

Summary
Activation of the heterotrimeric energy‐sensing kinase AMP‐activated protein kinase (AMPK) has been reported to improve experimental diabetic kidney disease. We examined the effect of type 1 diabetes in wild‐type (WT) mice and mice lacking the β1 subunit of AMPK (AMPK β1−/− mice), which have reduced AMPK activity in kidneys and other organs. Diabetes was induced using streptozotocin (STZ) and the animals followed up for 4 weeks. Hyperglycaemia was more severe in diabetic AMPK β1−/− mice, despite the absence of any difference in serum levels of insulin, adiponectin and leptin. There was no change in AMPK activity in the kidneys of diabetic WT mice by AMPK activity assay, or phosphorylation of either the αT172 activation site on the α catalytic subunit of AMPK or the AMPK‐specific phosphosite S79 on acetyl CoA carboxylase 1 (ACC1). Phosphorylation of the inhibitory αS485 site on the α subunit of AMPK was significantly increased in the WT diabetic mice compared to non‐diabetic controls. Despite increased plasma glucose levels in the diabetic AMPK β1−/− mice, there were fewer myofibroblasts in the kidneys compared to diabetic WT mice, as evidenced by reduced α‐smooth muscle actin (α‐SMA) protein by Western blot, mRNA by qRT‐PCR and fewer α‐SMA‐positive cells by immunohistochemical staining. Albuminuria was also reduced in the AMPK β1−/− mice. In contrast to previous studies, therefore, myofibroblasts were reduced in the kidneys of AMPK β1−/− diabetic mice compared to diabetic WT mice, despite increased circulating glucose, suggesting that AMPK can worsen renal fibrosis in type 1 diabetes.
Keywords: AMPK, diabetes, fibrosis, myofibroblast
1. INTRODUCTION
Diabetic kidney disease (DKD) is the leading cause of end‐stage renal failure worldwide.1 The cellular energy sensor, AMP‐activated protein kinase (AMPK), is an important therapeutic target in the management of diabetes and other metabolic disorders such as obesity and fatty liver disease.2, 3 Activation of AMPK by metformin is important for the anti‐diabetic and other metabolic benefits of this commonly used treatment for type II diabetes.4 Furthermore, direct activators of AMPK are currently being developed as potential therapeutics for patients with type II diabetes and metabolic disorders.3, 5 In the kidney, AMPK has been found to have a variety of important roles in physiology and pathophysiology, such as regulation of ion transport and podocyte function.6 Some investigators have suggested that activation of AMPK in the kidney might be protective against the development of diabetic kidney disease.7, 8, 9 Consequently, it has been suggested that AMPK activators might be useful in patients with type I diabetes so as to reduce progression of diabetic kidney disease.
AMPK is an αβγ heterotrimer with multiple isoforms existing for each subunit.10, 11 The β‐subunit of AMPK, existing as β1 and β2 isoforms, has regulatory functions derived from its carbohydrate binding and subunit interacting domains.12, 13 Biological distinctions between β1 and β2 AMPK heterotrimers include differences in subcellular localization and responses to pharmacological AMPK activators.14 While the kidney expresses both β1 and β2 subunits, we have demonstrated that β1 is predominant with AMPK β1−/− mice having 84% reduced total renal AMPK activity, making these mice useful for studying AMPK in the kidney.15 In addition, we have previously demonstrated that the α1 catalytic subunit is predominant in mouse kidneys, whereas complexes containing the α2 subunit contribute approximately 30% of the enzymatic activity.15 The aim of this present study was to determine the role of AMPK, in particular, the AMPK β1 subunit, in the early development of diabetic kidney disease associated with type 1 diabetes.
2. METHODS
2.1. Animal studies
Type I diabetes was induced by using the low‐dose mouse model of streptozotocin (STZ)‐induced diabetic nephropathy described by Tesch et al16 STZ selectively destroys the insulin‐producing beta islet cells of the pancreas, and this model produces modest elevations in albuminuria and serum creatinine and some of the early histological lesions associated with diabetic nephropathy.16 AMPK β1−/− mice on a C57BL/6 background have been previously described.17 They are not diabetic and have no physical phenotype. Male C57BL/6 mice (WT) and AMPK β1−/− mice were given daily intraperitoneal injections of 55 mg/kg STZ, dissolved in sodium citrate buffer, at 6 weeks of age for five consecutive days. The day of the fifth injection is then day 0 for the subsequent studies. Plasma glucose measurements were obtained by tail vein sampling at day 7 after the last injection. Mice with a plasma glucose level greater than 15 mmol/L were regarded as diabetic. The control groups received vehicle (sodium citrate buffer without STZ). Plasma glucose levels and body weights were monitored weekly. The mice were maintained with diabetes for 4 weeks, allowing sufficient time for any direct effect of the STZ on AMPK activity in the kidney to resolve. Timed urine collections were performed using metabolic cages (6 hours) on day 28. Eye bleeding for plasma creatinine, insulin, leptin and adiponectin levels was obtained at day 32, and mice were sacrificed and kidneys harvested for further assessment by histology or homogenized into lysates for protein and RNA. About 75% of male mice became diabetic with this model. Female mice are less susceptible to the development of diabetes with STZ and, therefore, were not used.
