

Abstract
The development of synthetic tools to introduce saccharide derivatives into functionally complex molecules is of great interest, particularly in the field of drug discovery. Herein, we report a new route toward highly functionalized, arylated saccharides, involving a nickel-catalyzed cross-coupling of photoredox-generated saccharyl radicals with a range of aryl- and heteroaryl bromides, triggered by an organic photocatalyst. In contrast with existing methods, the mild reaction conditions achieve arylation of saccharide motifs while leaving available the anomeric carbon, thus providing access to a class of arylated glycosides underexplored until now. To demonstrate the potential of this strategy in late-stage functionalization, a variety of structurally complex molecules incorporating saccharide moieties were synthesized.
Keywords: Photoredox; Nickel Catalysis; 1,4-Dihydropyridines; Glycosylation
Graphical Abstract
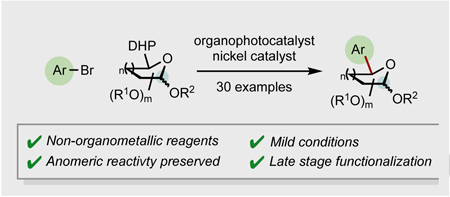
Keep the anomeric! The development of synthetic tools to introduce saccharide derivatives into functionally complex molecules is of great interest, particularly in the field of drug discovery. Herein, we report a new route toward highly functionalized, arylated saccharides, involving a nickel-catalyzed cross-coupling of photoredox-generated saccharyl radicals with a range of aryl- and heteroaryl bromides, triggered by an organic photocatalyst. In contrast with existing methods, the mild reaction conditions achieve arylation of saccharide motifs while leaving available the anomeric carbon, thus providing access to a class of arylated glycosides underexplored until now. To demonstrate the potential of this strategy in late-stage functionalization, a variety of structurally complex molecules incorporating saccharide moieties were synthesized.
The incorporation of saccharide derivatives into lead compounds represents an attractive way to diversify drug candidate scaffolds by modulating crucial parameters related to the in vivo efficacy of a therapeutic drug (e.g., solubility, membrane transport, pharmacodynamics or pharmacokinetics).1 Notably, “reverse aryl C-glycosides”2 comprise a class of saccharides that have proven to be efficient antibiotics,3 antitumor agents4 or inhibitors for diabetes.2d, 5 These glycosides have the particularity of bearing an aromatic moiety directly attached to the carbohydrate through a C-C bond, differentiating them from the usual O-glycosides, thus leading to better stability to both enzymatic and acidic hydrolysis, while preserving excellent biological efficacy.
Several routes toward anomeric, arylated saccharides have been reported,2 such as Friedel-Crafts reactions or nucleophilic additions of organometallic reagents (e.g., organolithium or Grignard reagents) to suitable electrophiles. However, such methods suffer from serious drawbacks, including harsh acidic or basic conditions, low functional group tolerance, and undesired side-products arising from elimination or epimerization processes.
Recently, the transition metal-catalyzed cross-coupling of in situ generated glycosyl radicals6 (from the corresponding glycosyl chloride or bromide) with organometallic reagents (aryl- or alkenylzinc and magnesium reagents) has emerged as one of the most efficient ways to access aryl C-saccharides (Scheme 1). This transformation has been achieved in the presence of nickel,7 cobalt8 or iron9 catalysts, affording the desired saccharide derivatives in moderate to good yields. Alternatively, pyranosylstannanes could be coupled with aryl halides in the presence of a palladium catalyst.10 Despite these improvements, such reaction conditions remain limited in terms of functional group tolerance and operational simplicity as they involve highly reactive organometallic species that need to be freshly prepared and suffer from β-elimination.8a Furthermore, stoichiometric amounts of metal waste are generated. Finally, these processes rely on substitution reactions at the anomeric center, thus preventing the substrate from being further functionalized by traditional chemistry at the anomeric carbon.
Scheme 1.

Transition metal-catalyzed synthesis of aryl C-glycosides: classical vs. non-classical anomeric arylation.
