

Abstract
EndMT is an intricate cellular differentiation process whereby endothelial cells detach and migrate away from the endothelium and, to varying extents, decrease endothelial properties and acquire mesenchymal features. First described in developing heart valves as an epithelial mesenchymal transformation, EndMT begins in response to an external signal, often transforming growth factor-β (TGFβ). The endothelial cells lose luminal-abluminal polarity, extend filopodia and migrate into extravascular space where they take up residence, in the case of heart valves, as valve interstitial cells. Properly controlled EndMT is essential for heart valve development: too little and valves fail to form; too much and the valves thicken and cannot close properly. EndMT appears to persist past embryonic development as endothelial cells expressing EndMT markers can be detected in vivo in adult ovine and human valves. In vitro, valve endothelial cells treated with TGFβ undergo robust EndMT; hence valve endothelial cells can serve as a prototype for revealing regulators of EndMT.
Reactivation of EndMT in post-natal settings has emerged as a potential mechanism for adaptation to new physiologic settings and at the same time for maladaptive responses to disease1. This is exemplified by EndMT in cardiac valves, which are lined with a specialized endothelium that originates from FLK1+ (a.k.a. VEGFR2+) progenitor cells in cardiac mesoderm, distinct from vascular endothelium2. Although endocardial and vascular endothelium are molecularly similar2, endocardial endothelial cells exhibit a distinct plasticity, which may provide valve endothelial cells with unique capabilities for adaptation and function over a lifetime.
Keywords: Endothelial-to-mesenchymal transition, mitral valve, transforming growth factor-β, endothelial cells
Short summary
The mitral valve, lined with a unique endothelium, provides an exemplary setting to unravel the causes and nuances of endothelial-to-mesenchymal transition (EndMT), and may provide guideposts for understanding EndMT more broadly as its reactivation in post-natal settings has emerged as a potential mechanism for adaptation to both physiologic and pathologic settings.
EndMT in mitral valves
EndMT can be detected in 1–2% of endothelial cells in healthy adult valves from sheep and humans 3. In vitro studies with valve endothelial cells provide supporting evidence for post-development EndMT capability 3, and thus we speculated that EndMT persists at a low level throughout life in order to replenish valve interstitial cells, major producers of the valve extracellular matrix needed to insure durability and function. In contrast, EndMT is not seen in adult murine valves when analyzed by in vivo endothelial lineage tracing4, indicating that species, models and experimental tools are important variables. We found EndMT increased, in vivo, when ovine mitral valve leaflets were exposed to mechanical stretch designed to mimic the tethering imparted on the leaflets when the left ventricle enlarges after myocardial infarction; EndMT occurred with little evident TGFβ or leukocyte infiltration, and coincided with an increase in leaflet area 5, 6. We postulated that mechanical stretch-induced EndMT produced new valve interstitial cells, which in turn increased leaflet area in the large animal model. This appeared to be purposeful EndMT that reflected an adaptive capability in ovine mitral valves.
In contrast, when a myocardial infarction was added to the mechanical stretch model, potentially maladaptive EndMT ensued in the mitral valve leaflets along with a constellation of cellular changes, including excessive collagen deposition, sub-endothelial localized TGFβ and leukocytes 6. This apparently unchecked EndMT was diminished, along with the collagen, TGFβ and leukocytes, in animals treated with Losartan7, an angiotensin receptor-1 antagonist that can indirectly block TGFβ signaling. These studies indicate Losartan could be used to prevent post-infarction thickening of mitral valve leaflets, which can impair leaflet closure and lead to mitral regurgitation, a serious complication that leads to heart failure and doubles mortality.
Further insight on the excessive, maladaptive EndMT came from a second ovine model – an inferior myocardial infarction model in which mitral regurgitation develops. Surveying for inflammatory cells at 6 months post-infarction, we unexpectedly found the CD45 protein tyrosine phosphatase expressed in mitral valve endothelial cells undergoing EndMT3. The CD45+/VE-cadherin+/αSMA+ cells increased from 1% in sham animals to 17% in animals with inferior myocardial infarction, and were positively correlated with mitral regurgitation and inversely correlated with ejection fraction. Hematopoietic cell markers CD11b and CD14 were not detected. In vitro, TGFβ induced CD45 mRNA and protein in mitral valve endothelial cells, along with collagens and endogenous TGFβs, and increased cellular migration (a hallmark of EndMT). None of the changes were seen in TGFβ-treated arterial endothelial cells 3. A CD45 protein tyrosine phosphatase inhibitor blocked the EndMT and fibrotic markers and reduced migration of TGFβ-treated valve endothelial cells.