2.2. Ethical approval
All experiments were approved by the Austin Health Animal Ethics Committee.
2.3. Serum and urine biochemistry
Urinary albumin excretion was quantified as urine albumin:creatinine ratio (ACR, mg/mmol). Urinary albumin was measured by an enzyme‐linked immunosorbent assay (ELISA) kit (Bethyl Laboratories, Montgomery, TX), while plasma and urine creatinine concentration was measured by HPLC as previously described.18, 19 Commercial ELISA kits were used to measure insulin (ALPCO Diagnostics, Salem, New Hampshire), adiponectin (Abnova, Taipei City, Taiwan) and leptin (R&D systems, Sapphire Bioscience Pty Ltd. Waterloo, NSW, Australia), according to the manufacturer‘s instructions.
2.4. AMPK activity assay
Kidney AMPK (α1 and α2) activity was measured by a SAMS (water‐soluble AMP‐activated protein kinase substrate) kinase activity assay, as previously described.20 AMPK activity was also assessed using Western blots for phosphorylation of the activation site T172 in the α subunit of AMPK and S79 in ACC1, which is a well‐known substrate for AMPK.
2.5. Antibodies for Western blotting and immunohistochemistry
The following antibodies were used: anti‐α‐SMA (Sigma, mouse mAb), anti‐E‐cadherin (Abcam, rabbit polyclonal Ab) and anti‐β‐actin (Cell Signaling Technology, rabbit mAb clone 13E5). Antibodies against AMPK subunits, ACC1 and related phosphoantibodies have previously been described.21 The anti‐β1 rabbit mAb was from Epitomics.
2.6. Immunohistochemistry
Kidneys were perfusion fixed with 4% paraformaldehyde (BDH, Poole, UK), processed and embedded in paraffin. Four‐micrometre sections were cut on a rotary microtome (microTex), and immunohistochemistry performed as previously described. Briefly, sections were dewaxed and endogenous peroxidase activity inhibited using 3% H2O2. Non‐specific binding was blocked with 10% bovine serum albumin (BSA) before incubation overnight with monoclonal antibody (mAb). Slides were then incubated in secondary antibody conjugated to horseradish peroxidase (HRP) (DAKO Corporation, Carpinteria, CA). This was followed by a peroxidase anti‐peroxidase antibody step and development using liquid 3,3‐diaminobenzidine (DAKO). Sections were counterstained using Harris's haematoxylin and mounted in DePeX.
2.7. Western blotting analysis
Kidneys were snap frozen in liquid nitrogen and lysates prepared, as previously described.15 Western blots were then performed again as described. In blots for AMPK, lysates were first immunoprecipitated with a mixture of anti‐α1 and anti‐α2 AMPK antibodies. For blots of other antigens, cell lysates were run on SDS‐PAGE gels. Briefly, samples were separated by SDS‐PAGE and transferred to polyvinylidene difluoride membrane (Immobilon‐P, Millipore, Bedford, MA). The membranes were blocked in 10% BSA and then incubated in primary antibody. Optimal antibody concentration and duration of incubation were determined for each antibody. The membrane was incubated in FITC‐conjugated secondary antibody (Dako, Glostrup, Denmark). Antibody complexes were detected with anti‐FITC antibody conjugated with HRP (Roche Diagnostics, Basil, Switzerland) followed by enhanced chemiluminescence with the Western Lightning System (PerkinElmer, MA). If the membrane was to be probed with another primary antibody, antibody bound to the membrane was stripped by incubation in Reblot stripping solution (Chemicon, MA). Quantification of Western blots was performed by densitometry with analysis using ImageJ software (NIH, Bethesda, MD).
