

Abstract
Introduction:
Commonly used scoring systems rely on blood counts, histological and cytological examination of bone marrow and peripheral blood, and cytogenetic assessments to estimate prognosis of patients with myelodysplastic syndromes (MDS) and guide therapy decisions. Next generation sequencing (NGS) has identified recurrent genetic abnormalities in up to 90% of patients with MDS and may provide important information regarding the pathogenesis of the disease, diagnostic and prognostic evaluation, and therapy selection.
Areas covered:
Herein, the authors review the role of NGS in diagnosis, treatment, and prognosis of MDS at various disease stages, and discuss advantages and caveats of incorporating molecular genetics in routine management of MDS. While a vast majority of patients harbor recurrent mutations implicated in MDS pathogenesis, similar mutations can be detected in otherwise healthy individuals with other hematologic malignancies. Besides establishing a diagnosis, NGS may be used to monitor minimal residual disease following treatment.
Expert opinion:
As more targeted therapies become available, assessment of genetic mutations will become central to individualized therapy selection and may improve diagnostic accuracy and further guide management for each patient. However, multiple challenges remain before NGS can be incorporated into routine clinical practice.
Keywords: genetics, myelodysplastic syndrome, MDS, mutations, next generation sequencing, NGS
1. Introduction
Myelodysplastic syndromes (MDS) are a spectrum of clonal hematopoietic stem cell neoplasms characterized by morphologic dysplasia, ineffective hematopoiesis, cytopenias, recurrent genetic mutations and cytogenetic abnormalities, and an increased risk of progression to acute myeloid leukemia (AML) (1).The recognition of the clinical, morphologic, immunophenotypic, and cytogenetic heterogeneity of MDS has led to the development of complex diagnostic criteria and diverse risk stratification models such as the 1997 International Prognostic Scoring System (IPSS), the updated Revised IPSS (IPSS-R), the dynamic WHO-based Prognostic Scoring System (WPSS) and the MD Anderson Cancer Center Score (MDAS) to estimate survival and guide the approach to treatment (2–5).
While the 2016 WHO classification of MDS has recognized a number of recurrent chromosomal abnormalities, their role remained largely limited to the risk stratification of MDS subtypes in respect to survival and incidence of progression to AML (10, 11). Deletion of the long arm of chromosome 5 is a notable exception and is used to define a specific entity, MDS with isolated del(5q) (12–14). The 2016 WHO classification also recognized the importance of recurrent gene mutations found in patients with MDS, but only required the assessment of SF3B1 to define a subtype of MDS (refractory anemia with ring sideroblasts which is defined as dysplasia, <5% bone marrow blasts, and ≥5% ringed sideroblasts in the presence or ≥15% ringed sideroblasts in the absence of SF3B1 mutations, respectively) (15). Some somatic mutations have been shown to be associated with specific morphological features and overall prognosis. For example, mutations in ASXL1, RUNX1, TP53, and SRSF2 were associated with severe dysplasia of granulocytes while mutations in RUNX1, TP53, and NRAS are associated with severe thrombocytopenia(16, 17). Similarly, mutations in TP53, EZH2, ETV6, RUNX1, and ASXL1 have been associated with poorer outcomes(18). Some mutations have been linked to augmented treatment response; for example mutations in TET2 and DNMT3A predicted a better response to hypomethylating agents in patients with MDS (19–21). Interestingly, the predictive value of DNMT3A mutations for response to HMAs in AML patients is controversial and seems to vary in the frontline setting compared with relapsed/refractory patients which might be due to the genetic heterogeneity and the acquisition of additional mutations during the disease course (22, 23).
Overall, more than 50 genes with recurrent somatic mutations have been identified, with somatic mutations in at least one gene found in up to 90% patients (18, 24, 25). These mutations affect various key functions of DNA biology ranging from epigenetic processes such as DNA methylation and histone modifications, to DNA repair, and mRNA splicing (25–27). Table 1 provides an overview of these mutations. However, the role of genetics in classification, diagnosis, prognostication, and treatment of MDS remains controversial (26, 28, 29).