CD45 is not normally expressed in endothelial cells so its presence in valve endothelial cells is perplexing. It may be that the plasticity of valve endothelial cells presents a pliable chromatin landscape, permissive for transcription of genes such as CD45. Once synthesized, CD45 may cause any number of perturbations in the mitral valve endothelial cells. That CD45 can fine tune signaling is clear from its role in hematopoietic cells. We speculate that TGFβ-induced CD45 drives mitral valve endothelial cells into fibrotic, collagen-producing mesenchymal cells (Figure 1), however further experiments are needed to understand the full extent of EndMT, the functionality of the resulting cells and to explore possible paracrine effects on resident valve interstitial cells, which may also transition into fibrotic cells.
Figure 1 –
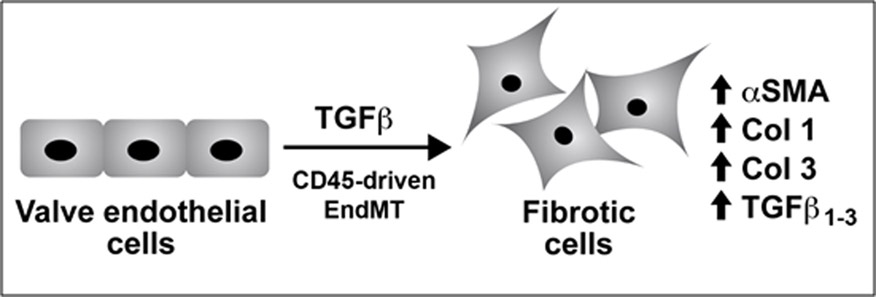
TGFβ-treated valve endothelial cells show hallmarks of EndMT: loss of cell-cell junctions and cobblestone morphology, increased α-SMA and increased migration. Collagens 1 and 3 and endogenous TGFβ1–3 are increased, suggesting a transition to a fibrotic phenotype. CD45, a protein tyrosine phosphatase, is also increased, and blocking its phosphatase activity reduces EndMT markers, collagens and TGFβ1–3. More studies are need to understand the functionality of cells generated from CD45-driven EndMT.
The notion of adaptive versus excessive EndMT prompts many questions. In vitro TGFβ-treated valve endothelial cells appear to model excessive EndMT, but what incites EndMT in vivo? Inflammatory cytokines together with TGFβ and altered shear stress can stimulate EndMT in vascular endothelial cells and therefore it is likely that pro-inflammatory cytokines provoke EndMT in valvular endothelial cells as well. Are endogenous shut off mechanisms, such as the histone deacetylase-3-mediated silencing of TGFβ seen in heart development8, thwarted by cytokines and altered shear forces? An imperfect off-switch might cause EndMT to go too far, to overshoot, which could then produce fibrotic cells. Can the overshoot be reversed, asseen with Losartan7:if so, reversal of EndMT might be used to reduce leaflet thickening in ischemic mitral regurgitation. Studies are needed to determine if there is a specific combination of factors that drives endothelial cells directly and/or irreversibly to a fibrotic phenotype. And to what extent is a latent endothelial phenotype maintained, as indicated by reduced but still detectable expression of endothelial markers? Do these cells return more readily to endothelial function compared to cells that have fully transitioned to endothelial marker-negative mesenchymal cells?
Partial versus complete EndMT
Partial EndMT has been implicated at the onset of angiogenesis9 which makes sense because the initial steps of EndMT and angiogenesis are similar: endothelial cells depart from the endothelial monolayer and migrate into the extravascular milieu. The angiogenic transition is partial and reversed: the cells resume endothelial functions as nascent blood vessels take shape. In contrast, full-course EndMT creates extracellular matrix-producing mesenchymal cells with substantially diminished endothelial functions such as cytokine-induced leukocyte adhesion. Hughes and colleagues revealed a molecular overlap between partial and complete EndMT by showing Slug9, a key transcriptional factor required for EndMT, is also required to form endothelial sprouts. In contrast, tumor endothelial cells appear constitutively poised for mesenchymal transition. Studies from a decade ago revealed tumor endothelial cells co-express endothelial and mesenchymal markers and show a propensity for vascular calcification 10. More recently, tumor endothelial cells in glioblastoma multiforme were shown to lack endothelial receptors such as VEGFR2 and express mesenchymal markers such as N-cadherin and α-smooth muscle actin (αSMA), indicating EndMT in tumors may negate certain anti-endothelial therapies 11. In vivo pulmonary hypertension studies identified two sets of genetically-labeled: partial EndMT-derived cells showed progenitor cell qualities while pulmonary endothelial cells that underwent complete EndMT showed increased proliferation and migration12 providing an example of divergent EndMT outcomes.