2.8. Real‐time qRT‐PCR
Total RNA was purified from whole mouse kidney or liver samples using TRIzol reagent (Invitrogen) in accordance with the manufacturer's instructions. RNA quality and quantity was determined using spectrophotometry and reverse‐transcribed using the high‐capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). Real‐time PCR using primers for fibronectin (5′CGAGGTGACAGAGACCACAA3′; 5′CTGGAGTCAAGCCAGACACA3′), collagen IV (5′AAAGGGAGAAAGAGGCTTGC3′; 5′CTCCCTTTGTACCGTTGCAT3′), α‐smooth muscle actin (5′CAGGCA TGGA TGGCA TCAA TCAC3′; 5′ACTCTAGCTGTGAAGTCAGTGTCG3′) and housekeeping genes β‐actin (5′CGGGATCCCCGCCCTAGGCACCAGGGTG 3′; 5′GGAATTAGGCTGGGGTGTTGAAGGTCTCAAA 3′) and 18s (5′AGTCCCTGCCCTTTGTACACA 3′; 5′GATCCGAGGGCCTCACTAAAC 3′) was performed on a Stratagene MX‐3000 with the Solis Biodyne EvaGreen master mix (Tartu, Estonia) according to the manufacturer's instructions. Primer efficiency was measured using standard dilution, and the Pfaffl method was used to calculate relative expression. Results were expressed as fold expression relative to control WT mice.
2.9. Mouse embryo fibroblasts
Mouse embryonic fibroblasts (MEFs) were isolated from wild‐type (WT) or AMPK β1floxed/β2 knockout mice and maintained in culture as previously described.21
2.10. Statistics
Statistics were performed using InStat version 3.05 (GraphPad Software, San Diego, CA). Data are presented as means ± 1 SD. Multiple group means were compared by ANOVA followed by a post hoc test. Comparison of means from two groups was performed by unpaired t test. P values < 0.05 were considered significant.
3. RESULTS
3.1. Physical and biochemical effects of type 1 diabetes in AMPK β1 WT and AMPK β1−/− mice
After one month of type I diabetes, body weights were significantly reduced in both diabetic WT and diabetic β1−/− mice compared with non‐diabetic control mice (Figure 1A). There was, however, no difference in kidney weight relative to body weight between any of the groups of mice (data not shown). Plasma glucose levels were significantly increased in diabetic AMPK β1−/− mice at days 7, 14, 21 and 28 compared with diabetic WT mice (Figure 1B). As expected, diabetic animals had lower insulin levels but, despite the glucose difference, diabetic AMPK β1−/− mice had similar serum insulin concentrations compared with diabetic WT mice (Figure 2A). Serum adiponectin levels were indistinguishable between diabetic WT and diabetic AMPK β1−/− mice (Figure 2B). Control AMPK β1−/− mice had reduced adiponectin levels compared with control WT mice (P < 0.05) (Figure 2B). Serum leptin was reduced in diabetic mice compared to controls (P < 0.001) but there was no difference between diabetic WT and diabetic AMPK β1−/− mice (Figure 2C).
Figure 1.
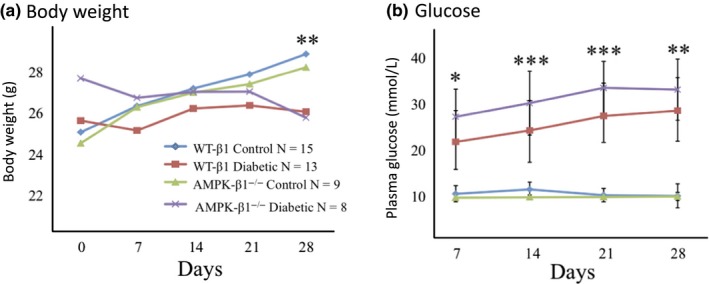
Body weight and plasma glucose WT and AMPK β1−/− mice. (A) Weekly body weight (g) up to 28 days after diabetes induction. WT control n = 15, WT diabetic n = 13, AMPK β1−/− control n = 9, AMPK β1−/− diabetic n = 8. **P < 0.01 diabetic vs non‐diabetic. Mean + SD. (B) Weekly plasma glucose levels following STZ treatment. WT control n = 10, WT diabetic n = 12, AMPK β1−/− control n = 6, AMPK β1−/− diabetic n = 13. *P < 0.05, **P < 0.01, ***P < 0.001, diabetic WT vs diabetic AMPK β1−/− mice. Mean + standard deviation [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 2.
