abstract
Despite their phenotypic heterogeneity, most human prion diseases belong to two broadly defined groups: Creutzfeldt-Jakob disease (CJD) and Gerstmann-Sträussler-Scheinker disease (GSS). While the structural characteristics of the disease-related proteinase K-resistant prion protein (resPrPD) associated with the CJD group are fairly well established, many features of GSS-associated resPrPD are unclear. Electrophoretic profiles of resPrPD associated with GSS variants typically show 6–8 kDa bands corresponding to the internal PrP fragments as well as a variable number of higher molecular weight bands, the molecular nature of which has not been investigated. Here we have performed systematic studies of purified resPrPD species extracted from GSS cases with the A117V (GSSA117V) and F198S (GSSF198S) PrP gene mutations. The combined analysis based on epitope mapping, deglycosylation treatment and direct amino acid sequencing by mass spectrometry provided a conclusive evidence that high molecular weight resPrPD species seen in electrophoretic profiles represent covalently-linked multimers of the internal ~ 7 and ~ 8 kDa fragments. This finding reveals a mechanism of resPrPD aggregate formation that has not been previously established in prion diseases.
Electronic supplementary material
The online version of this article (10.1186/s40478-019-0734-2) contains supplementary material, which is available to authorized users.
Keywords: Creutzfeldt-Jakob disease, Prion protein, Aggregate formation, Multimers, Mass spectrometry, Epitope mapping
Introduction
A well-known feature of human prion diseases is the presence of three distinct etiologic forms - sporadic, inherited and acquired by infection. A further complicating factor is the great phenotypic variability within each of these etiologically distinct groups. For example, the sporadic form alone encompasses seven major disease phenotypes [15, 16, 39], and many different variants have been reported for inherited prion diseases [20, 21]. This phenotypic variability is believed to be directly related to (and likely encoded in) distinct strains of the disease-related prion protein (PrPD). However, the nature and extent of specific structural differences between phenotype-specific PrPD strains remain poorly understood [15].
According to the classification based on electrophoretic profiles of the proteinase K (PK)-resistant PrPD (resPrPD), most cases of sporadic and inherited prion diseases fall into two broadly defined groups: Creutzfeldt-Jakob disease (CJD) and Gerstmann-Sträussler-Scheinker disease (GSS). CJD is characterized by the presence of relatively large resPrPD fragments which, depending on the N-terminus, are classified as type 1 (typically starting at residue G82) and type 2 (starting at residue S97) [15, 26]. Both fragment types extend to the C-terminus and include the glycosylphosphatidylinositol (GPI) anchor [16, 39]. Electrophoretic profiles of these fragments include bands of approximately 30 and 27 kDa (representing the di- and mono-glycosylated forms) as well as bands of 21 and 19 kDa which represent the un-glycosylated form in resPrPD types 1 and 2, respectively. Collectively, these three resPrPD fragments are commonly referred to as PrP 27–30 [8]. A remarkably different electrophoretic profile is observed in GSS, where the most prominent and by far best characterized resPrPD species is a 6–8 kDa fragment encompassing internal residues from within the ~ 70–150 region [12, 25, 27, 33, 34]. Higher molecular weight (hmw) bands of variable estimated molecular weights have also been reported, occasionally prompting the hypothesis that they represent multimers. However, the molecular nature of the PrP fragments giving rise to these bands remains enigmatic [12, 17, 23].
To bridge this gap, here we have performed detailed analysis of purified resPrPD preparations from GSS cases harboring the A117V (GSSA117V) and F198S (GSSF198S) PrP mutations. Our data demonstrate that high molecular weight species seen in electrophoretic profiles of GSSA117V and GSSF198S resPrPD represent covalently-linked multimers of the same internal PrP fragments that are present (as monomers) in the ~ 7 and ~ 8 kDa bands.
Materials and methods
Reagents and antibodies
β–mercaptoethanol, Dithiothreitol, Laemmli Sample Buffer, Non-fat dry milk, Sodium dodecyl sulfate (SDS), Tris Buffered Saline (TBS), Tris/Glycine/SDS buffer, Tris/Glycine Buffer, Tween 20 and 15% Criterion Tris-HCl polyacrylamide precast gels were purchased from Bio-Rad Laboratories (Hercules, CA, USA). Benzonase, Calcium chloride, Complete Ultra Protease Inhibitor Cocktail Tablets, Dulbecco’s PBS (D-PBS), Kodak Biomax MR and XAR films, NaCl, Nonidet P-40, N-Lauroylsarcosine sodium salt solution (Sarkosyl), Phenylmethanesulfonyl fluoride (PMSF), Polyvinylidene difluoride (PVDF) membrane (Immobilon-P), Proteinase K, Sodium deoxycholate and Tris-HCl came from MilliporeSigma (Burlington, MA, USA) whereas Ethylenediaminetetraacetic acid (EDTA) from Promega (Madison, WI, USA). Glycerol-free PNGase F was from New England Biolabs Inc. (Ipswich, MA, USA); Acetonitrile, Colloidal Blue Staining Kit, Ethanol, Formic Acid, 8 M Guanidine-HCl Solution, Methanol, Pierce ECL 2 Western Blotting Substrate and Trypsin from Thermo Fisher Scientific Inc. (Waltham, MA, USA).
The following antibodies (Abs) were used in the study: 8B4 (to human PrP residues 36–43) [22], SAF32 (to human octarepeat region) [14] (Cayman Chemical, Ann Arbor, MI, USA), 3F4 (to human PrP residues 106–110) [19, 42], F89 (to human PrP residues 139–142) (Thermo Fisher Scientific Inc., Waltham, MA, USA), L42 (to human PrP residues 145–150) [18, 38] (R-Biopharm, AG, Darmstadt, Germany) and SAF70 (to human PrP residues 156–162) [14]. Secondary Ab was sheep anti-mouse IgG, HRP-linked whole antibody (GE Healthcare Life Sciences, Chicago, IL, USA).