In this context, the development of straightforward, modular, and operationally simple conditions to access C-arylated saccharides remains an unsolved challenge and, specifically, methods that would preserve the anomeric carbon to afford non-classical “reverse aryl C-glycosides” are particularly scarce. Indeed, despite their attractive biological properties,11 few synthetic efforts have been carried out in this regard.12 Other approaches toward the synthesis of “reverse aryl C-glycosides” have been reported, such as the [4+2] cycloaddition between Danishefsky’s dienes and aromatic aldehydes, yielding glycal derivatives,13 or the addition of organozinc reagents to 4α-epoxypyranosides (Scheme 2).14 However, these strategies are restricted to pyranoses, lack modularity, and require several extra steps to obtain the desired “reverse aryl C-glycoside.”
Scheme 2.

Previous reports forming reverse C-aryl glycals and glycosides via cycloadditions and organozinc addition to epoxides.
In recent years, photoredox/nickel cross-coupling reactions have drawn extensive attention from the chemistry community.15 Such processes allow the cross-coupling of Csp3 nucleophiles under mild conditions by invoking a single-electron transmetalation pathway. Taking advantage of these modular and operationally simple conditions would allow access to virtually unexplored, non-classical, arylated saccharides. This approach would be a chemoselective strategy wherein arylation is directed to one of the two potential anomeric positions, one being masked by the DHP.12
With the goal of accessing glycosyl radicals that could be engaged in such dual catalytic processes, we turned our attention to 4-alkyl-1,4-dihydropyridine derivatives (DHPs).16 These species have proven to afford Csp3-centered alkyl radicals efficiently upon SET oxidation. In addition to being bench stable and easy to handle, these compounds tolerate high functionalization levels owing to the mild reaction conditions required for their preparation from the corresponding aldehyde. A wide range of 4-glycosyl-1,4-DHPs was accessed from commercially available pentose and hexose derivatives via deprotection and oxidation chemistry to form the aldehyde. Using Tripathi and coworker’s procedure, the monosaccharide DHPs were accessed via the reported acid-catalyzed condensation reaction.17 It is worth mentioning that this synthetic pathway accommodated a broad range of carbohydrate derivatives (e.g., ribonucleoside 1g, furanoses and pyranoses).
Next, the feasibility of the photoredox/nickel cross-coupling reaction between DHPs and 2-bromonaphthalene was studied by means of microscale high-throughput experimentation.18 Results from the screening revealed that the organic photocatalyst 4CzIPN (excited state Ered = +1.35 V vs SCE)19 was extremely efficient in oxidatively cleaving the DHP [Ered (1a) = +1.20 V vs SCE],18 delivering the desired glycosyl radical. Among the advantages of 4CzIPN compared to traditional iridium-based photocatalyst are its lower cost ($4.7/mmol vs $140.0/mmol),19 which provides significant benefit when it comes to industrial application. After further screening, the best yields were obtained in the presence of 2 mol % of 4-CzIPN photocatalyst, 5 mol % of NiBr2•dme and 7 mol % of dMeObpy as a ligand in acetone at room temperature for 24 h. As expected, control experiments showed that all parameters were essential for the transformation to proceed.
Next, the generality of the reaction with respect to the DHP derivative was explored. As illustrated in Figure 1, the reaction was remarkably tolerant of substitution in the saccharide scaffold, providing the desired products in moderate to high yields. Noteworthy, good to excellent diastereoselectivity was observed for certain furanosyl units (2a, 2d, and 2e), likely because of steric interactions with the adjacent substituent, an effect observed by Nakamura and coworkers in their recent iron-catalyzed arylation of halosugars.9 Excellent dr has also been observed in the field of photoredox/Ni catalysis when 2-methylcyclopentyltrifluoroborate was coupled with aryl bromide, affording exclusively the trans product.20
Figure 1.

Carbohydrate scope under optimized conditions: 2-bromonaphthalene (1.0 equiv), 1 (1.5 equiv), 4CzIPN (2 mol %), NiBr2•dme (10 mol %), dMeObpy (14 mol %), acetone, blue LEDs, rt, 24 h. aStandard conditions followed by TBAF addition (1 M in THF, 6 equiv). Ar = 2-naphthyl.
The steric control on the L-arabinofuranose and D-xylofuranose derivatives was made evident when comparing the effect of the vicinal substituent, where small substituents (e.g., MeO and F, 2b and 2c, respectively) afforded poor drs, whereas sterically encumbered, TBS-protected moieties afforded excellent steric control (9:1 dr). D-Ribofuranosyl DHP afforded excellent dr. X-Ray crystallography confirmed the retention of configuration (2f). Likewise, uridine-derived DHP afforded excellent diastereoselectivity, although in low yields; again, this highlights the prevalent role of steric interactions. Additionally, aryl pyranose 2h generated excellent dr with the aryl group cis to the dimethyl acetal protecting group (confirmed by X-ray crystallography). Not suprisingly, the more flexible radical leading to 2i afforded lower diastereoselectivity.6, 21, 22 Finally, the TBS protecting group choice allowed the formation of unprotected derivative 2e upon TBAF addition to the crude reaction mixture of 2d.