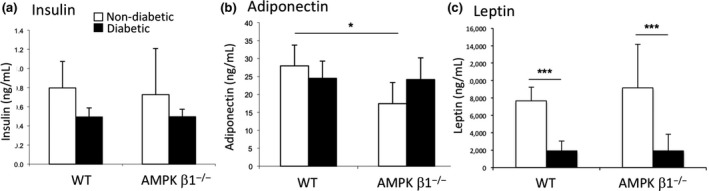
Serum insulin (A), adiponectin (B) and leptin (C) concentration WT and AMPK β1−/− mice 28 days after induction of diabetes as measured by ELISA. Control WT and AMPK β1−/− mice (white bars), diabetic WT and diabetic AMPK β1−/− mice (black bars). n = 3‐7 per group. (A) Insulin: WT control n = 8, WT diabetic n = 7, AMPK β1−/− control n = 8, AMPK β1−/− diabetic n = 6. B, Adiponectin: WT control n = 3, WT diabetic n = 7, AMPK β1−/− control n = 3, AMPK β1−/− diabetic n = 6. C, Leptin: WT control n = 3, WT diabetic n = 6, AMPK β1−/− control n = 3, AMPK β1−/− diabetic n = 6. *P < 0.05, ***P < 0.001. Mean + standard deviation
3.2. AMPK expression, phosphorylation and activity in kidneys from diabetic and non‐diabetic WT and AMPK β1−/− mice
There was no significant difference in expression of the α1 or α2 catalytic subunits between any of the groups despite the absence of a detectable β subunit (Figure 3A; densitometry not shown). We have previously demonstrated that murine kidneys express both the β1 and β2 units at levels detectable by Western blot, but AMPK activity is reduced by 70% in AMPK β1−/− mice.15 Phosphorylation of the αT172 site associated with AMPK activation was similar in mice of all groups (Figure 3A,B). Phosphorylation of the αS485 site, which has been described as a negative regulatory site,22 was significantly increased in diabetic WT mice compared to control WT mice (P < 0.05; Figure 3A,C).
Figure 3.
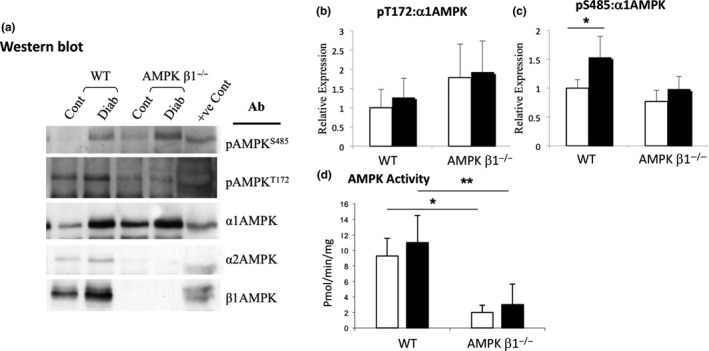
AMPK phosphorylation in kidneys from diabetic and non‐diabetic WT and AMPK β1−/− mice 28 days after induction of diabetes. A Lysates were immunoprecipitated with a mixture of anti‐α1 and anti‐α2 AMPK antibodies, and then blotted and probed with antibodies against pT172, pS485, both α subunits of AMPK, and the β1 subunit. B, Densitometric analysis of Western blots showing relative expression of pαT172. WT control n = 3, WT diabetic n = 5, AMPK β1−/− control n = 3, AMPK β1−/− diabetic n = 5. C, Densitometric analysis of Western blots showing relative expression of pαS485. n = 3‐5. D, AMPK activity assay in kidneys from diabetic and non‐diabetic WT and AMPK β1−/− mice 28 days after induction of diabetes. Kidney lysates were immunoprecipitated with both α1 and α2 AMPK antibodies and the immunoprecipitates assayed by SAMS assay. (B) Adiponectin: WT control n = 3, WT diabetic n = 6, AMPK β1−/− control n = 3, AMPK β1−/− diabetic n = 5. *P < 0.05, **P < 0.01. Non‐diabetic (white bars), diabetic (black bars)
In the kidney lysates, control and diabetic WT mice did not have any significant change in AMPK activity (Figure 3D). As anticipated, AMPK β1−/− mice had significantly reduced AMPK activity compared with WT mice in both control (P < 0.05) and diabetic (P < 0.01) groups in the kidney (Figure 3D).
3.3. ACC expression and phosphorylation in kidneys from diabetic and non‐diabetic WT and AMPK β1−/− mice
There was no difference in total ACC protein expression (Figure 4A; densitometry not shown). Phosphorylation of ACC at S79 was significantly reduced in both diabetic and non‐diabetic AMPK β1−/− mice compared to diabetic and non‐diabetic WT mice (P < 0.01 and P < 0.001, respectively; Figure 4A,B). There was no difference between diabetic and non‐diabetic WT mice.
Figure 4.