Brain tissues
All frozen brain tissues were obtained from the National Prion Disease Pathology Surveillance Center (NPDPSC). Frontal cortex samples from two cases each of GSS A117V–129V (129MV and 129VV) and F198S-129V (129MV) were used. All cases were used for immunoblotting, but only one case of GSSA117V and GSSF198S, both with 129MV genotype, were used for mass spectrometry. A case of sCJDMV1 was used as control.
Preparation of brain homogenates
Brain homogenates (BH) were prepared as previously described [13].
Proteinase K digestion
Samples were incubated for 1 h at 37 °C with different amounts of PK, as follows: GSSA117V: purified PrPD (PK 2 U/ml); GSSF198S: BH (PK 5 U/ml), purified PrPD (PK 10 U/ml); sCJDMV1: purified PrPD (PK 10 U/ml). The reaction was stopped by the addition of 3 mM PMSF.
Epitope mapping
The optimal PK concentrations and the volumes of purified GSSF198S, GSSA117V and sCJDMV1 resPrPD loaded into the gel were selected to obtain a clear visibility of all the bands with 3F4. Recombinant human PrP full-length (23–231) and truncated (90–231) species were used as molecular weight markers.
Methanol/ethanol precipitation
Methanol precipitation was performed as previously described [13]. For ethanol precipitation the same procedure was followed with the exception of 9 volumes of pre-chilled ethanol instead of 5 volumes used to dilute the sample.
PrP deglycosylation
Samples were treated with glycerol-free PNGase F in accordance with manufacturer’s instructions.
Electrophoresis and immunoblot
Protein samples were run on 15% Criterion Tris-HCl polyacrylamide precast gels and then subjected to immunoblot (60 V for 2 h) using Immobilon-P PVDF membranes. After 1 h blocking in 5% non-fat dry milk in TBS with 0.1% Tween 20 (TBS-T), the membranes were incubated with primary Ab overnight at 4 °C; they were then washed with TBS-T, incubated for 1 h at RT with the secondary HRP-conjugated Ab and then washed again prior to developing by enhanced chemiluminescence reaction using ECL 2 western blotting substrate, as per manufacturer’s instructions. Kodak MR and XAR films were used to capture the signal.
ResPrPD purification
ResPrPD purification was performed as reported in Bolton et al. [7] and adapted by Zou et al. [40]. Rotor SW55Ti (Beckman Coulter, Brea, CA, USA) was used for ultracentrifugation.
In-gel trypsin digestion of purified resPrPD
In-gel trypsin digestion was performed according to an established protocol [31]. Briefly, the protein bands of interest (visualized by Colloidal Coomassie blue staining) were excised from the gel, cut into small cubes and destained. The destained gel pieces were then dehydrated and followed by reduction/alkylation step. For digestion, trypsin solution (15 ng/μl) was added and gel pieces were incubated overnight at 37 °C with shaking. The supernatant was then transferred into a new Eppendorf tube and the remaining peptides in gel pieces were extracted, supernatants were pooled, concentrated and stored at − 80 °C until analyzed by Nano LC-MS.
Nano LC-MS/MS
Nanospray LC-MS-MS analysis was performed using an LTQ Orbitrap XL mass spectrometer equipped with nanoelectrospray source (Thermo Scientific, San Jose, CA, USA). Trypsin-digested samples were loaded onto a C-18 trap column (to remove salts) and separated on a C-18 column connected to an emitter. Separation was performed using a Dionex UltiMate 3000 system (Thermo Scientific, San Jose, CA, USA) and a gradient of acetonitrile in water containing 0.1% formic acid. The flow rate was 300 nl/min. The mass spectrometer was externally calibrated using a Pierce LTQ ESI positive ion calibration solution (Thermo Scientific, catalog number 88322). Full scan experiments were acquired in the m/z 300–1800 range at a resolution of 30,000 (FWHM at m/z 400). The following source settings were used: spray voltage = 4.2 kV; capillary temperature = 200 °C. Data-dependent MSn (n = 2) were acquired at ITMS using collision induced dissociation (CID); the top 14 intense ions were subjected for further fragmentation. Calculation of elemental formulae was performed on the mono-isotopic peak of each ion cluster using Xcalibur software v2.2 with a mass tolerance of 3 to 5 ppm. MS/MS raw files were searched using MASCOT Deamon engine against the database containing sequence of human prion protein 129M/V with mutation A117V or F198S. Trypsin/P search parameters for Mascot peptide identification included one missed tryptic cleavage, fixed carbamidomethylation (+ 57 Da, Cys), and variable oxidation (+ 16 Da, Met). Mass tolerances of 2.0 and 1.0 Da were used for parent and monoisotopic fragment ions, respectively. The resulting files generated by MASCOT were used for peptide identification with the constraints that only MASCOT ion scores greater than 10 were considered. The percentage of 129M and 129V PrP in resPrPD samples was calculated by the spectral counting method [4].