The next step was to explore the limitations with respect to the (hetero)aryl bromide partner (Figure 2). Aryl bromides bearing electron-withdrawing groups exhibited excellent reactivity, affording the corresponding product in good yields and high diastereoselectivities (2j-2k, 2m-2p). In addition, electron-neutral and electron-rich aryl bromides were tolerated (2q- 2t). Notably, a pinacol boronic ester (2r) was well accommodated, providing a handle for further functionalization.23 Although excellent functional group tolerance was observed, we hypothesize that the diminished yields with electron-neutral aryl bromides (2s and 2t) result from a challenging oxidative addition. To access more complex structures, (hetero)aryl bromides, such as pyridine moieties (2u and 2v), oxadiazoles (2w and 2y) and thiophene derivatives (2x and 2z), were successfully employed. Interestingly, carbonyl groups, which could react in the presence of organometallic reagents, pose no problem (2n, 2o and 2x).
Figure 2.

Exploration of the aryl and heteroaryl bromide scope.
To explore further the chemical diversity accessible through this method, several structurally complex molecules were engaged under the developed reaction conditions (Figure 3). Coupling with a steroid derivative (3a) proved successful, thus offering a privileged substructure of use in medicinal chemistry.24 Recognizing the potential for late-stage functionalization, we introduced functional group-dense aryl bromides from Merck’s chemistry informer library containing drug-like motifs.25 A quinoxalinedione derivative, which is encountered in ionotropic glutamate receptor antagonists,26
Figure 3.

Late-stage C-glycosylation of functionally dense aryl bromides (see Supporting Information for experimental details).
exhibited good reactivity under optimized conditions to furnish pyranose product 3b with excellent diastereocontrol. Even the more complex 3-bromothiophene derivative bearing a guanidine motif, belonging to the family of aspartic protease inhibitors,27
afforded the desired products in high yield (3c). In addition, a furanosyl residue could be introduced into the loratadine scaffold (3d) under the standard reaction conditions.
Based on previous reports on the reactivity of DHPs under Ni/photoredox dual catalytic conditions,16c,d a plausible mechanism is outlined in Scheme 3. This pathway involves the initial photoexcitation of the 4CzIPN photocatalyst (Ered = +1.35 V vs SCE),18 which undergoes reductive quenching with the DHP derivative [Ered (1a) = + 1.20 V vs SCE]. The resulting radical cation A rapidly fragments to generate an aromatized pyridine derivative along with the corresponding saccharyl radical B. This latter species would first add to the active Ni(0) catalyst, thus producing a Ni(I)-saccharyl complex C that would undergo oxidative addition with the aryl bromide to afford the corresponding Ni(III) complex D.28 Subsequent reductive elimination would take place to yield the cross-coupling product 2 and a Ni(I) species E. This latter complex would then be reduced to Ni(0) F with the reduced form of the 4CzIPN photocatalyst (Ered = + 1.21 V vs SCE),18 thus regenerating both active catalysts for subsequent catalytic cycles.
Scheme 3.

Putative mechanism for Ni/photoredox dual catalysis for the synthesis of arylated C-saccharides.
It is worth noting that although the saccharide backbone plays a key role in the observed diastereomeric ratios, leading in certain cases to substrate control products (e.g., 2f–2h), it is evident when looking at other examples that the aryl bromide is playing a role as well (e.g., 2k and 2s), where different substitution patterns in the aromatic backbone lead to different drs in the presence of the same saccharyl radical. Previous mechanistic studies suggest the high-valent Ni(III) species D ultimately dictates the observed diastereoselectivity after the irreversible reduction elimination;29 therefore, we sought to improve the lower drs by identifying suitable ligands.7 Bidentate and tridentate ligands were screened with both Ni(0) and Ni(II) species.18 Modifying the bipyridine backbone by replacing electron-donating methoxy groups with bulkier, less electron-rich, tert-butyl substituents afforded excellent diastereoselectivities, improving 2w from 3.3:1 dr to >20:1, for example. Alternatively, the use of phenanthroline resulted in a >20:1 dr for hexose 1,4-dihydropyridine 1i, whereas the previously successful dtbbpy showed no improvement. Although the subtleties dictating the diastereoselectivity remain elusive, it is clear that diastereoselectivities can be improved on a case-by-case basis, if needed, through effective screening efforts. Further mechanistic studies are ongoing to shed light on these ligand effects.
