

Abstract
Rhombencephalosynapsis (RES) is a unique cerebellar malformation characterized by fusion of the cerebellar hemispheres with partial or complete absence of a recognizable cerebellar vermis. Subsets of patients also have other brain malformations such as midbrain fusion with aqueductal stenosis, characteristic craniofacial features (prominent forehead, flat midface, hypertelorism, ear abnormalities), and somatic malformations (heart, kidney, spine and limb defects). Similar to known genetic brain malformations, the RES cerebellar malformation is highly stereotyped, yet no genetic causes have not been identified. Here, we outline our current understanding of the genetic basis for RES, discuss limitations, and outline future approaches to identifying the causes of this fascinating brain malformation.
Keywords: RES, genetics, cerebellum, neuroimaging
1. INTRODUCTION
1.1. History
Rhombencephalosynapsis (RES) is a unique cerebellar malformation in which the vermis is deficient or absent and the hemispheres are fused across the midline. It was first described in the autopsy report of a 28-year old male in 1914 (Obersteiner, 1914). In the early 1980s, Gomez and Lopez defined a congenital anomaly syndrome in a series of patients characterized by RES, craniosynostosis, dysmorphic facial features, parietal-occipital scalp alopecia, and trigeminal anesthesia (Gomez, 1979; Lopez-Hernandez, 1982), subsequently called Gomez-Lopez-Hernandez syndrome (GLH). More recently, the GLH diagnosis has also been applied to patients with RES and alopecia, in the absence of trigeminal anesthesia or obvious craniofacial features (Sukhudyan et al., 2010). Since these original reports, more than 150 additional individuals with RES have been described in the literature (Barth, 2008; Pasquier et al., 2009; Tully et al., 2012).
1.2. Epidemiology
As with most brain malformations, the true prevalence of RES is unknown. Historically, it has been thought to be exceedingly rare. However, in a series of 3,000 consecutive pediatric patients who underwent neuroimaging, RES was observed in 0.13% (Sener, 2000). We care for ~20 patients with RES from a maximal catchment area of ~5,000,000 people, which yields a conservative prevalence estimate of ~½50,000. In addition, we now enroll ~10 patients annually in our research cohort, likely due to the advent of improved MRI technology and increased recognition by clinicians.
1.3. Neuroanatomical features
RES is characterized by a deficient or absent vermis with hemispheres that are fused across the midline (Figure 1). The cerebellar tonsils and deep cerebellar nuclei can also be fused. The severity of RES can be graded by the amount of remaining vermis (Ishak et al., 2012). Milder forms of RES are often associated with normal or even large cerebellar size with both inferior and superior cerebellar ectopia. In contrast, severe and complete RES are often associated with cerebellar hypoplasia. The midbrain is often fused across the midline (mesencephalosynapsis) which is recognized by fused colliculi and sometimes associated with evidence of aqueductal obstruction. Absent septum pellucidum, dysplastic corpus callosum, and absent olfactory bulbs are also frequently noted. Posterior holoprosencephaly with fusion of the occipital lobes has been observed, though this feature is quite rare (2/115 patients in our cohort). The presence of the following features on pre- or postnatal imaging should prompt close scrutiny for RES: aqueductal stenosis, Chiari I malformation, substantial central cerebellar white matter on sagittal view, rounded fastigial point, absent posterior cerebellar notch (incisura), absent septum, and/or ventriculomegaly with a small cerebellum. A large fetal autopsy series confirmed all of these imaging findings, identified frequent Purkinje cell heterotopia, and found that the aqueductal obstruction was commonly due to atresia or forking, presumably a developmental rather than acquired abnormality (Pasquier, et al. 2009).
FIGURE 1.