Results
The characteristics of resPrPD in GSSF198S and GSSA117V were examined and compared with those of sCJDMV1 resPrPD, used as control, by combining epitope mapping with enzymatic deglycosylation on immunoblots of purified resPrPD preparations (Fig. 1). Overall, GSSF198S and GSSA117V showed similar electrophoretic profiles that were easily distinguishable from the profile of sCJDMV1 (Fig. 1). GSSF198S and GSSA117V displayed low mw bands of ~ 8 and ~ 7 kDa as well as two broad bands comprised between 17-20 23–24 kDa for GSSF198S, and 16–18 and 22–23 kDa for GSSA117V; additional bands and smears of higher mw were also present more prominently in GSSF198S (Fig. 1). By contrast, resPrPD profile in sCJDMV1 included the typical three glycoform bands of ~ 30, ~ 27, and ~ 21 kDa of resPrPD type 1. Two additional unique characteristics further distinguished GSSA117V and GSSF198S from sCJDMV1. Virtually all resPrPD bands associated with these two GSS conditions were readily detected only with Abs to epitopes within the PrP ~ 51–150 region, but not with Abs recognizing more C-terminal epitopes (Fig. 1, panels b-f). Furthermore, GSSA117V and GSSF198S resPrPD (as well as resPrPD from sCJDMV1) showed no immunoreactivity with 8B4 Ab that recognizes the N-terminal epitope 36–43 (Fig. 1, panel a). This finding excludes the presence of full length resPrPD in these two GSS variants, and is at variance with the presence of full-length resPrPD reported in GSSH187R [12]. In addition to epitope mapping, a second major distinguishing feature was that, in contrast to sCJDMV1 resPrPD, deglycosylation had no significant effect on the electrophoretic mobility of any of the major resPrPD bands observed in GSSF198S and GSSA117V, strongly suggesting that resPrPD populating these bands is not glycosylated (Fig. 1). In a separate experiment carried out in GSSF198S, we also observed that the resPrPD profile did not change after pretreatment of samples with strong denaturants such as guanidine hydrochloride and urea (Additional file 1: Figure S1). Collectively, these data suggest that at least two hmw bands seen in immunoblots of GSSA117V and GSSF198S resPrPD represent covalently-linked multimers of the ~ 7 and ~ 8 kDa fragments (Fig. 1 and Additional file 1: Figure S1).
Fig. 1.

Epitope mapping combined with deglycosylation of purified resPrPD associated with GSSF198S, GSSA117V and sCJDMV1 used as control. PNGase F-treated or untreated, PK-resistant PrPD (resPrPD) purified (see Materials and Methods) from GSSF198S, GSSA117V and sCJDMV1 control were blotted and probed with the Abs to PrP denoted, with their epitopes, at the top of each panel. Panel a: Only full-length (PK-untreated) recombinant PrP (23–231) was detected by this Ab to the proximal N-terminal region confirming the absence of resPrPD with complete N-terminus. Panels b to e: Both GSSF198S and GSSA117V resPrPD conformers were selectively detected by the same Abs to PrP N- and C-terminal regions; both also showed no major variation of the resPrPD banding pattern following deglycosylation indicating that most of the resPrPD is unglycosylated in both conditions. Although overall similar, the two resPrPD profiles differed especially in the higher molecular weight region suggesting a distinct repertoire, or relative proportions, of polymers in the two GSS variants. The GSS banding patterns clearly differed from the three-band pattern of sCJDMV1, two of which are glycosylated (See Results for detailed description). Panel c includes the bands of both 23–231 and 90–231 recombinant PrP which have been used as molecular weight markers. In b the portion of the panel with the GSSA117V samples required longer exposure. In panel f, * indicates the 13 kDa component of the glycosylated and anchor bearing PrP 12/13 C-terminus fragments with N-termini at residues 162–167 and 154–156, respectively [24, 41]
To definitely assess whether the higher molecular bands represented multimers (likely covalently-linked) of the ~ 7 and ~ 8 kDa internal fragments, as our epitope mapping and glycosylation study strongly suggested, we performed trypsin in-gel digestion of protein fragments on individual electrophoretic bands of purified resPrPD from GSSA117V and GSSF198S (~ 7, 16–18 and 22–23 kDa in the case of GSSA117V and ~ 8, 17–20 and 23–24 kDa in the case of GSSF198S) and did amino acid sequencing using mass spectrometry (Nano LC-MS).
The tryptic digest of the ~ 7 kDa resPrPD fragment extracted from GSSA117V and analyzed by Nano LC-MS revealed the presence of peptides exclusively from the central region of PrP between residues 78 and 152, with no detectable fragments from other parts of the protein. Trypsin is known to cleave polypeptide chains at the carboxyl side of lysine (K) or arginine (R). As shown in Fig. 2, seven potential trypsin cleavage sites are available within the central region of PrP between residues 70 and 153. Nano LC-MS analysis of the GSSA117V ~ 7 kDa fragment using the Mascot Deamon searching engine identified fourteen trypsin-generated peptides with N-termini before the first potential trypsin cleavage site, two internal tryptic fragments, and twelve trypsin-generated peptides with N-terminus at the cleavage site following R136 (Fig. 2a and Additional file 2: Table S1). Altogether, these data demonstrated that in GSSA117V the ~ 7 kDa fragment encompassed residues within the 78–152 region and had ragged N- and C-termini corresponding to residues 78/82/85–88 and 141–152, respectively. This is generally consistent with previous MALDI-derived data from extracts of PrP amyloid plaque cores, even though our Nano LC-MS analysis (which is more accurate) revealed that the N-terminus of this fragment may extend as far as up to residue 78 versus residue 85 previously reported based on the MALDI analysis [33].
Fig. 2.