Table 1.
Genes tested by current next generation sequencing panels
| Epigenetic regulators | IDH1, IDH2, EZH2, ASXL1, DNMT3A, TET2, SUZ12, KDM6A/UTX |
| Spliceosomal genes | SF3B1, U2AF1, SRSF2, ZRSR2 |
| Cytoplasmic tyrosine kinases | JAK1, JAK2, JAK3, ABL1 |
| Receptor tyrosine kinases | FLT3, KIT |
| Signaling molecules | CBL, CBLB, NRAS, KRAS, HRAS |
| Cytokine receptors | MPL, IL7R, CSF3R |
| Transcriptional factors | GATA1, GATA2, CEBPA, ETV6, RUNX1, STAT3, PAX5, IKZF1 |
| Tumor suppressors | TP53, WT1 |
| Phosphatase | PTPN11 |
| Other | FBXW7, CREBBP, NOTCH1, BCOR, NPM1 |
With proliferation of high throughput gene sequencing methods, gene mutation profiling is becoming increasingly employed to provide insights on pathogenesis and heterogeneity of MDS, aid with diagnosis, enhance prognostication and risk stratification tools, and steer clinical management and development of new drugs targeting specific mutations (26, 29, 30). In this review, we discuss advantages and challenges of incorporating next generation sequencing (NGS) in the routine management of MDS.
2. Next generation sequencing (NGS)
Next generation sequencing (NGS), a high throughput system used to sequence millions of DNA fragments simultaneously, has enabled us to analyze genetic information ranging from individual genes to coding exomes and entire genomes at a reduced cost. These DNA fragments are compared to those in a reference genome to help identify genetic variability and detect mutations (31–34). NGS can detect a wide range of mutations and DNA variations such as substitutions, insertions, deletions, inversions, translocations, and even mosaic variations which may be present in only a small percentage of cells. With the help of NGS, somatic mutations relevant to solid and hematologic cancer development have been identified and validated (33). NGS is proving to be a powerful tool that may be applied to improve diagnosis, risk stratification, prognosis, and therapeutic decision making in the realm of malignant disease including MDS (29, 33, 35).
The advent of NGS has led to the discovery of more than 50 recurrently mutated genes in 80–90% of MDS patients. Many of these mutations may have prognostic values independent of an identified cytogenetic aberration or an assigned IPSS risk category (36–38). For example, isolated deletion of the long arm of chromosome 20 (del(20q)) is associated with a favorable prognosis according to IPSS-R. However, it is often found in patients with WTI1, SRSF2, U2AF1, and ASXL1 mutations, all of which are associated with less favorable outcomes (36, 39, 40). Even patients with normal karyotypes and cytogenetics may harbor gene mutations that portend unfavorable outcomes. For example, in one study 75% of 196 patients with normal karyotypes had equal frequencies of mutations in DNMT3A and ASXL1, both of which were associated with poor outcomes (39). The high throughput NGS-based testing that is rapidly adopted in clinical practice is usually grouped into hotspot panels, actionable gene panels, disease-focused panels, and all the way to whole exome/genome sequencing (31, 34).
Variant allele frequency (VAF) is a marker of the size of the cell subpopulation in relationship to the entire cell population that carries the mutation compared to the entire cell population. Sensitivities of VAF as low as 0.03% have been described which enables us to detect very small clonal populations (41). However, the clinical relevance of this finding is not well-studied and further standardization with regard to which genes should be included in sequencing panels and which clonal size a clinically meaningful finding.