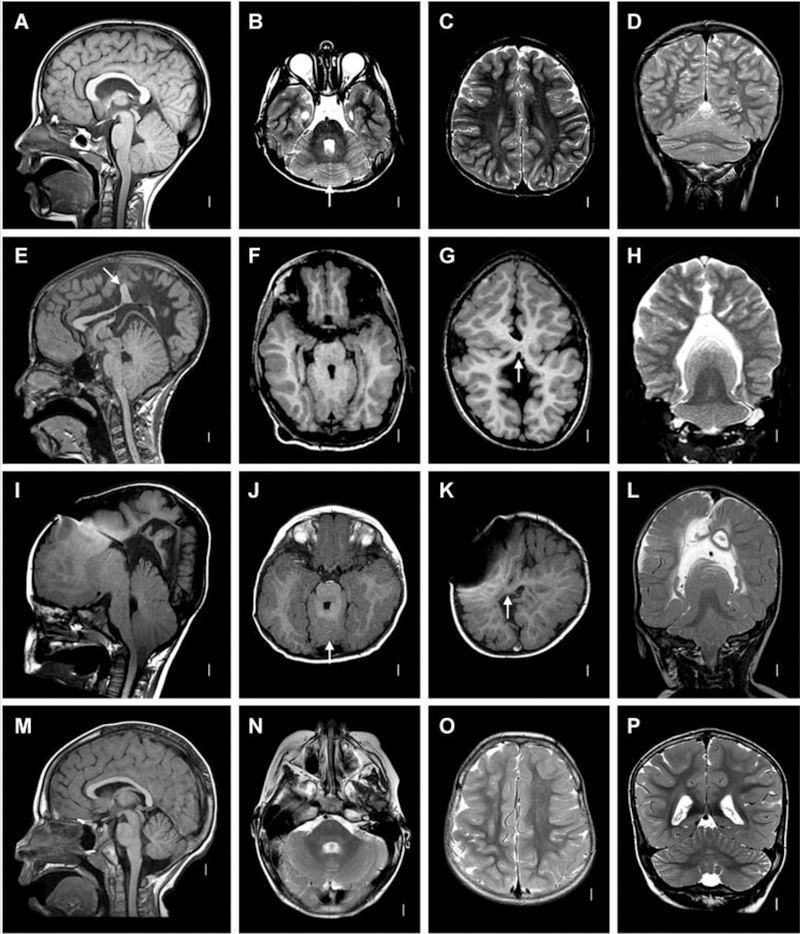
Postnatal radiographic features of RES. A–D: Patient 48. Midline sagittal T1-weighted image through the cerebellum demonstrates hemispheric rather than vermian configuration (A). Axial T2 through the cerebellum shows fusion of white matter across the midline and a keyhole-shaped 4th ventricle (B). Axial T2 through though the cerebral hemispheres shows normal anatomy (C). Coronal T2 showing continuity of cerebellar folia across the midline without an intervening vermis (D). E–H: Patient 46. Midline sagittal T1-weighted image demonstrating a towering cerebellum with hemispheric architecture. Note the absence of a visible aqueduct. This patient had severe congenital hydrocephalus. Many of the supratentorial abnormalities (arrow) are likely a consequence of distortion from hydrocephalus and subsequent decompression (E). Axial T1 through the cerebellum demonstrating similar findings as the patient above (F). Axial T1 through the cerebral hemispheres shows an area of white matter continuity suspicious for mild HPE, but likely representing post-hydrocephalus distortion (G). Coronal T2 showing a towering cerebellum with upward displacement through the tentorial notch (H). I–L: Patient 14. I, Mid-sagittal and axial T1 demonstrating similar findings to Patient 46. This patient also had severe congenital hydrocephalus (J). Axial T1 showing area suspicious for HPE (arrow), though post-hydrocephalic distortion makes this difficult to confirm (K). Coronal T2 demonstrating similar findings to Patient 46 (L). M–P: normal brain. Reproduced from Tully, et al. (2012) with permission.
Several reports have described RES in combination with spina bifida and Chiari II malformation (Hasan, Shopra, Purohit, & Sharma, 2017; Sener & Dzelzite, 2003)); however, based on our careful review of the images in these papers and comparison to our cohort of patients with RES and many patients with Chiari II malformation at our institution, we believe that the appearance is due to compression of the cerebellum rather than actual fusion of the hemispheres, and that RES and Chiari II are unrelated malformations. That said, 5/115 (4%) individuals in our cohort have been diagnosed with tethered cord in the absence of spina bifida, and we consider this to be a true association.
1.4. Clinical features
Prenatally, RES is diagnosed by the imaging findings described above, and should be considered in the context of aqueductal stenosis or ventriculomegaly with a small cerebellum (Figure 2). Postnatally, patients present with hypotonia and motor developmental delays that lead to neuroimaging. “Figure-of-eight” head shaking is particularly suggestive of RES (Accogli & Srour, 2018; Tully et al., 2013), and other neurological features include poor balance, ataxia, and abnormal eye movements. Trigeminal anesthesia is seen in a small subset of patients. The full range of cognitive outcome is observed, from profound intellectual disability to normal or even exceptional intelligence (IQ ≥130). Attention deficit hyperactivity symptoms are common. Although more severe neuroimaging findings and additional congenital malformations are associated with higher risk for more severely abnormal cognitive outcome, no features are strongly predictive in a given patient.
FIGURE 2.