Mass spectrometry (MS)-based sequencing of PrP fragments in the material extracted from individual gel bands of two GSS variants. GSSA117V resPrPD (a) and GSSF198S resPrPD (b). The material present in these bands was subjected to trypsin digestion, followed by MS analysis of tryptic fragments. The fragments identified by MS for species extracted from individual gel bands are shown as blue, red and green lines. Amino acid sequence within the 70-152 region is shown above the lines. Residues marked in red represent potential cleavage sites in the relevant region of PrP; 129M/V polymorphic residues are marked in blue
A similar sequencing analysis of the 16–18 kDa and 22–23 kDa bands revealed the presence of essentially identical tryptic fragments, with the exception that the most N-terminal fragment started at residue 82, and the most C-terminal fragment ended at residue 150 (Fig. 2a). Remarkably, we could not detect any tryptic peptides from the region C-terminal to residue 150. Given that numerous peptides from the latter region are readily detectable by MS upon proteolytic digestion of resPrPD from sCJD cases and mouse prion strains [1, 30], one can definitely conclude from these data that the 16–18 and 22–23 kDa species in GSSA117V resPrPD indeed represent oligomers (likely trimers and tetramers, respectively) of internal fragments encompassing residues within the 82–150 region. It is of note that fragments in the oligomers are somewhat shorter compared to those in the ~ 7 kDa band.
MS-based sequencing analysis of protein present in the resPrPD ~ 8 kDa band from GSSF198S demonstrated that this band contained rugged internal resPrPD fragments from the 70–152 region, with N-termini between residues 70 and 90 and C-termini between residues 141 and 152 (Fig. 2b and Additional file 2: Table S2). Again, MS analysis did not reveal the presence of peptides from parts of PrP other than the central region between residues 70 and 152. Previous sequencing studies of the ~ 8 kDa fragment (or fragments of similar kDa) extracted from PrP amyloid plaques and analyzed by Edman degradation chemistry alone or combined with automated sequencing identified the major N-terminus at residues G58, G74 and G81 while the C-terminus was reported at residue 150 [27, 34, 35].
Very similar internal fragments were identified in higher molecular bands of 17–20 and 23–24 kDa in GSSF198S resPrPD, with the exception that the longest of these fragments had N-termini at residue 74 and 78 in the 17–20 and 23–24 kDa bands, respectively (Fig. 2b). Importantly, akin to the finding for GSSA117V resPrPD, no peptides from the region C-terminal to residue 152 were detected by MS in tryptic digests of protein in these two higher molecular weight bands. Thus, also in GSSF198S resPrPD, the latter bands contained covalently-linked oligomers of the internal PrP fragments within the 74–78/142–152 region, which, as in GSSA117V, are somewhat shorter than the ~ 8 kDa monomer.
Using Nano LC-MS we also determined the relative representation in resPrPD of the 129M and 129V polymorphic forms of the prion protein. Consistent with a previous report, resPrPD from GSSA117V was invariably 100% 129V, with no detectable 129M polymorph [33]. By contrast, and at variance with a previous report, in GSSF198S resPrPD, all three bands examined consistently showed the presence of both polymorphic variants, with a large dominance (76–88%) of the 129V polymorph (Fig. 3) [35].
Fig. 3.
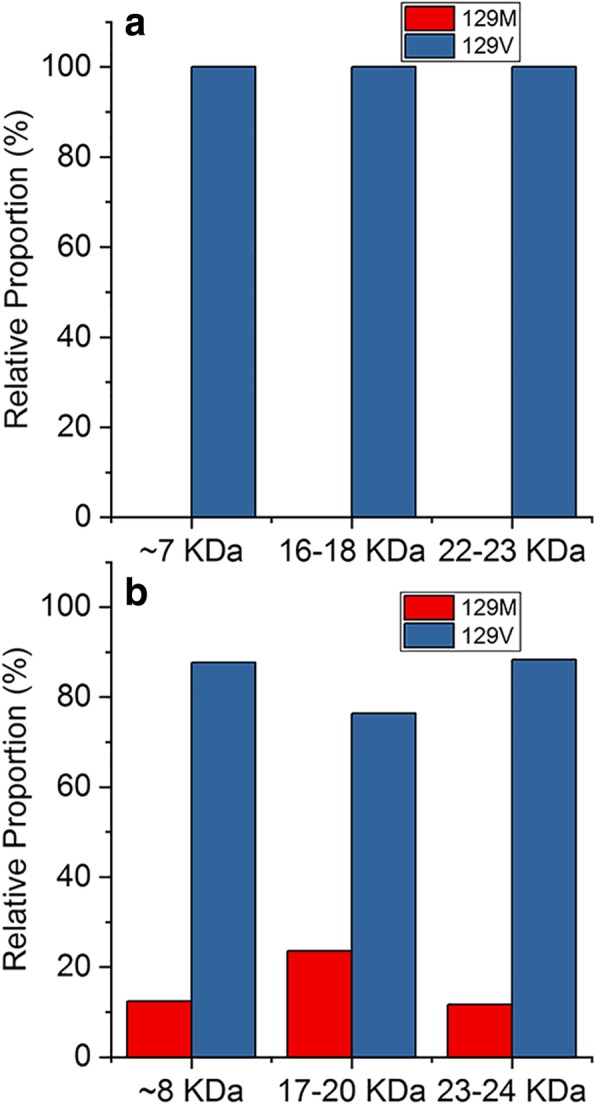
Relative abundance of 129M and 129V PrP variants in resPrPD associated with GSSA117V and GSSF198S. a: GSSA117V; b: GSSF198S. The relative abundance of resPrPD with M or V at residue 129 reflects the representation of the PrP mutation, which is coupled with the 129V in both GSS variants. Approximately 10–25% of resPrPD could be identified as non-mutated (wild type) in GSSF198S while only mutated resPrPD could be detected in GSSA117V. The relative populations were determined by mass spectrometry using the spectral counting method
Discussion
The present results indicate that the mechanism of resPrPD aggregation in GSSA117V and GSSF198S involves formation of covalently-linked multimers of the ~7-8 kDa internal fragments. Furthermore, it has been previously shown by Edman degradation chemistry that the 7 and 14 kDa fragments in GSSH187R resPrPD share the N-terminus, suggesting that formation of covalently-linked multimers may also take place in the latter GSS variant [12]. PrP bands suggestive of dimerization have been reported in cell systems and brain tissues [13, 29]. However, the resistance to PK digestion of these PrP species and, more importantly, their molecular nature have not been determined. In this context, the present data provide the strongest evidence to date for the presence of covalently cross-linked species of resPrPD in prion diseases, suggesting that these previously unrecognized species may play a major role in the pathogenesis of human prion diseases. Of note, covalent crosslinking has been reported for toxic aggregates of α-synuclein and Aβ involved in Parkinson’s and Alzheimer’s diseases, respectively [2, 3, 9, 10, 32].