A productive, partial EndMT appears to happen in the “artery reassembly” reported by Das and colleagues13. The group discovered a novel mechanism in injured neonatal hearts that serves to build collateral arterial vessels. Lineage tracing and state-of-the-art imaging revealed non-coalesced arterial endothelial cells moving out from arterial tips and migrating along capillaries where they subsequently formed collaterals. Hence, for a transient period of time, the arterial endothelial exhibit EndMT behaviors: they leave an established endothelium, extend filopodia and migrate through extracellular. It would be fascinating to learn if the collateral-bound arterial endothelial cells show molecular features of partial EndMT, and if these cells might provide guideposts for purposeful EndMT in tissue regeneration.
EndMT in cardiovascular diseases
EndMT has been implicated in several cardiovascular diseases, in particular atherosclerosis where TGFβ, inflammation and altered shear stress drive EndMT in vascular endothelium. (EndMT in cardiovascular disease is covered beautifully in a recent review by Kovacic and colleagues1.) An elegant example is the work by Evrard and colleagues where they applied inducible endothelial lineage tracing and single cell imaging to distinguish partial from completely transitioned mesenchymal cells derived from the endothelium in atherosclerotic aortas14. Their quantification showed that 3–9% of the intimal plaque cells were endothelial-derived, indicating a robust process. Importantly, the extent of EndMT was correlated with plaque instability, and results from the murine model were extended to human atherosclerotic plaques.
EndMT has also been described in venular vessel segments moved to an arterial position to create a grafted vessels in mice. In such a setting, EndMT-derived smooth muscle cells could contribute to building an arterial-appropriate smooth muscle layer, however excessive TGFβ-mediated EndMT in grafted vessels resulted in intimal thickening. Hence, this may be an example of the precarious balance between purposeful and maladaptive EndMT. EndMT has also been implicated in cerebral cavernous malformations (CCM), a relatively common vascular malformation that occurs in both sporadic and inherited forms. Disassembled cellular junctions seen in CCM may reflect the first step of EndMT. In another setting, EndMT appeared to be the source of sub-endocardial fibrotic tissue known to accumulate in endocardial fibroelastosis, a devastating condition associated with congenital heart defects. In summary, EndMT can disrupt the endothelium and contribute unwanted fibroblastic cells in a number of cardiovascular diseases.
Experimental approaches
Our understanding of EndMT has grown by leaps and bounds with the wealth of new tools and enhanced experimental capabilities. Inducible endothelial lineage tracing in murine models is particularly useful for distinguishing mesenchymal cells derived from embryonic versus post-natal EndMT4, 14. In vitro studies complement and extend in vivo findings, and enable mechanistic experiments that can sharpen hypotheses for further testing in vivo. Yet meticulous attention to detail is needed to tease out mechanisms that drive EndMT. Purified endothelial cells are essential – the culture must be free of smooth muscle cells, pericytes and fibroblasts. Even a small number of contaminating cells can lead to an appearance of EndMT because TGFβ increases α-SMA, a commonly used marker for EndMT, in fibroblasts, for example. Indeed, a small number of mesenchymal cells are likely to be present in most primary endothelial cultures such as human umbilical vein endothelial cells. In contrast to valve endothelial cells, TGFβ-driven EndMT is not readily detected in highly purified vascular endothelial cells in vitro3. Further, EndMT appears to be a far less robust process in vascular endothelial cells; perhaps additional factors, stimuli and time is needed to achieve partial or complete EndMT in non-valvular endothelial cells. Gain of mesenchymal and loss of endothelial markers should be combined with functional assays that measure contractility (mesenchymal cell) and cytokine-induced leukocyte adhesion (endothelial) to determine the extent to which cells have transitioned. In summary, a full toolbox of experimental approaches – from in vivo animal studies to in vitro experiments to analyses of human tissues and cells – is needed to understandhow EndMT contributes to adaptive and maladaptive responses to disease.
Future Directions
Conceptual and practical insights from new research on EndMT and endothelial plasticity may teach us how to manipulate EndMT. For example, can purposeful EndMT be incited to rebuild, remodel or regenerate tissues and organs? Can maladaptive EndMT be prevented or reversed? That both partial and full EndMT begin with similar cellular activities and transcriptional regulators (e.g. Slug) underscores the need for experiments that can track cellular phenotypes and analyze cellular functions, and thereby distinguish between the two. In heart valves, we need to understand the how healthy new interstitial cells arise and how transition to fibrosis is kept in check, as this may guide us towards new approaches for treating valve diseases. Losartan, perhaps through indirect dampening down of excessive TGFβ, would be an excellent starting point based on its ability to prevent EndMT and disorganized collagen deposition in mitral valves after myocardial infarction.
Acknowledgments
I thank Dr. Robert A. Levine, Cardiac Ultrasound Laboratory, Massachusetts General Hospital, Harvard Medical School, Boston MA and Dr. Christopher C. W. Hughes, Department of Molecular Biology and Biochemistry, University of California, Irvine for helpful comments and suggestions for this article.