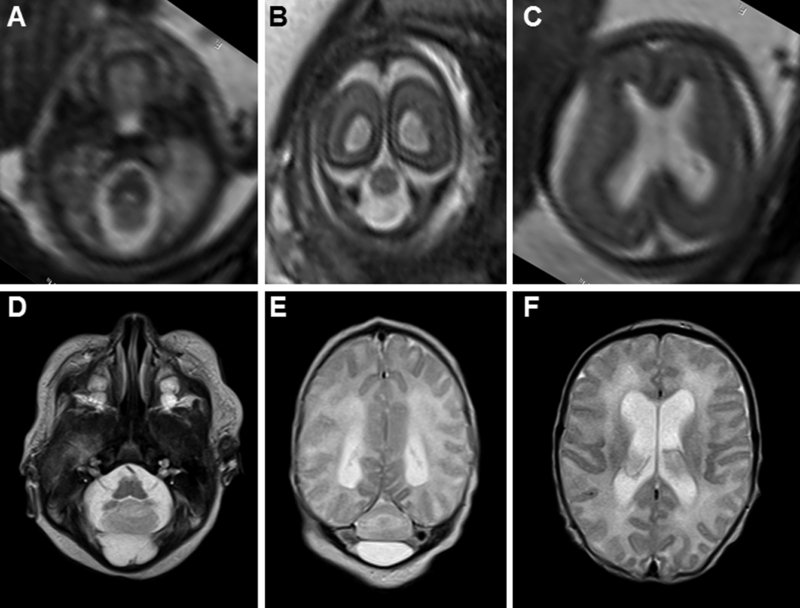
Pre- and post-natal brain MRI features of RES. (A–C) Fetal MRI at 21 and 4/7 weeks gestational age demonstrates severe cerebellar hypoplasia (A and B) and mild ventriculomegaly (C). (D-F) Neonatal MRI of the same patient demonstrates severe cerebellar hypoplasia (D and E) and mild ventriculomegaly (F) in the same patient.
Various non-neurological features have been described in patients with RES (Table I). The majority of patients have parietal scalp alopecia associated with the GLH diagnosis. Many patients have atypical craniofacial features including hypertelorism, flat midface (or midface retrusion), prominent forehead and low-set, posteriorly rotated ears (Figure 3). In our cohort, 4 patients have unilateral craniofacial microsomia (Rush et al., 2013; Pasquier et al. 2009). Some patients have features of VACTERL association (vertebral anomalies, anal atresia, cardiac malformations, tracheoesophageal fistula, renal anomalies, radial dysplasia, or other limb defects), although only a few meet criteria for a VACTERL diagnosis. In 24% of patients, RES is diagnosed as an isolated brain malformation. Overall, our analysis of 115 individuals with RES demonstrates a continuous series of associated congenital anomalies that do not clearly separate into distinct syndromes.
Table I.
Clinical presentations associated with RES
|
total N |
male |
female |
GLH features | VACTERL features | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| alopeci a |
trigeminal anesthesia |
vertebra l |
rib | cardiac | GU/G I |
limb | renal | ||||
|
RES w/o alopecia or VACTERL features† |
28 | 16 | 12 | — | — | — | — | — | — | — | — |
| RES with alopecia | 44 | 27 | 17 | 44 | — | — | — | — | — | — | — |
|
RES with alopecia and trigeminal anesthesia‡ |
14 | 8 | 6 | 14 | 14 | — | — | — | — | — | — |
|
RES with VACTERL features§ +/− alopecia |
25 | 15 | 10 | 11 | 4 | 11 | 9 | 16 | 4 | 7 | 7 |
|
RES + HPE + VACTERL +/− alopecia |
2 | 1 | 1 | 1 | — | 1 | 1 | 1 | — | 1 | — |
|
unk (not enough info to diagnose) |
2 | — | 2 | — | — | — | — | — | — | — | — |
| Total N | 115 | 67 | 48 | 70 | 18 | 12 | 10 | 17 | 4 | 8 | 7 |
| Percentage | 58% | 42% | 61% | 16% | 10% | 9% | 15% | 3% | 7% | 6% | |
Legend:
2 males have craniofacial microsomia;
1 female has craniofacial microsomia;
each individual may have more than one feature;
GLH, Gomez-Lopez-Hernandez syndrome; HPE, holoprosencephaly; RES, rhombencephalosynapsis; VACTERL, vertebral defects, anal atresia, cardiac defects, trachea-esophageal fistula, renal anomalies, and limb abnormalities.
FIGURE 3.

Faces of patients with RES showing typical facial features including hypertelorism (A, G, I K, L), prominent forehead (all), flat midface/midface retrusion (all except D), alopecia (B, D, L, P), turricephaly (N and P), and ear abnormalities (low, position (B, J, P), posterior rotation (B, D, L, P), and microtia (N)). Adapted from Tully, et al. (2012) with permission.
2. EVIDENCE FOR GENETIC BASIS
Multiple observations are consistent with a genetic basis for RES (Table II). Most prominently, RES is similar to other highly stereotyped brain malformation conditions with known genetic causes, e.g. holoprosencephaly (Solomon, Gropman, & Muenke, 1993), Joubert syndrome (Parisi & Glass, 1993), megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome (Mirzaa, 1993). Importantly, the brain imaging features of RES are consistent and distinctive. In addition, RES is consistently associated with additional congenital malformations as described above. RES is not associated with prematurity or other prenatal events, and the pattern of associated anomalies is consistent with defects that arise early in embryogenesis.