This novel finding has important implications for understanding phenotypic variability in human prion diseases. Even though resPrPD aggregates associated with both CJD and GSS phenotypes have been shown to be transmissible, the dramatic difference between these species suggests fundamentally different structural mechanisms of prion protein conformational conversion in GSS as compared with those in CJD [6, 28]. Indeed, the absence of the constraints imposed by glycans and the GPI anchor (that are missing in ~ 7–8 kDa fragments and their covalently-linked multimers) is likely to allow for different types of resPrPD assemblies. A potential structural model for the ~ 7–8 kDa fragments and their multimers is provided by synthetic amyloid fibrils generated from PrP23–144, a protein matching the sequence of the C-terminally truncated PrP harbored in GSSY145Stop [11, 17]. These synthetic amyloid fibrils adopt a parallel in-register β-structure [36] that is fundamentally different from the 4-rung solenoid model proposed for the CJD-associated resPrPD [5, 37]. Remarkably, diseased mice inoculated with PrP23–144 fibrils accumulate GSS-like resPrPD aggregates (consisting of ~ 7 kDa fragment) that likely share the structural motif of amyloid fibrils used as the inoculum [11].
The nature of the covalent crosslinks between PrP fragments and the residues involved in these crosslinks are at present unknown. However, our data strongly suggest that at least two different types of linkages (between different residues) are likely involved in coupling the monomers that form trimers and tetramers, since a single linkage type would preclude mass spectrometric identification of all tryptic fragments from the ~ 80–150 region.
Although the basic structural characteristics and mode of aggregation of resPrPD in GSSA117V and GSSF198S appear to be similar, at least two features distinguish these two conditions. First, substantially more resPrPD populates the hmw regions of the immunoblot in GSSF198S than in GSSA117V, suggesting a stronger propensity for covalent polymerization in GSSF198S. Second, resPrPD associated with GSSA117V consists solely of fragments containing the 129V polymorphic form of PrP, whereas both polymorphs are present in GSSF198S, even though the 129V form predominates. Since pathogenic mutations in both GSS variants are on the background of 129V PrP, this implies that resPrPD in GSSF198S contains internal PrP fragments derived both from both the mutant as well as wild-type proteins. By contrast, the internal PrP fragments populating GSSA117V resPrPD represent exclusively the mutant protein. This suggests that the A117V mutation (which is within the PK-resistant fragments) may impede the templated conversion of wild type PrP. No such impediment would be expected in GSSF198S where the pathogenic mutation is outside the PK-resistant region.
Recently, a novel eight-residue insertion in the hydrophobic region of PrP has been associated with a GSS phenotype [23]. Brains of transgenic mice harboring the corresponding mouse insertion variation revealed the presence of a 8 kDa resPrPD fragment similar to those observed in other GSS variants as well as a 16 kDa thermolysin-resistant fragment mapping to residues ~ 23–155. Based on this finding and other data, it was suggested that the 8 kDa internal fragment derived from the N-terminal truncation of the 16 kDa fragment and that this mechanism was shared by other GSS variants. While this hypothesis may apply to the mouse model and, possibly, to this specific GSS insertion variant, our demonstration that hmw bands harbor oligomers of the ~ 7 and ~ 8 kDa fragments in GSSA117V and GSSF198S (and possibly GSSH187R) makes the proposed mechanism not applicable to these GSS variants.
Conclusion
We demonstrated for the first time that the formation of multimers of a small internal fragment is the primary mechanism of prion aggregation in GSSA117V and GSSF198S, two classical variants of Gerstmann-Sträussler-Scheinker disease (GSS). Prion aggregation by covalently-linked multimer formation from a small glycan- and GPI-free fragment is likely to allow for novel prion assembles. This finding opens new horizons and likely will stimulate new research. Covalently-linked multimers formation has been reported in other neurodegenerative diseases such as Parkinson’s and Alzheimer’s diseases.
Additional files
Figure S1. Further biochemical characterization of resPrPD associated with GSSF198S. Lane 1: PNGase F-deglycosylated resPrPD from brain homogenate immunoblotted with 3F4 following standard conditions. Lane 2: with additional boiling, freezing-thawing and sonication pre-deglycosylation; lane 3: 41 h PNGase F treatment; lanes 4–7: incubation with strong denaturants, 8 M urea (after ethanol or methanol precipitation, lanes 4, 5) and 8 M guanidine hydrochloride at 80 °C (after ethanol or methanol precipitation, lanes 6, 7) pre-deglycosylation. Treatments had no detectable effect on the resPrPD electrophoretic profile. (TIF 17802 kb)
Table S1. Tryptic peptides identified for ~ 7 KDa band in GSSA117V resPrPD. Residue at position 129 is marked in blue. Table S2. Tryptic peptides identified for ~ 8 KDa band in GSSF198S resPrPD. (PDF 122 kb)
Acknowledgements
The authors thank the CJD Foundation, the patients’ families and the National Prion Disease Pathology Surveillance Center, in particular Brian S. Appleby, MD, NPDPSC Director, as well as Mses. Janis Blevins, Katie Glisic and Mr. Aaron Foutz.