In summary, we disclosed the first general synthesis of non-classical, “reverse” aryl C-glycosides via Ni/photoredox dual catalysis using dihydropyridyl saccharide motifs as radical precursors. The optimized conditions provide straightforward access to a wide variety of highly functionalized, arylated saccharides. Further studies were conducted to improve observed diastereoselectivities by targeting the diastereo-determining step, reductive elimination from the high-valent Ni(III) complex. This new strategy could find broad applications in the field of medicinal chemistry research, affording structurally novel materials that have been largely underexplored.
Supplementary Material
Figure 4.

Exploring the ligand effect on the diastereoselectivity. Improving the catalyst-control.
Acknowledgements
We thank the NIH (R01 GM 113878) and NSF GOALI program (CHE-0848460) for supporting this program. J.K.M. was supported by the Bristol-Myers Squibb (BMS) Graduate Fellowship in Synthetic Organic Chemistry. We thank Dr. Charles W. Ross, III (UPenn) for obtaining HRMS values, Dr. George Furst for NMR assistance, and Dr. Patrick Carroll for obtaining X-ray crystallography data. Finally, we greatly appreciate the helpful insight provided by the referees.
Abbreviations
- DHP
1,4-dihydropyridine
- SET
Single Electron Transfer
- 4CzIPN
2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile
- dMeObpy
4–4′-dimethoxy-2–2′-bipyridine
Footnotes
Conflict of interest
The author declare no conflict of interest.
References
- [1].Gantt RW, Peltier-Pain P, Thorson S, J.Nat. Prod. Rep 2011, 28, 1811–1853 [DOI] [PubMed] [Google Scholar]
- [2].For selected books and reviews:; a) Jaramillo C, Knapp S, Synthesis 1994, 1–20; [Google Scholar]; b) Lee D, He M, Curr. Top. Med. Chem 2005, 5, 1333–1350; [DOI] [PubMed] [Google Scholar]; c) Bililign T, Griffith BR, Thorson JS, Nat. Prod. Rep 2005, 22, 742–760; [DOI] [PubMed] [Google Scholar]; d) Bokor É, Kun S, Goyard D, Tóth M, Praly J-P, Vidal S, Somsák L, Chem. Rev 2017, 117, 1687. [DOI] [PubMed] [Google Scholar]
- [3].a) Hacksell U, Daves GD, Progress in Medicinal Chemistry, Elsevier, Amsterdam, 1985; Vol. 22, 1–65; [DOI] [PubMed] [Google Scholar]; b) Buchanan JG, Progress in the Chemistry of Organic Natural Products, Springer, New York, 1985, 44, 243. [DOI] [PubMed] [Google Scholar]
- [4].For selected examples:; a) Hosoya T, Takashiro E, Matsumoto T, Suzuki K, J. Am. Chem. Soc 1994, 116, 1004–1015; [Google Scholar]; b) Parker KA, Koh Y, J. Am. Chem. Soc 1994, 116, 11149–11150; [Google Scholar]; c) Cai X, Ng K, Panesar H, Moon S-J, Paredes M, Ishida K, Hertweck C, Minehan TG, Org. Lett 2014, 16, 2962–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].For selected reviews and articles:; a) Fujita Y, Inagaki NJ, Diabetes Invest 2014, 5, 265–275; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang Y, Liu Z-P, Curr. Med. Chem 2016, 23, 832–849; [DOI] [PubMed] [Google Scholar]; c) Goodwin NC, Ding Z-M, Harrison BA, Strobel AL Harris M Smith AY Thompson W Xiong F Mseeh DJ Bruce D Diaz S Gopinathan L Li EO O’Neill M Thiel AGE Wilson KG Carson DR Powell D Rawlins B, J. Med. Chem 2017, 60, 710–721. [DOI] [PubMed] [Google Scholar]