Table II.
Evidence for and against a genetic basis for rhombencephalosynapsis
| Evidence | Genetic Feature |
Observation | Frequency | References |
|---|---|---|---|---|
| For | Neuroimaging | Stereotyped morphological cerebellar malformation | Required for diagnosis | Ishak (2012) |
| For | Chromosomal abnormalities | del 2q | Rare | Truwit (1991) |
| der(10)t(2;10)(p25.3;q26.3)(Tel 2p+, Tel 10q-) | Rare | Demurger (2013), Lespinasse (2004) | ||
| del 14q12-q21.2 | Rare | Demurger (2013) | ||
| del 16p11.2 | Rare | Demurger (2013) | ||
| del 16p13.11 | Rare | Demurger (2013) | ||
| Rearrangement of 22q13.3 and 10q26.3 (dup and del of 22q)† | Rare | Ramocki (2011) | ||
| Tetrasomy 9p | Rare | di Vera (2008) | ||
| Microduplication 1p | Rare | Pasquier (2009) | ||
| Microduplication 7p | Rare | Pasquier (2009) | ||
| For | Postzygotic Mutation | Discordant monozygotic twins Asymmetric craniofacial features |
Present | This report |
| Against | Recurrence | Affected siblings | Rare or absent | Pasquier (2009), Ramocki (2011)† |
| Against | Consanguinity | Affected siblings | None | Chemli (2007), de Mattos (2014), Gomy (2008), Pasquier (2009), Romanengo (1997), Toelle (2002) |
| Against | Vertical Transmission | Affected children | None | This report |
The authors also reported partial RES in conjunction with HPE in two half-sisters with a heterozygous intragenic deletion of ZIC2. However, the presence of RES has been disputed by Guleria (2011) and Tully (2012).
2.1. Family studies
Possible recurrence has been reported in only two families. Among 40 fetuses with RES examined through neonatal ultrasound, neuroimaging, and neuropathology, one pair of sibling fetuses was reported (Pasquier et al., 2009). In another family, two half-sisters were diagnosed with partial RES and holoprosencephaly, though the RES diagnosis for these sisters has been disputed in the literature (Guleria, 2011; Ramocki et al., 2011; Tully et al., 2012). RES has also been reported in at least four consanguineous families, though no recurrences were reported despite 10 unaffected siblings in one family (Chemli, Abroug, Tlili, & Harbi, 2007; de Mattos et al., 2014; Gomy et al., 2008; Pasquier et al., 2009; Romanengo, Tortori-Donati, & Di Rocco, 1997; Sandalcioglu et al., 2006; Toelle et al., 2002).
2.2. Prenatal risk factors
No evidence for extrinsic factors contributing to RES has been reported in the literature. In addition, we have interviewed mothers of children with RES in our research cohort and have not identified any obvious pregnancy exposures (medications, high fever, illicit drug or alcohol use, severe illness, trauma), though several mothers reported pre-existing diabetes or use of assisted reproduction.
2.3. Published genetic studies
Several studies have reported chromosomal rearrangements in patients with RES (Table II), though no recurrent copy number variants have emerged (Demurger et al., 2013; di Vera et al., 2008; Lespinasse et al., 2004; Pasquier et al., 2009; Ramocki et al., 2011; Truwit, Barkovich, Shanahan, & Maroldo, 1991). Partial RES was reported in one of 5 individuals with a de novo mutation in the CHAMP1 gene, but limited neuroimaging data were reported to substantiate this diagnosis (Hempel et al., 2015) and mutations in this gene have not been identified in other individuals with RES.
3. THEORIES REGARDING GENETIC MECHANISM OR CAUSE
3.1. Dominant inheritance
Most patients with RES are sporadic, suggesting de novo autosomal dominant mutations (germline or mosaic) are the likely cause (Chemli et al., 2007; Romanengo et al., 1997; Sandalcioglu et al., 2006; Toelle et al., 2002). Consistent with this genetic mechanism, several chromosomal abnormalities have been identified in children with RES. That said, no vertical transmission has been reported, and our research cohort includes 6 individuals with RES who have had 11 children without producing a child affected by RES. We have not observed an obviously elevated miscarriage rate, nor decreased fertility in our research cohort, although our numbers are small. Tissue specific mosaicism excluding the germline could also explain the lack of familial recurrence or vertical transmission observed for RES.
3.2. Recessive inheritance
The lack of recurrences, particularly in consanguineous families, contrasts starkly with typical recessive disorders for which consanguineous and non-consanguineous families with multiple affected offspring are frequently described.