Funding
This study was supported by the US National Institutes of Health Grants R01 NS083687, P01 AI106705 (to WKS and PG), P01 AI077774 (to PG), R01 NS103848 (to WKS) and The Charles S. Britton Fund (to PG).
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- Ab
Antibody
- BH
Brain homogenates
- CJD
Creutzfeldt-Jakob disease
- GPI
Glycosylphosphatidylinositol
- GSS
Gerstmann-Sträussler-Scheinker disease
- hmw
Higher molecular weight
- K
Lysine
- kDa
Kilodaltons
- M
Methionine
- MS
Mass spectrometry
- mw
Molecular weight
- Nano LC-MS/MS
Nano liquid chromatography mass spectrometry
- NPDPSC
National Prion Disease Pathology Surveillance Center
- PK
Proteinase K
- PMSF
Phenylmethanesulfonyl fluoride
- PrPD
Disease-related prion protein
- PVDF
Polyvinylidene difluoride
- R
Arginine
- resPrPD
Disease-related proteinase K-resistant prion protein
- SDS
Sodium dodecyl sulfate
- TBS
Tris buffered saline
- V
Valine
Authors’ contributions
Conception and design of the project: LC, WKS, PG. Acquisition of data: LC, XX, SKN, JL, IC. Analysis and interpretation of the data: LC, XX, SKN, SN, WKS, PG. Writing of the manuscript: LC, XX, SKN, WKS, PG. Revision of the manuscript: LC, XX, SKN, JL, IC, BG, SN, WKS, PG. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All procedures were performed under protocols approved by the Institutional Review Board at Case Western Reserve University. Written informed consent for research was obtained from all patients or legal guardians according to the Declaration of Helsinki. All patients’ data and samples were coded and handled in accordance with NIH guidelines to protect patients’ identities.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Aguilar-Calvo P, Xiao X, Bett C, Erana H, Soldau K, Castilla J, Nilsson KP, Surewicz WK, Sigurdson CJ. Post-translational modifications in PrP expand the conformational diversity of prions in vivo. Sci Rep. 2017;7:43295. doi: 10.1038/srep43295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Hilaly YK, Biasetti L, Blakeman BJ, Pollack SJ, Zibaee S, Abdul-Sada A, Thorpe JR, Xue WF, Serpell LC. The involvement of dityrosine crosslinking in alpha-synuclein assembly and deposition in Lewy bodies in Parkinson's disease. Sci Rep. 2016;6:39171. doi: 10.1038/srep39171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Hilaly YK, Williams TL, Stewart-Parker M, Ford L, Skaria E, Cole M, Bucher WG, Morris KL, Sada AA, Thorpe JR, Serpell LC. A central role for dityrosine crosslinking of amyloid-beta in Alzheimer's disease. Acta Neuropathol Commun. 2013;1:83. doi: 10.1186/2051-5960-1-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bantscheff M, Lemeer S, Savitski MM, Kuster B. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal Bioanal Chem. 2012;404:939–965. doi: 10.1007/s00216-012-6203-4. [DOI] [PubMed] [Google Scholar]
- 5.Baskakov IV, Caughey B, Requena JR, Sevillano AM, Surewicz WK, Wille H. The prion 2018 round tables (I): the structure of PrP(Sc) Prion. 2019;13:46–52. doi: 10.1080/19336896.2019.1569450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bishop MT, Will RG, Manson JC. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A. 2010;107:12005–12010. doi: 10.1073/pnas.1004688107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolton DC, Bendheim PE, Marmorstein AD, Potempska A. Isolation and structural studies of the intact scrapie agent protein. Arch Biochem Biophys. 1987;258:579–590. doi: 10.1016/0003-9861(87)90380-8. [DOI] [PubMed] [Google Scholar]
- 8.Bolton DC, Meyer RK, Prusiner SB. Scrapie PrP 27-30 is a sialoglycoprotein. J Virol. 1985;53:596–606. doi: 10.1128/jvi.53.2.596-606.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borsarelli CD, Falomir-Lockhart LJ, Ostatna V, Fauerbach JA, Hsiao HH, Urlaub H, Palecek E, Jares-Erijman EA, Jovin TM. Biophysical properties and cellular toxicity of covalent crosslinked oligomers of alpha-synuclein formed by photoinduced side-chain tyrosyl radicals. Free Radic Biol Med. 2012;53:1004–1015. doi: 10.1016/j.freeradbiomed.2012.06.035. [DOI] [PubMed] [Google Scholar]
- 10.Brinkmalm G, Hong W, Wang Z, Liu W, O'Malley TT, Sun X, Frosch MP, Selkoe DJ, Portelius E, Zetterberg H, Blennow K, Walsh DM. Identification of neurotoxic cross-linked amyloid-beta dimers in the Alzheimer's brain. Brain. 2019;142:1441–1457. doi: 10.1093/brain/awz066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi JK, Cali I, Surewicz K, Kong Q, Gambetti P, Surewicz WK. Amyloid fibrils from the N-terminal prion protein fragment are infectious. Proc Natl Acad Sci U S A. 2016;113:13851–13856. doi: 10.1073/pnas.1610716113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colucci M, Moleres FJ, Xie ZL, Ray-Chaudhury A, Gutti S, Butefisch CM, Cervenakova L, Wang W, Goldfarb LG, Kong Q, Ghetti B, Chen SG, Gambetti P. Gerstmann-Straussler-Scheinker: a new phenotype with 'curly' PrP deposits. J Neuropathol Exp Neurol. 2006;65:642–651. doi: 10.1097/01.jnen.0000228198.81797.4d. [DOI] [PubMed] [Google Scholar]