- [6].Togo H, He W, Waki Y, Yokoyama M, Synlett 1998, 7, 700–717. [Google Scholar]
- [7].Gong H, Gagné MR, J. Am. Chem. Soc 2008, 130, 12177–12183. [DOI] [PubMed] [Google Scholar]
- [8].(a) Nicolas L, Angibaud P, Stansfield I, Bonnet P, Meerpoel L, Reymond S, Cossy J, Angew. Chem. Int. Ed 2012, 51, 11101–11104; Angew. Chem. 2012, 124, 11263–11266; [DOI] [PubMed] [Google Scholar]; b) Nicolas L, Izquierdo E, Angibaud P, Stansfield I, Meerpoel L, Reymond S, Cossy J, J. Org. Chem 2013, 78, 11807–11814. [DOI] [PubMed] [Google Scholar]
- [9].Adak L, Kawamura S, Toma G, Takenaka T, Isozaki K, Takaya H, Orita A, Li HC, Shing TKM, Nakamura M J.Am. Chem. Soc 2017, 139, 10693–10701. [DOI] [PubMed] [Google Scholar]
- [10].a) Zhu F, Rodriguez J, Yang T, Kevlishvili I, Miller E, Yi D, O’Neill S, Rourke MJ, Liu P, Walczak MA, J. Am. Chem. Soc 2017, 139, 17908–17922; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhu F, Rourke MJ, Yang T, Rodriguez J, Walczak MA, J. Am. Chem. Soc 2016, 138, 12049–12052. [DOI] [PubMed] [Google Scholar]
- [11].For selected examples:; a) Goodwin NC, Mabon R, Harrison BA, Shadoan MK, Almstead ZY, Xie Y, Healy J, Buhring LM, DaCosta CM, Bardenhagen J, Mseeh F, Liu Q, Nouraldeen A, Wilson AGE, Kimball SD, Powell DR, Rawlins DB, J. Med. Chem 2009, 52, 6201–6204; [DOI] [PubMed] [Google Scholar]; b) Župančić N, Ban Ž, Matić J, Saftić D, Glavaš-Obrovac L, Žinić B, Croat. Chem. Acta 2015, 88, 43–52; [Google Scholar]; c) Goodwin NC, Ding Z-M, Harrison BA, Strobel ED, Harris AL, Smith M, Thompson AY, Xiong W, Mseeh F, Bruce DJ, Diaz D, Gopinathan S, Li L, O’Neill E, Thiel M, Wilson AGE, Carson KG, Powell DR, Rawlins DB, J. Med. Chem 2017, 60, 710–721. [DOI] [PubMed] [Google Scholar]
- [12].a) Hashmi IA, Feist H, Michalik M, Reinke H, Peseke K, J. Carbohyd. Chem 2006, 25, 19–32; [Google Scholar]; b) Hashmi IA, Ali FI, Fiest H, Peseke K, Synth. Commun 2010, 40, 1243–1247; [Google Scholar]; c) Kaliappan KP, Subrahmanyam AV, Org. Lett 2007, 9, 1121–1124. [DOI] [PubMed] [Google Scholar]; d) Chakraborty S; Das G; Ghosh S; Mal D Org. Biomol. Chem 2016, 14, 10636–10647. [DOI] [PubMed] [Google Scholar]
- [13].a) Bednarski M, Danishefsky S, J. Am. Chem. Soc 1986, 108, 7060–7067; [Google Scholar]; b) Berkowitz DB, Danishefsky SJ, Schulte GK, J. Am. Chem. Soc 1992, 114, 4518–4529; [Google Scholar]; c) Helliwell M, Phillips IM, Pritchard RG, Stoodley RJ, Tetrahedron Lett 1999, 40, 8651–8655. [Google Scholar]
- [14].Cheng G, Fan R, Hernández-Torres JM, Boulineau FP, Wei A, Org. Lett 2007, 9, 4849–4852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].For selected reviews:; a) Prier CK, Rankic DA, MacMillan DWC, Chem. Rev 2013, 113, 5322–5363; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tellis JC, Kelly CB, Primer DN, Jouffroy M, Patel NR, Molander GA, Acc. Chem. Res 2016, 49, 1429–1439; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cavalcanti LN, Molander GA, Top. Curr. Chem 2016, 374, 1–23. [DOI] [PubMed] [Google Scholar]