3.3. Discordant monozygotic twin and implications for cause
Our research cohort includes two pairs of monozygotic (MZ) twins discordant for RES. Discordant MZ twins have been reported for multiple developmental disorders and result from three types of mechanisms. First, and most commonly, discordant MZ twins have been associated with postzygotic (mosaic) mutations for disorders in which the gene is known and mosaicism proven, including other neurocutaneous syndromes (Lederer, Rack, Boulanger, Battisti, & Verellen-Dumoulin, 2012; Vogt et al., 2011; Zwijnenburg, Meijers-Heijboer, & Boomsma, 2010). Second, discordant MZ twins have been reported with 11p14 imprinting disorders, Beckwith-Wiedemann and Russell-Silver syndromes (Tierling et al., 2011; Yamazawa, Kagami, Fukami, Matsubara, & Ogata, 2008). Finally, both MZ and dizygotic twins have increased risk for structural defects associated with presumed prenatal vascular perfusion deficiency (Hoyme, Higginbottom, & Jones, 1981).
3.4. Genomic investigations to date
To identify the genetic cause(s) of RES, we have performed extensive analyses on our cohort of 115 individuals with RES (Table III). Primarily, we have used exome sequencing (N=59 probands, including 29 parent-proband trios), since this method has successfully identified the genetic bases for >4,000 Mendelian phenotypes (Chong et al., 2015). At least 41 individuals with RES were specifically screened for copy number variation. We also performed whole genome sequencing in one discordant MZ twin pair and in one parent-fetus trio using DNA isolated from midline cerebellum of the fetus. In all experiments, we looked for rare (<0.01 minor allele frequency in population databases), predicted-deleterious variants using de novo dominant and recessive modes of inheritance. Surprisingly, our analyses have not identified a single gene with variants meeting these criteria in more than one family.
Table III.
Summary of genomic investigations
| Method | Study Design | DNA Source | N |
|---|---|---|---|
| Chromosome array | Proband | Blood | 41 |
| Exome | Proband | Blood or Saliva | 28 |
| Exome | Trio | Blood or Saliva | 29 |
| Exome and Genome | Trio | Cerebellum | 1 |
| Exome and Genome | Discordant MZ Twin Pair and parents | Blood | 1 |
N, number of families/probands/affected individuals; MZ, monozygotic
3.5. Hypotheses about mechanism
Our working hypothesis is that RES is a Mendelian disorder. If this hypothesis is correct, a number of possibilities may explain why extensive genomic investigations have failed to identify the genetic bases for RES (Table IV). Most studies utilize DNA isolated from blood or saliva. Thus, a postzygotic mutation that is absent from these tissues is undetectable in our cohort. Since exome sequencing only interrogates the 1% of the genome that encodes proteins, DNA variants that are outside of the coding region, or that are not readily detectable using this assay (e.g. structural variants, repeat expansions) remain unexplored in RES. Larger cohorts than are currently available will be required to address genetic heterogeneity, complex inheritance, and gene-environment interactions. Transcript-level changes that either do not impact DNA sequence or are incorrectly predicted to be non-deleterious have not been investigated. Though no hypotheses regarding a non-genetic etiology for RES have been proposed, this relationship remains incompletely explored.
Table IV.
Alternative hypotheses not addressed by current genomic investigations
| Feature | Rationale for failure |
|---|---|
| Postzygotic mutation | Alternate allele present at low levels or absent from blood and/or saliva |
| Variants not detected by exome sequencing | Non-coding DNA variants Complex structural rearrangements |
| Genetic heterogeneity | Small sample size |
| Incomplete penetrance, variable expressivity, and non-Mendelian inheritance | Small sample size |
| Epigenetic causes | Not due to DNA variants |
In the absence of known genetic causes, several possible mechanisms for RES have been proposed based on the brain malformation and associated clinical features. For instance, one popular hypothesis is that RES results from a defect in dorsal-ventral axis formation (Sarnat, 2000; Yachnis, 2002) similar to some forms of holoprosencephaly. In contrast, RES could be due to other abnormalities in specification of cerebellar vermis cell fates. Given the phenotypic overlap with VACTERL, the two disorders may share common mechanisms; however, little more is known about the pathophysiology of VACTERL. One intriguing paradigm for birth defects caused by gene-environment interactions is vertebral defects resulting partial loss of Notch pathway function combined with in utero hypoxia (Sparrow et al., 2012), particularly because RES is associated with vertebral defects.