- 13.Cracco L, Notari S, Cali I, Sy MS, Chen SG, Cohen ML, Ghetti B, Appleby BS, Zou WQ, Caughey B, Safar JG, Gambetti P. Novel strain properties distinguishing sporadic prion diseases sharing prion protein genotype and prion type. Sci Rep. 2017;7:38280. doi: 10.1038/srep38280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feraudet C, Morel N, Simon S, Volland H, Frobert Y, Creminon C, Vilette D, Lehmann S, Grassi J. Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem. 2005;280:11247–11258. doi: 10.1074/jbc.M407006200. [DOI] [PubMed] [Google Scholar]
- 15.Gambetti P, Cali I, Notari S, Kong Q, Zou WQ, Surewicz WK. Molecular biology and pathology of prion strains in sporadic human prion diseases. Acta Neuropathol. 2011;121:79–90. doi: 10.1007/s00401-010-0761-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213–239. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]
- 17.Ghetti B, Piccardo P, Zanusso G. Dominantly inherited prion protein cerebral amyloidoses - a modern view of Gerstmann-Straussler-Scheinker. Handb Clin Neurol. 2018;153:243–269. doi: 10.1016/B978-0-444-63945-5.00014-3. [DOI] [PubMed] [Google Scholar]
- 18.Gretzschel A, Buschmann A, Langeveld J, Groschup MH. Immunological characterization of abnormal prion protein from atypical scrapie cases in sheep using a panel of monoclonal antibodies. J Gen Virol. 2006;87:3715–3722. doi: 10.1099/vir.0.81816-0. [DOI] [PubMed] [Google Scholar]
- 19.Kascsak RJ, Rubenstein R, Merz PA, Tonna-DeMasi M, Fersko R, Carp RI, Wisniewski HM, Diringer H. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol. 1987;61:3688–3693. doi: 10.1128/jvi.61.12.3688-3693.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kong Q, Surewicz WK, Petersen RB, et al. (2004). Inherited prion diseases. In: SB Prusiner (Ed.), Prion Biology and Diseases, 2nd edition, Cold Springs Harbor Laboratory Press, New York. p. 673.
- 21.Ladogana A, Kovacs GG. Genetic Creutzfeldt-Jakob disease. Handb Clin Neurol. 2018;153:219–242. doi: 10.1016/B978-0-444-63945-5.00013-1. [DOI] [PubMed] [Google Scholar]
- 22.Li R, Liu T, Wong BS, Pan T, Morillas M, Swietnicki W, O'Rourke K, Gambetti P, Surewicz WK, Sy MS. Identification of an epitope in the C terminus of normal prion protein whose expression is modulated by binding events in the N terminus. J Mol Biol. 2000;301:567–573. doi: 10.1006/jmbi.2000.3986. [DOI] [PubMed] [Google Scholar]
- 23.Mercer RCC, Daude N, Dorosh L, Fu ZL, Mays CE, Gapeshina H, Wohlgemuth SL, Acevedo-Morantes CY, Yang J, Cashman NR, Coulthart MB, Pearson DM, Joseph JT, Wille H, Safar JG, Jansen GH, Stepanova M, Sykes BD, Westaway D. A novel Gerstmann-Straussler-Scheinker disease mutation defines a precursor for amyloidogenic 8 kDa PrP fragments and reveals N-terminal structural changes shared by other GSS alleles. PLoS Pathog. 2018;14:e1006826. doi: 10.1371/journal.ppat.1006826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Notari S, Strammiello R, Capellari S, Giese A, Cescatti M, Grassi J, Ghetti B, Langeveld JP, Zou WQ, Gambetti P, Kretzschmar HA, Parchi P. Characterization of truncated forms of abnormal prion protein in Creutzfeldt-Jakob disease. J Biol Chem. 2008;283:30557–30565. doi: 10.1074/jbc.M801877200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parchi P, Chen SG, Brown P, Zou W, Capellari S, Budka H, Hainfellner J, Reyes PF, Golden GT, Hauw JJ, Gajdusek DC, Gambetti P. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Straussler-Scheinker disease. Proc Natl Acad Sci U S A. 1998;95:8322–8327. doi: 10.1073/pnas.95.14.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parchi P, Zou W, Wang W, Brown P, Capellari S, Ghetti B, Kopp N, Schulz-Schaeffer WJ, Kretzschmar HA, Head MW, Ironside JW, Gambetti P, Chen SG. Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci U S A. 2000;97:10168–10172. doi: 10.1073/pnas.97.18.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piccardo P, Liepnieks JJ, William A, Dlouhy SR, Farlow MR, Young K, Nochlin D, Bird TD, Nixon RR, Ball MJ, DeCarli C, Bugiani O, Tagliavini F, Benson MD, Ghetti B. Prion proteins with different conformations accumulate in Gerstmann-Straussler-Scheinker disease caused by A117V and F198S mutations. Am J Pathol. 2001;158:2201–2207. doi: 10.1016/S0002-9440(10)64692-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pirisinu L, Di Bari MA, D'Agostino C, Marcon S, Riccardi G, Poleggi A, Cohen ML, Appleby BS, Gambetti P, Ghetti B, Agrimi U, Nonno R. Gerstmann-Straussler-Scheinker disease subtypes efficiently transmit in bank voles as genuine prion diseases. Sci Rep. 2016;6:20443. doi: 10.1038/srep20443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Priola SA, Caughey B, Wehrly K, Chesebro B. A 60-kDa prion protein (PrP) with properties of both the normal and scrapie-associated forms of PrP. J Biol Chem. 1995;270:3299–3305. doi: 10.1074/jbc.270.7.3299. [DOI] [PubMed] [Google Scholar]