- [16].For selected examples:; a) Nakajima K, Nojima S, Sakata K, Nishibayashi Y, ChemCatChem 2016, 8, 1028–1032; [Google Scholar]; b) Chen W, Liu Z, Tian J, Li J, Ma J, Cheng X, Li G, J. Am. Chem. Soc 2016, 138, 12312–12315; [DOI] [PubMed] [Google Scholar]; c) Nakajima K, Nojima S, Nishibayashi Y, Angew. Chem. Int. Ed 2016, 55, 14106–14110; Angew. Chem. 2016, 128, 14312–14316; [DOI] [PubMed] [Google Scholar]; d) Gutiérrez-Bonet Á, Tellis JC, Matsui JK, Vara BA, Molander GA, ACS Catal 2016, 6, 8004–8008; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Gutiérrez-Bonet Á, Remeur C, Matsui JK, Molander GA, J. Am. Chem. Soc 2017, 139, 12251–12258; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Buzzetti L, Prieto A, Roy SR, Melchiorre P, Angew. Chem., Int. Ed 2017, 56, 15039–15043; Angew. Chem. 2017, 129, 15235–15239; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Verrier C, Alandini N, Pezzetta C, Moliterno M, Buzzetti L, Hepburn HB, Vega-Penaloza A, Silvi M, Melchiorre P, ACS Catal. 2018, 8, 1062–1066; [Google Scholar]; h) Zhang H-H, Yu S, J. Org. Chem 2017, 82, 995–1006; [DOI] [PubMed] [Google Scholar]; For a review see:; i) Huang W, Cheng X, Synlett 2017, 28, 148–158. [Google Scholar]
- [17].Tewari N, Dwivedi N, Tripathi RP, Tetrahedron Lett 2004, 45, 9011–9014. [Google Scholar]
- [18].See the Supporting Information for further details.
- [19].Luo J, Zhang J, ACS Catalysis 2016, 6, 873–877. [Google Scholar]
- [20].Primer DN, Karakaya I, Molander GA, J. Am. Chem. Soc 2015, 137, 2195–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].For selected reviews on the conformation of glycosyl radical species:; a) Dupuis J, Giese B, Rüegge D, Fischer H, Korth H-G, Sustmann R , Angew. Chem. Int. Ed 1984, 23, 896–898; Angew. Chem. 1984, 96, 887–888; [Google Scholar]; b) Giese B, Angew. Chem. Int. Ed 1989, 28, 969–980; Angew. Chem. 1989, 101, 993–1004; [Google Scholar]; c) Beckwith ALJ, Duggan, Tetrahedron 1998, 54, 6919–6928; [Google Scholar]; d) Rychnovsky SD, Powers JP, LePage TJ, J. Am. Chem. Soc 1992, 114, 8375–8384. [Google Scholar]
- [22].Absolute configurations were determined either by comparison with the literature data or by X-ray crystal structure analysis when possible (see Supporting Information for further details).
- [23].Yamashita Y, Tellis JC, Molander GA, Proc. Natl. Acad. Sci 2015, 112, 12026–12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ono M, Takamura C, Sugita F, Masuoka C, Yoshimitsu H, Ikeda T, Nohara T, Chem. Pharm. Bull 2007, 55, 551–556. [DOI] [PubMed] [Google Scholar]
- [25].Kutchukian PS, Dropinski JF, Dykstra KD, Li B, DiRocco DA, Streckfuss EC, Campeau L-C, Cernak T, Vachal P, Davies IW, Krska SW, Dreher SD, Chem. Sci 2016, 7, 2604–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Turski L, Schneider HH, Neuhaus R, McDonald F, Jones GH, Löfberg B, Schweinfurth H, Huth A, Krüger M, Ottow E, Restor. Neurol. Neurosci 2000, 17, 45–59. [PubMed] [Google Scholar]
- [27].Iserloh U, Gu H, Qian G, Tadesse D, Lai G, Duo J, Heterocyclic aspartyl protease inhibitors . PCT Int. Appl WO2008/103351 A2, 2008. [Google Scholar]
- [28].a) Anderson TJ, Jones GD, Vicic DA, J. Am. Chem. Soc 2004, 126, 8100–8101; [DOI] [PubMed] [Google Scholar]; b) Jones GD, McFarland C, Anderson TJ, Vicic DA, Chem. Commun 2005, 33, 4211–4213. [DOI] [PubMed] [Google Scholar]
- [29].Gutierrez O, Tellis JC, Primer DN, Molander GA, Kozlowski MC, J. Am. Chem. Soc 2015, 137, 4896–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.