4. FUTURE DIRECTIONS
Ongoing genomic investigations will incorporate strategies to address the limitations of current genetic approaches. Postzygotic mutations could be identified by deep sequencing of accessible affected tissue, such as skin from areas of alopecia. Whole genome sequencing, including long-read sequencing, could be leveraged to detect structural rearrangements and non-coding variation. Larger collaborations could be established to recruit additional individuals and to share existing data to identify potential shared genetic causes. Incomplete penetrance and non-Mendelian inheritance mechanisms could be addressed by assessing enrichment for risk alleles that may be more common in population databases than anticipated for rare conditions. Altered gene regulation could be probed through RNA sequencing of affected or unaffected tissues to examine both transcript level nucleotide changes and gene expression differences. Additional epigenetic approaches could also uncover changes not associated with nucleotide variation. Finally, larger RES cohorts with rich prenatal history data are required to evaluate possible environmental exposures that could underlie RES etiology.
Brief bio:
Dr. Aldinger is a developmental geneticist and neurobiologist investigating the molecular and cellular diversity underlying typical and atypical brain development in order to improve the health and well-being of children.
Ms. Dempsey is a research coordinator with a master of public health degree in epidemiology. She collects, maintains, and analyzes clinical phenotype data and enjoys interacting with families participating in the University of Washington hindbrain research program.
Dr. Tully is a pediatric neurologist at Seattle Children’s Hospital with a clinical focus on brain malformations. Her research program investigates how differences in brain structure correlate with outcome.
Ms. Grout worked in Dr. Dan Doherty’s lab as a research scientist for three years after graduating from the University of Washington. She is currently pursuing a Master’s in Bioinformatics at Oregon Health and Science University.
Ms. Mehaffey is a bioinformaticist and molecular geneticist focused on testing and implementing innovative approaches to genomic data analysis. Presently, she is using her experience to find novel genetic causes for epileptic encephalopathies and congenital hindbrain malformations.
Dr. Dobyns is a physician-scientist who studies the nature and causes of human developmental disorders. His work includes studies of human brain malformations, early childhood epilepsy, intellectual disability and autism, and most recently to developmental vascular disorders including vascular malformations and prenatal or childhood strokes. He is trained and Board Certified in both Medical Genetics and Child Neurology, and leads a research group on developmental disorders at SCRI.
Dr. Doherty is a Developmental-Behavioral Pediatrician committed to improving the lives of children with neurodevelopmental disabilities and their families. He runs a neurogenetics laboratory focused on defining human hindbrain malformation conditions, identifying their causes, understanding their mechanisms, and developing better treatments.
REFERENCES
- Accogli A, & Srour M (2018). Teaching Video NeuroImages: Figure 8 head-shaking stereotypy in rhombencephalosynapsis. Neurology, 90(20), e1832–e1833. doi: 10.1212/WNL.0000000000005531 [DOI] [PubMed] [Google Scholar]
- Barth PG (2008). Rhombencephalosynapsis. Handb Clin Neurol, 87, 53–65. doi: 10.1016/S0072-9752(07)87004-7 [DOI] [PubMed] [Google Scholar]
- Chemli J, Abroug M, Tlili K, & Harbi A (2007). Rhombencephalosynapsis diagnosed in childhood: clinical and MRI findings. Eur J Paediatr Neurol, 11(1), 35–38. doi: 10.1016/j.ejpn.2006.09.007 [DOI] [PubMed] [Google Scholar]
- Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, . . . Bamshad MJ (2015). The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am J Hum Genet, 97(2), 199–215. doi: 10.1016/j.ajhg.2015.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mattos VF, Graziadio C, Machado Rosa RF, Lenhardt R, Alves RP, Trevisan P, . . . Zen PR (2014). Gomez-Lopez-Hernandez syndrome in a child born to consanguineous parents: new evidence for an autosomal-recessive pattern of inheritance? Pediatr Neurol, 50(6), 612–615. doi: 10.1016/j.pediatrneurol.2014.01.035 [DOI] [PubMed] [Google Scholar]