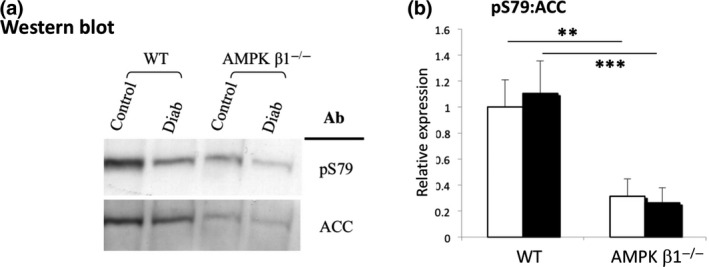
ACC phosphorylation in kidneys from diabetic and non‐diabetic WT and AMPK β1−/− mice 28 days after induction of diabetes. (A) Western blot of pACC1 S79, and total ACC1/2 expression in kidneys from diabetic and non‐diabetic WT and AMPK β1−/− mice 28 days after induction of diabetes. Lysates were immunoprecipitated with streptavidin, and then blotted and probed with streptavidin and antibodies against pS79. (B) Densitometric analysis of Western blots showing relative expression of pS79. WT control n = 3, WT diabetic n = 5, AMPK ββ1−/− control n = 3, AMPK β1−/− diabetic n = 5. **P < 0.01, ***P < 0.001. Non‐diabetic (white bars), diabetic (black bars)
3.4. Creatinine clearance and urinary albumin excretion
There was no difference in creatinine clearance between any of the groups, although there was a trend to higher levels in the diabetic WT mice compared with non‐diabetic WT mice (Figure 5A). The urinary albumin/creatinine ratio (ACR), a measure of albumin excretion in the urine, was increased in the diabetic WT mice compared to non‐diabetic WT mice (Figure 5B). ACR was also significantly lower in the diabetic AMPK β1−/− mice compared with WT diabetic mice (P < 0.01).
Figure 5.
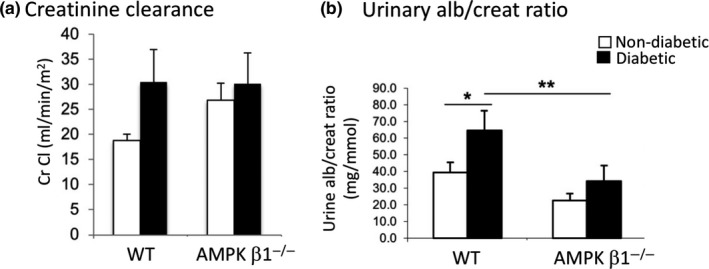
(A) Creatinine clearance measured by HPLC. WT control and AMPK β1−/− control mice (white bars), diabetic WT and diabetic AMPK β1−/− mice (black bars). Mean + SD. (B) Urinary albumin/creatinine ratio (ACR). WT control and AMPK β1−/− control mice (white bars), diabetic WT and diabetic AMPK β1−/− mice (black bars). WT control n = 3, WT diabetic n = 7, AMPK β1−/− control n = 3, AMPK β1−/− diabetic n = 5. *P < 0.05, **P < 0.01. Mean + SD
3.5. Alpha smooth muscle actin (α‐SMA) in control and diabetic WT and AMPK β1−/− mice
Characteristic histological changes of diabetic nephropathy were not observed in either the diabetic WT or diabetic AMPK β1−/− mice due to the short duration of diabetes. An early finding in diabetic kidney disease with STZ‐induced diabetic mice is increased numbers of myofibroblasts.23 After four weeks of hyperglycaemia, immunohistochemical staining for myofibroblasts, detected by α‐SMA staining, was reduced in diabetic AMPK β1−/− mice relative to diabetic WT mice (Figure 6A). In control WT mouse kidneys, α‐SMA was found mainly in the blood vessels of control WT mouse kidneys. In comparison, interstitial staining for α‐SMA was reduced in kidneys from diabetic AMPK β1−/− mice and similar to kidneys from non‐diabetic AMPK β1−/− mice (Figure 6A). Consistent with the immunohistochemistry, Western blot analysis on kidneys from diabetic AMPK β1−/− mice demonstrated significantly less α‐SMA than diabetic WT mice (Figure 6B), confirmed by densitometric analysis of Western blots (Figure 5C) (P < 0.05 relative to diabetic WT mice). E‐cadherin was unchanged (Figure 6B).
Figure 6.
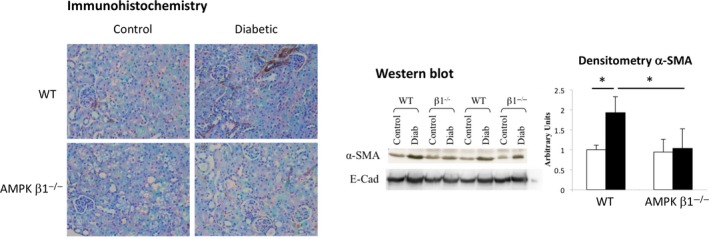
Expression of α‐smooth muscle actin (α‐SMA) in diabetic and non‐diabetic WT and AMPK β1−/− mice 28 days after induction of diabetes. (A) Immunodetection of α‐smooth muscle actin (α‐SMA) in kidney sections. Original magnification: × 400. Enlarged for clarity. (B) Western blots for α‐SMA and E‐cadherin. C, α‐SMA blots were quantitated by densitometry. E‐cadherin showed no difference and is not shown. WT control n = 3, WT diabetic n = 5, AMPK β1−/− control n = 3, AMPK β1−/− diabetic n = 5. *P < 0.05. Mean + standard deviation [Colour figure can be viewed at wileyonlinelibrary.com]
α‐SMA mRNA expression in the kidney was significantly reduced in diabetic AMPK β1−/− mice compared with diabetic WT mice (Figure 7A), consistent with the Western blot and immunohistochemical data. Other markers of fibrosis in diabetic renal injury were also measured by qRT‐PCR, including collagen IV mRNA expression, which was significantly reduced in diabetic AMPK β1−/− mice compared with diabetic WT mice (Figure 7B). There was no change in fibronectin (Figure 7C).