- 30.Safar JG, Xiao X, Kabir ME, Chen S, Kim C, Haldiman T, Cohen Y, Chen W, Cohen ML, Surewicz WK. Structural determinants of phenotypic diversity and replication rate of human prions. PLoS Pathog. 2015;11:e1004832. doi: 10.1371/journal.ppat.1004832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 32.Souza JM, Giasson BI, Chen Q, Lee VM, Ischiropoulos H. Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem. 2000;275:18344–18349. doi: 10.1074/jbc.M000206200. [DOI] [PubMed] [Google Scholar]
- 33.Tagliavini F, Lievens PM, Tranchant C, Warter JM, Mohr M, Giaccone G, Perini F, Rossi G, Salmona M, Piccardo P, Ghetti B, Beavis RC, Bugiani O, Frangione B, Prelli F. A 7-kDa prion protein (PrP) fragment, an integral component of the PrP region required for infectivity, is the major amyloid protein in Gerstmann-Straussler-Scheinker disease A117V. J Biol Chem. 2001;276:6009–6015. doi: 10.1074/jbc.M007062200. [DOI] [PubMed] [Google Scholar]
- 34.Tagliavini F, Prelli F, Ghiso J, Bugiani O, Serban D, Prusiner SB, Farlow MR, Ghetti B, Frangione B. Amyloid protein of Gerstmann-Straussler-Scheinker disease (Indiana kindred) is an 11 kd fragment of prion protein with an N-terminal glycine at codon 58. EMBO J. 1991;10:513–519. doi: 10.1002/j.1460-2075.1991.tb07977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tagliavini F, Prelli F, Porro M, Rossi G, Giaccone G, Farlow MR, Dlouhy SR, Ghetti B, Bugiani O, Frangione B. Amyloid fibrils in Gerstmann-Straussler-Scheinker disease (Indiana and Swedish kindreds) express only PrP peptides encoded by the mutant allele. Cell. 1994;79:695–703. doi: 10.1016/0092-8674(94)90554-1. [DOI] [PubMed] [Google Scholar]
- 36.Theint T, Nadaud PS, Aucoin D, Helmus JJ, Pondaven SP, Surewicz K, Surewicz WK, Jaroniec CP. Species-dependent structural polymorphism of Y145Stop prion protein amyloid revealed by solid-state NMR spectroscopy. Nat Commun. 2017;8:753. doi: 10.1038/s41467-017-00794-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vazquez-Fernandez E, Vos MR, Afanasyev P, Cebey L, Sevillano AM, Vidal E, Rosa I, Renault L, Ramos A, Peters PJ, Fernandez JJ, van Heel M, Young HS, Requena JR, Wille H. The structural architecture of an infectious mammalian prion using Electron Cryomicroscopy. PLoS Pathog. 2016;12:e1005835. doi: 10.1371/journal.ppat.1005835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vorberg I, Buschmann A, Harmeyer S, Saalmuller A, Pfaff E, Groschup MH. A novel epitope for the specific detection of exogenous prion proteins in transgenic mice and transfected murine cell lines. Virology. 1999;255:26–31. doi: 10.1006/viro.1998.9561. [DOI] [PubMed] [Google Scholar]
- 39.Zerr I, Parchi P. Sporadic Creutzfeldt-Jakob disease. Handb Clin Neurol. 2018;153:155–174. doi: 10.1016/B978-0-444-63945-5.00009-X. [DOI] [PubMed] [Google Scholar]
- 40.Zou W, Colucci M, Gambetti P, Chen SG (2003) Characterization of prion proteins. in: Methods in Molecular Biology-Neurogenetics: Methods and Protocols. Edited by Potter, N.T., pp. 305-314. Humana Press Inc., Totowa. 2002. [DOI] [PubMed]
- 41.Zou WQ, Capellari S, Parchi P, Sy MS, Gambetti P, Chen SG. Identification of novel proteinase K-resistant C-terminal fragments of PrP in Creutzfeldt-Jakob disease. J Biol Chem. 2003;278:40429–40436. doi: 10.1074/jbc.M308550200. [DOI] [PubMed] [Google Scholar]
- 42.Zou WQ, Langeveld J, Xiao X, Chen S, McGeer PL, Yuan J, Payne MC, Kang HE, McGeehan J, Sy MS, Greenspan NS, Kaplan D, Wang GX, Parchi P, Hoover E, Kneale G, Telling G, Surewicz WK, Kong Q, Guo JP. PrP conformational transitions alter species preference of a PrP-specific antibody. J Biol Chem. 2010;285:13874–13884. doi: 10.1074/jbc.M109.088831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Further biochemical characterization of resPrPD associated with GSSF198S. Lane 1: PNGase F-deglycosylated resPrPD from brain homogenate immunoblotted with 3F4 following standard conditions. Lane 2: with additional boiling, freezing-thawing and sonication pre-deglycosylation; lane 3: 41 h PNGase F treatment; lanes 4–7: incubation with strong denaturants, 8 M urea (after ethanol or methanol precipitation, lanes 4, 5) and 8 M guanidine hydrochloride at 80 °C (after ethanol or methanol precipitation, lanes 6, 7) pre-deglycosylation. Treatments had no detectable effect on the resPrPD electrophoretic profile. (TIF 17802 kb)
Table S1. Tryptic peptides identified for ~ 7 KDa band in GSSA117V resPrPD. Residue at position 129 is marked in blue. Table S2. Tryptic peptides identified for ~ 8 KDa band in GSSF198S resPrPD. (PDF 122 kb)
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.