- Demurger F, Pasquier L, Dubourg C, Dupe V, Gicquel I, Evain C, . . . David V (2013). Array-CGH Analysis Suggests Genetic Heterogeneity in Rhombencephalosynapsis. Mol Syndromol, 4(6), 267–272. doi: 10.1159/000353878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Vera E, Liberati M, Celentano C, Calabrese G, Guanciali-Franchi PE, Morizio E, & Rotmensch S (2008). Rhombencephalosynapsis in a severely polymalformed fetus with non-mosaic tetrasomy 9p, in intracytoplasmic-sperm-injection pregnancy. J Assist Reprod Genet, 25(11–12), 577–580. doi: 10.1007/s10815-008-9257-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez MR (1979). Cerebellotrigeminal and focal dermal dysplasia: a newly recognized neurocutaneous syndrome. Brain Dev, 1(4), 253–256. [DOI] [PubMed] [Google Scholar]
- Gomy I, Heck B, Santos AC, Figueiredo MS, Martinelli CE Jr., Nogueira MP, & Pina-Neto JM (2008). Two new Brazilian patients with Gomez-Lopez-Hernandez syndrome: reviewing the expanded phenotype with molecular insights. Am J Med Genet A, 146A(5), 649–657. doi: 10.1002/ajmg.a.32173 [DOI] [PubMed] [Google Scholar]
- Guleria S (2011). ZIC2 mutations are seen in holoprosencephaly and not partial rhombencephalosynapsis. Am J Med Genet A, 155A(11), 2901; author reply 2902. doi: 10.1002/ajmg.a.34282 [DOI] [PubMed] [Google Scholar]
- Hasan A, Shopra S, Purohit DK, & Sharma S (2017). Rhombencephalosynapsis and Chiari II Malformation with Spinal Deformaties. The Journal of Spinal Surgery, 4(3), 123–125. [Google Scholar]
- Hempel M, Cremer K, Ockeloen CW, Lichtenbelt KD, Herkert JC, Denecke J, . . . Lessel D (2015). De Novo Mutations in CHAMP1 Cause Intellectual Disability with Severe Speech Impairment. Am J Hum Genet, 97(3), 493–500. doi: 10.1016/j.ajhg.2015.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyme HE, Higginbottom MC, & Jones KL (1981). Vascular etiology of disruptive structural defects in monozygotic twins. Pediatrics, 67(2), 288–291. [PubMed] [Google Scholar]
- Ishak GE, Dempsey JC, Shaw DW, Tully H, Adam MP, Sanchez-Lara PA, . . . Doherty D (2012). Rhombencephalosynapsis: a hindbrain malformation associated with incomplete separation of midbrain and forebrain, hydrocephalus and a broad spectrum of severity. Brain, 135(Pt 5), 1370–1386. doi: 10.1093/brain/aws065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederer D, Rack K, Boulanger S, Battisti O, & Verellen-Dumoulin C (2012). Discordant monozygotic twins for macrocephaly-capillary malformation. Am J Med Genet A, 158A(6), 1509–1511. doi: 10.1002/ajmg.a.35382 [DOI] [PubMed] [Google Scholar]
- Lespinasse J, Testard H, Nugues F, Till M, Cordier MP, Althuser M, . . . Jouk PS (2004). A submicroscopic unbalanced subtelomeric translocation t(2p;10q) identified by fluorescence in situ hybridization: fetus with increased nuchal translucency and normal standard karyotype with later growth and developmental delay, rhombencephalosynapsis (RES). Ann Genet, 47(4), 405–417. doi: 10.1016/j.anngen.2004.07.005 [DOI] [PubMed] [Google Scholar]
- Lopez-Hernandez A (1982). Craniosynostosis, ataxia, trigeminal anaesthesia and parietal alopecia with pons-vermis fusion anomaly (atresia of the fourth ventricle). Report of two cases. Neuropediatrics, 13(2), 99–102. [DOI] [PubMed] [Google Scholar]
- Mirzaa G (1993). MPPH Syndrome; In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, & Amemiya A (Eds.), GeneReviews((R)). Seattle (WA). [PubMed] [Google Scholar]
- Obersteiner H (1914). Ein Kleinhirn ohne Wurm. Arb Neurol Inst (Wien), 21, 124–136. [Google Scholar]
- Parisi M, & Glass I (1993). Joubert Syndrome; In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, & Amemiya A (Eds.), GeneReviews((R)). Seattle (WA). [PubMed] [Google Scholar]
- Pasquier L, Marcorelles P, Loget P, Pelluard F, Carles D, Perez MJ, . . . Laquerriere A (2009). Rhombencephalosynapsis and related anomalies: a neuropathological study of 40 fetal cases. Acta Neuropathol, 117(2), 185–200. doi: 10.1007/s00401-008-0469-9 [DOI] [PubMed] [Google Scholar]
- Ramocki MB, Scaglia F, Stankiewicz P, Belmont JW, Jones JY, & Clark GD (2011). Recurrent partial rhombencephalosynapsis and holoprosencephaly in siblings with a mutation of ZIC2. Am J Med Genet A, 155A(7), 1574–1580. doi: 10.1002/ajmg.a.34029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanengo M, Tortori-Donati P, & Di Rocco M (1997). Rhombencephalosynapsis with facial anomalies and probable autosomal recessive inheritance: a case report. Clin Genet, 52(3), 184–186. [DOI] [PubMed] [Google Scholar]