Figure 7.
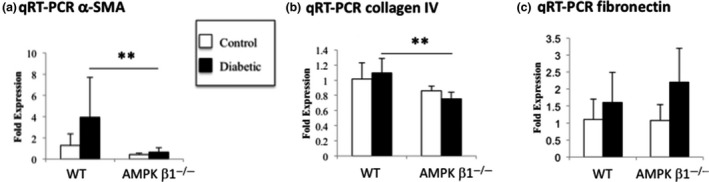
Expression of α‐smooth muscle actin (α‐SMA), collagen IV and fibronectin in diabetic and non‐diabetic WT and AMPK β1−/− mice 28 days after induction of diabetes. A, α‐SMA, B, collagen IV and C, fibronectin mRNA in kidneys from diabetic and non‐diabetic WT and AMPK β1−/− mice 28 days after induction of diabetes. (B) Adiponectin: WT control n = 3, WT diabetic n = 6, AMPK β1−/− control n = 3, AMPK β1−/− diabetic n = 5. **P < 0.01. Mean + standard deviation
3.6. AMPK and α‐SMA expression in mouse embryonic fibroblasts (MEFs)
The relationship between AMPK and α‐SMA expression was also examined in MEFs comparing WT with AMPK β1floxed/β2 knockout mice, which have low to negligible levels of AMPK expression.21 Consistent with the kidney findings, in AMPK‐deficient MEFs, expression of α‐SMA was reduced to 34% of the levels seen in WT MEFs (Figure 8A,B; P = 0.004), whereas E‐cadherin expression was unchanged (Figure 8C).
Figure 8.
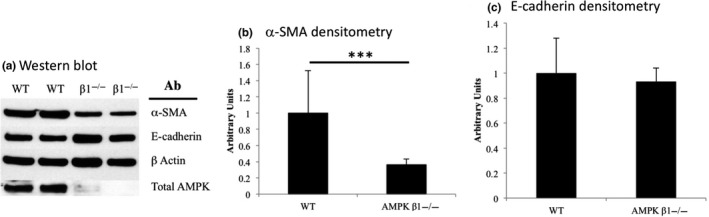
A, Expression of α‐SMA, E‐cadherin and AMPK in WT and AMPK −/− MEFs by Western blot. N = 6 per group. Densitometric analysis showed that there was a significant reduction in α‐SMA (B) but not E‐cadherin (C) in AMPK −/− MEFs. AMPK was undetectable or only weakly seen in AMPK −/− MEFs (A). ***P < 0.005
4. DISCUSSION
In the present study, AMPK β1−/− mice had reduced renal myofibroblast formation in a murine model of early type 1 diabetic nephropathy. The apparent protective effect of AMPK β1 deficiency was seen despite the presence of increased hyperglycaemia. This contrasts with previous in vitro studies performed in tubular epithelial cells and fibroblasts showing that pharmacological activation of AMPK reduces myofibroblast transformation associated with TGF‐β1 signalling in vitro.24, 25 In addition, other investigators have correlated reduced renal AMPK activity with nephropathy and shown that AICAR, a pharmacological activator of AMPK, has beneficial effects on the kidneys of diabetic mice.26 It was unexpected, therefore, to observe reduced renal myofibroblasts expression in the diabetic AMPK β1−/− mice, consistent with a beneficial effect of AMPK deficiency in this context. This could suggest a specific role for the AMPK β1 subunit in early diabetic kidney pathology in this model.
An in vivo precedent for specific effects for individual AMPK subunits in renal fibrosis is found in the unilateral ureteric obstruction (UUO) model of kidney fibrosis. Several groups have reported that increased signalling through the AMPK pathway with AMPK activators such as AICAR and metformin was associated with reduced fibrosis in the UUO model.27, 28 Despite this, the absence of the α1 subunit of AMPK in the UUO model has been associated with reduced fibrosis.29 In contrast, the absence of the α2 subunit increased fibrosis 30 suggesting that activation of AMPK by drugs reduces fibrosis which is probably due to activation of AMPK complexes containing the α2 subunit.