Sources of Funding – Supported by HL141917 from NHLBI of the National Institutes of Health The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- EndMT
endothelial to mesenchymal transition
- TGFβ
transforming growth factor-β
- VEGFR2
vascular endothelial growth factor receptor-2
- αSMA
α-smooth muscle actin
- CCM
cerebral cavernous malformation
Footnotes
Disclosures - none
References
- 1.Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G and Baker AH. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73:190–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakano A, Nakano H, Smith KA and Palpant NJ. The developmental origins and lineage contributions of endocardial endothelium. Biochim Biophys Acta. 2016;1863:1937–47. [DOI] [PubMed] [Google Scholar]
- 3.Bischoff J, Casanovas G, Wylie-Sears J, Kim DH, Bartko PE, Guerrero JL, Dal-Bianco JP, Beaudoin J, Garcia ML, Sullivan SM, Seybolt MM, Morris BA, Keegan J, Irvin WS, Aikawa E and Levine RA. CD45 Expression in Mitral Valve Endothelial Cells After Myocardial Infarction. Circ Res. 2016;119:1215–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim AJ, Alfieri CM and Yutzey KE. Endothelial Cell Lineage Analysis Does Not Provide Evidence for EMT in Adult Valve Homeostasis and Disease. Anat Rec (Hoboken). 2019;302:125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dal-Bianco JP, Aikawa E, Bischoff J, Guerrero JL, Handschumacher MD, Sullivan S, Johnson B, Titus JS, Iwamoto Y, Wylie-Sears J, Levine RA and Carpentier A. Active adaptation of the tethered mitral valve: insights into a compensatory mechanism for functional mitral regurgitation. Circulation. 2009;120:334–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dal-Bianco JP, Aikawa E, Bischoff J, Guerrero JL, Hjortnaes J, Beaudoin J, Szymanski C, Bartko PE, Seybolt MM, Handschumacher MD, Sullivan S, Garcia ML, Mauskapf A, Titus JS, Wylie-Sears J, Irvin WS, Chaput M, Messas E, Hagege AA, Carpentier A, Levine RA and Leducq Transatlantic Mitral N. Myocardial Infarction Alters Adaptation of the Tethered Mitral Valve. J Am Coll Cardiol. 2016;67:275–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartko PE, Dal-Bianco JP, Guerrero JL, Beaudoin J, Szymanski C, Kim DH, Seybolt MM, Handschumacher MD, Sullivan S, Garcia ML, Titus JS, Wylie-Sears J, Irvin WS, Messas E, Hagege AA, Carpentier A, Aikawa E, Bischoff J, Levine RA and Leducq Transatlantic Mitral N. Effect of Losartan on Mitral Valve Changes After Myocardial Infarction. J Am Coll Cardiol. 2017;70:1232–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewandowski SL, Janardhan HP and Trivedi CM. Histone Deacetylase 3 Coordinates Deacetylase-independent Epigenetic Silencing of Transforming Growth Factor-beta1 (TGF-beta1) to Orchestrate Second Heart Field Development. J Biol Chem. 2015;290:27067–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welch-Reardon KM, Wu N and Hughes CC. A role for partial endothelial-mesenchymal transitions in angiogenesis? Arterioscler Thromb Vasc Biol. 2015;35:303–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudley AC, Khan ZA, Shih SC, Kang SY, Zwaans BM, Bischoff J and Klagsbrun M. Calcification of multipotent prostate tumor endothelium. Cancer Cell. 2008;14:201–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang M, Liu T, Ma P, Mitteer RA Jr., Zhang Z, Kim HJ, Yeo E, Zhang D, Cai P, Li C, Zhang L, Zhao B, Roccograndi L, O’Rourke DM, Dahmane N, Gong Y, Koumenis C and Fan Y c-Met-mediated endothelial plasticity drives aberrant vascularization and chemoresistance in glioblastoma. J Clin Invest. 2016;126:1801–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki T, Carrier EJ, Talati MH, Rathinasabapathy A, Chen X, Nishimura R, Tada Y, Tatsumi K and West J. Isolation and characterization of endothelial-to-mesenchymal transition cells in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2018;314:L118–L126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Das S, Goldstone AB, Wang H, Farry J, D’Amato G, Paulsen MJ, Eskandari A, Hironaka CE, Phansalkar R, Sharma B, Rhee S, Shamskhou EA, Agalliu D, de Jesus Perez V, Woo YJ and Red-Horse K. A Unique Collateral Artery Development Program Promotes Neonatal Heart Regeneration. Cell. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman KR, d’Escamard V, Li JR, Hadri L, Fujitani K, Moreno PR, Benard L, Rimmele P, Cohain A, Mecham B, Randolph GJ, Nabel EG, Hajjar R, Fuster V, Boehm M and Kovacic JC. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nature communications. 2016;7:11853. [DOI] [PMC free article] [PubMed] [Google Scholar]