- Rush ET, Adam MP, Clark RD, Curry C, Hartmann JE, Dobyns WB, & Olney AH (2013). Four new patients with Gomez-Lopez-Hernandez syndrome and proposed diagnostic criteria. Am J Med Genet A, 161A(2), 320–326. doi: 10.1002/ajmg.a.35817 [DOI] [PubMed] [Google Scholar]
- Sandalcioglu IE, Gasser T, van de Nes JA, Menken U, Stolke D, & Wiedemayer H (2006). Fusion of the cerebellar hemispheres ventral to the brainstem: a rare hindbrain-related malformation. Childs Nerv Syst, 22(1), 73–77. doi: 10.1007/s00381-004-1065-5 [DOI] [PubMed] [Google Scholar]
- Sarnat HB (2000). Molecular genetic classification of central nervous system malformations. J Child Neurol, 15(10), 675–687. doi: 10.1177/088307380001501007 [DOI] [PubMed] [Google Scholar]
- Sener RN (2000). Unusual MRI findings in rhombencephalosynapsis. Comput Med Imaging Graph, 24(4), 277–282. [DOI] [PubMed] [Google Scholar]
- Sener RN, & Dzelzite S (2003). Rhombencephalosynapsis and a Chiari II malformation. J Comput Assist Tomogr, 27(2), 257–259. [DOI] [PubMed] [Google Scholar]
- Solomon BD, Gropman A, & Muenke M (1993). Holoprosencephaly Overview; In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, & Amemiya A (Eds.), GeneReviews((R)). Seattle (WA). [PubMed] [Google Scholar]
- Sparrow DB, Chapman G, Smith AJ, Mattar MZ, Major JA, O’Reilly VC, . . . Dunwoodie SL (2012). A mechanism for gene-environment interaction in the etiology of congenital scoliosis. Cell, 149(2), 295–306. doi: 10.1016/j.cell.2012.02.054 [DOI] [PubMed] [Google Scholar]
- Sukhudyan B, Jaladyan V, Melikyan G, Schlump JU, Boltshauser E, & Poretti A (2010). Gomez-Lopez-Hernandez syndrome: reappraisal of the diagnostic criteria. Eur J Pediatr, 169(12), 1523–1528. doi: 10.1007/s00431-010-1259-7 [DOI] [PubMed] [Google Scholar]
- Tierling S, Souren NY, Reither S, Zang KD, Meng-Hentschel J, Leitner D, . . . Walter J. (2011). DNA methylation studies on imprinted loci in a male monozygotic twin pair discordant for Beckwith-Wiedemann syndrome. Clin Genet, 79(6), 546–553. doi: 10.1111/j.1399-0004.2010.01482.x [DOI] [PubMed] [Google Scholar]
- Toelle SP, Yalcinkaya C, Kocer N, Deonna T, Overweg-Plandsoen WC, Bast T, . . . Boltshauser E (2002). Rhombencephalosynapsis: clinical findings and neuroimaging in 9 children. Neuropediatrics, 33(4), 209–214. doi: 10.1055/s-2002-34498 [DOI] [PubMed] [Google Scholar]
- Truwit CL, Barkovich AJ, Shanahan R, & Maroldo TV (1991). MR imaging of rhombencephalosynapsis: report of three cases and review of the literature. AJNR Am J Neuroradiol, 12(5), 957–965. [PMC free article] [PubMed] [Google Scholar]
- Tully HM, Dempsey JC, Ishak GE, Adam MP, Curry CJ, Sanchez-Lara P, . . . Dobyns WB(2012). Beyond Gomez-Lopez-Hernandez syndrome: recurring phenotypic themes in rhombencephalosynapsis. Am J Med Genet A, 158A(10), 2393–2406. doi: 10.1002/ajmg.a.35561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully HM, Dempsey JC, Ishak GE, Adam MP, Mink JW, Dobyns WB, . . . Doherty D(2013). Persistent figure-eight and side-to-side head shaking is a marker for rhombencephalosynapsis. Mov Disord, 28(14), 2019–2023. doi: 10.1002/mds.25634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt J, Kohlhase J, Morlot S, Kluwe L, Mautner VF, Cooper DN, & Kehrer-Sawatzki H (2011). Monozygotic twins discordant for neurofibromatosis type 1 due to a postzygotic NF1 gene mutation. Hum Mutat, 32(6), E2134–2147. doi: 10.1002/humu.21476 [DOI] [PubMed] [Google Scholar]
- Yachnis AT (2002). Rhombencephalosynapsis with massive hydrocephalus: case report and pathogenetic considerations. Acta Neuropathol, 103(3), 301–304. doi: 10.1007/s004010100454 [DOI] [PubMed] [Google Scholar]
- Yamazawa K, Kagami M, Fukami M, Matsubara K, & Ogata T (2008). Monozygotic female twins discordant for Silver-Russell syndrome and hypomethylation of the H19-DMR. J Hum Genet, 53(10), 950–955. doi: 10.1007/s10038-008-0329-4 [DOI] [PubMed] [Google Scholar]
- Zwijnenburg PJ, Meijers-Heijboer H, & Boomsma DI (2010). Identical but not the same: the value of discordant monozygotic twins in genetic research. Am J Med Genet B Neuropsychiatr Genet, 153B(6), 1134–1149. doi: 10.1002/ajmg.b.31091 [DOI] [PubMed] [Google Scholar]