Previous work from our laboratory has shown that, in the kidney, the β1 subunit of AMPK is associated with a significant reduction in activity of heterotrimers containing the α1 but not the α2 subunit.15 Taken together, the data suggest that reduction in the renal expression of AMPK α1β1 complexes appears to protect against the development of fibrosis.
There was no change in AMPK activity detected by AMPK activity assay or activating phosphorylation of αT172 in the diabetic WT kidneys compared with non‐diabetic WT kidneys. These data contrast to those from another study in type 1 diabetic mice 26 that demonstrated a reduction in AMPK activity determined by reduced phosphorylation of αT172. We are unable to account for the difference between the studies, although the current work was performed at a significantly earlier stage after induction of diabetes, at 4 weeks compared with 24 weeks. AMPK activity in the SAMS assay could be affected by phosphorylation of the inhibitory αS485 phosphosite in the α subunit,22 which would decrease AMPK activity. As outlined, phosphorylation of S485 in the α subunit was increased in diabetic WT mice, incidentally confirming studies performed in cultured vascular smooth muscle cells showing that hyperglycaemia itself increases phosphorylation of αS485.31
Following STZ treatment, diabetic AMPK β1−/− mice had significantly higher plasma glucose levels but similar levels of insulin to diabetic WT mice. The difference is unlikely to be due to greater insulin resistance, as Dzamko et al have previously described reduced insulin resistance in AMPK β1−/− mice.17 An alternative explanation for the exaggerated hyperglycaemia seen in diabetic AMPK β1−/− mice is altered cellular metabolism, with a shift to increased gluconeogenesis and reduced glycolysis, consistent with known AMPK biology.10
In this study, AMPK β1 subunit deficiency reduced myofibroblast accumulation in the diabetic kidney, despite more severe hyperglycaemia. This indicates that the absence of the β1 subunit of AMPK, like the absence of the α1 but not the α2 subunit in the UUO model of renal fibrosis, is associated with reduced fibrosis in the early stage of diabetic kidney disease. This may have implications for the effect of novel AMPK activators that have been reported to have specificity for specific AMPK β isoforms.32
Choy S‐W, Fraser SA, Katerelos M, et al. Absence of the β1 subunit of AMP‐activated protein kinase reduces myofibroblast infiltration of the kidneys in early diabetes. Int. J. Exp. Path. 2019;100:114–122. 10.1111/iep.12313
REFERENCES
- 1. Svensson M, Sundkvist G, Arnqvist HJ, et al. Signs of nephropathy may occur early in young adults with diabetes despite modern diabetes management: results from the nationwide population‐based Diabetes Incidence Study in Sweden (DISS). Diabetes Care. 2003;26:2903‐2909. [DOI] [PubMed] [Google Scholar]
- 2. Boyle JG, Salt IP, McKay GA. Metformin action on AMP‐activated protein kinase: a translational research approach to understanding a potential new therapeutic target. Diabet Med. 2010;27:1097‐1106. [DOI] [PubMed] [Google Scholar]
- 3. Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009;9:407‐416. [DOI] [PubMed] [Google Scholar]
- 4. Zhou G, Myers R, Li Y, et al. Role of AMP‐activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167‐1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cool B, Zinker B, Chiou W, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403‐416. [DOI] [PubMed] [Google Scholar]
- 6. Hallows KR, Mount PF, Pastor‐Soler NM, Power DA. Role of the energy sensor AMP‐activated protein kinase in renal physiology and disease. Am J Physiol Renal Physiol. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eid AA, Ford BM, Block K, et al. AMP‐activated protein kinase (AMPK) negatively regulates Nox4‐dependent activation of p53 and epithelial cell apoptosis in diabetes. J Biol Chem. 2010;285:37503‐37512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee MJ, Feliers D, Mariappan MM, et al. A role for AMP‐activated protein kinase in diabetes‐induced renal hypertrophy. Am J Physiol Renal Physiol. 2007;292:F617‐F627. [DOI] [PubMed] [Google Scholar]
- 9. Sharma K, Ramachandrarao S, Qiu G, et al. Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest. 2008;118:1645‐1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hardie DG. Keeping the home fires burning: AMP‐activated protein kinase. J R Soc Interface. 2018;15:20170774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89:1025‐1078. [DOI] [PubMed] [Google Scholar]
- 12. Iseli TJ, Walter M, van Denderen BJ, et al. AMP‐activated protein kinase beta subunit tethers alpha and gamma subunits via its C‐terminal sequence (186‐270). J Biol Chem. 2005;280:13395‐13400. [DOI] [PubMed] [Google Scholar]
- 13. Polekhina G, Gupta A, Michell BJ, et al. AMPK beta subunit targets metabolic stress sensing to glycogen. Curr Biol. 2003;13:867‐871. [DOI] [PubMed] [Google Scholar]
- 14. Sanz P, Rubio T, Garcia‐Gimeno MA. AMPKbeta subunits: more than just a scaffold in the formation of AMPK complex. FEBS J. 2013;280:3723‐3733. [DOI] [PubMed] [Google Scholar]
- 15. Mount PF, Gleich K, Tam S, et al. The outcome of renal ischemia‐reperfusion injury is unchanged in AMPK‐beta1 deficient mice. PLoS ONE. 2012;7:e29887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tesch GH, Allen TJ. Rodent models of streptozotocin‐induced diabetic nephropathy. Nephrology. 2007;12:261‐266. [DOI] [PubMed] [Google Scholar]
- 17. Dzamko N, van Denderen BJ, Hevener AL, et al. AMPK beta1 deletion reduces appetite, preventing obesity and hepatic insulin resistance. J Biol Chem. 2010;285:115‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dunn SR, Qi Z, Bottinger EP, Breyer MD, Sharma K. Utility of endogenous creatinine clearance as a measure of renal function in mice. Kidney Int. 2004;65:1959‐1967. [DOI] [PubMed] [Google Scholar]
- 19. Tikellis C, Bialkowski K, Pete J, et al. ACE2 deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes. 2008;57:1018‐1025. [DOI] [PubMed] [Google Scholar]
- 20. Mount PF, Hill RE, Fraser SA, et al. Acute renal ischemia rapidly activates the energy sensor AMPK but does not increase phosphorylation of eNOS‐Ser1177. Am J Physiol Renal Physiol. 2005;289:F1103‐F1115. [DOI] [PubMed] [Google Scholar]
- 21. Davies M, Fraser SA, Galic S, et al. Novel mechanisms of Na+ retention in obesity: phosphorylation of NKCC2 and regulation of SPAK/OSR1 by AMPK. Am J Physiol Renal Physiol. 2014;307:F96‐F106. [DOI] [PubMed] [Google Scholar]
- 22. Horman S, Vertommen D, Heath R, et al. Insulin antagonizes ischemia‐induced Thr172 phosphorylation of AMP‐activated protein kinase alpha‐subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem. 2006;281:5335‐5340. [DOI] [PubMed] [Google Scholar]
- 23. Li J, Qu X, Bertram JF. Endothelial‐myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin‐induced diabetic mice. Am J Pathol. 2009;175:1380‐1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mishra R, Cool BL, Laderoute KR, Foretz M, Viollet B, Simonson MS. AMP‐activated protein kinase inhibits transforming growth factor‐beta‐induced Smad3‐dependent transcription and myofibroblast transdifferentiation. J Biol Chem. 2008;283:10461‐10469. [DOI] [PubMed] [Google Scholar]
- 25. Thakur S, Viswanadhapalli S, Kopp JB, et al. Activation of AMP‐activated protein kinase prevents TGF‐beta1‐induced epithelial‐mesenchymal transition and myofibroblast activation. The American Journal of Pathology. 2015;185:2168‐2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dugan LL, You YH, Ali SS, et al. AMPK dysregulation promotes diabetes‐related reduction of superoxide and mitochondrial function. J Clin Invest. 2013;123:4888‐4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cavaglieri RC, Day RT, Feliers D, Abboud HE. Metformin prevents renal interstitial fibrosis in mice with unilateral ureteral obstruction. Mol Cell Endocrinol. 2015;412:116‐122. [DOI] [PubMed] [Google Scholar]
- 28. Chen KH, Hsu HH, Lee CC, et al. The AMPK agonist AICAR inhibits TGF‐beta1 induced activation of kidney myofibroblasts. PLoS ONE. 2014;9:e106554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mia S, Federico G, Feger M, et al. Impact of AMP‐activated protein kinase alpha1 deficiency on tissue injury following unilateral ureteral obstruction. PLoS ONE. 2015;10:e0135235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Qiu S, Xiao Z, Piao C, et al. AMPKalpha2 reduces renal epithelial transdifferentiation and inflammation after injury through interaction with CK2beta. J Pathol. 2015. [DOI] [PubMed] [Google Scholar]
- 31. Ning J, Xi G, Clemmons DR. Suppression of AMPK activation via S485 phosphorylation by IGF‐I during hyperglycemia is mediated by AKT activation in vascular smooth muscle cells. Endocrinology. 2011;152:3143‐3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guigas B, Viollet B. Targeting AMPK: from ancient drugs to new small‐molecule activators. EXS. 2016;107:327‐350. [DOI] [PubMed] [Google Scholar]