3. Next generation sequencing in clinical practice in MDS diagnosis
When clinico-pathologic data does not lead to an unequivocal diagnosis, NGS can provide additional differentiating information to improve diagnostic accuracy (29, 42). For instance, it can be particularly challenging to distinguish hypoplastic MDS (hMDS) from aplastic anemia (AA) because of overlapping morphologic features (43–45). The distinction between the two entities is crucially important to inform clinical management as biology, prognosis, treatment (e.g. probability of response to immune suppressive therapy), and the risk of evolution to AML differ (46, 47). Additionally, the presence of somatic mutations (ASXL1, DNMT3A, BCOR, TET2, and MPL) in AA patients was associated with a higher risk of progression to MDS (48). Yoshizato et al performed extensive sequencing of genes associated with myeloid malignancies in 439 patients with AA, detecting somatic mutations and clonal hematopoiesis in 36% of screened patients (45). The most frequently mutated genes were BCOR, BCOR1, PIGA, DNMT3A, and ASXL1, overall accounting for 77% of mutations. Of note, the clonal dynamics were variable during the disease course adding an additional challenge to using them to predict therapeutic response and prognosis (45). Although there is a substantial overlap with patients with hMDS, where mutations in ASXL1 and TET2, RUNX1 and SF3B1 have been identified, these mutations are more prevalent and the VAF tends to be larger (18, 25, 36, 43, 44).
In certain cases, patients present with cytopenias and undergo a thorough work up including bone marrow evaluation with cytogenetic analysis with no apparent diagnosis. This condition is identified as “idiopathic cytopenias of undetermined significance” (ICUS) and is usually observed over time without any dedicated therapy (49). The absence of clonal mutations in these patients has a high negative predictive value for MDS or other myeloid malignancy (26). Patients with ICUS and concurrent mutations in certain driver genes such as TET2, DNMT3A, or ASXL1 have a higher risk of progression to myeloid malignancies, a state called “clonal cytopenias of undetermined significance” (CCUS) (50–53). Malcovati et al demonstrated that patients with CCUS can have a 14-fold higher risk of progressing to MDS or AML compared to unmutated ICUS depending on the number of mutations and the size of the mutant clones (52). It is important to note that not only the mere presence of a mutation but the VAF of a particular mutation is an important predictive marker. If the VAF of a known driver mutation in a patient with clonal hematopoiesis is >2%, this constitutes a diagnosis of clonal hematopoiesis of indeterminate potential (CHIP) which has been linked with an annual progression rate to other hematologic malignancies including AML and MDS of 0.5–1% (26, 53, 54). Therefore, detecting clonal mutations in ICUS via NGS can stratify patients based on the predicted increased risk in the absence of an MDS diagnosis and can influence the clinician to employ more vigilant follow-up. The drivers underlying the progression of CHIP to hematologic malignancies are not fully understood, but it is believed that changes in clonal size and the increased number of mutations, also known as clonal complexity, contribute to this transition (55). It has been shown that mutations in epigenetic modifiers (TET2, ASXL1, DNMT3A) and genes regulating splicing processes (SF3B1, SRSF2) are encountered earlier in MDS development while acquisition of mutations in TP53, NRAS, or GATA2 occur later and have been linked to the progression from CCUS to MDS (38, 52, 56). It remains to be seen if the serial monitoring of VAF and emergence of new mutations can be used to guide setting of follow-up intervals, clinical management decisions and to even initiate treatment before the emergence of overt clinical disease manifestations. The term “indeterminate potential” in the acronym CHIP emphasizes the diagnostic uncertainty associated with this condition. More than 10% of patients older than 70 years meet diagnostic criteria of CHIP (53, 54, 57). CHIP has similar clonal sizes to MDS and MDS/MPN, but with lower mutational burden. The most frequently mutated genes in CHIP are ASXL1, DNMT3A and TET2, which are also encountered early in the pathogenesis of MDS (25, 36). Certain somatic mutations like in SRSF2, SETPB1, CBL and PTNPN11, EZH2, RUNX1, and TP53 are much less common or even absent in CHIP as compared to MDS and MDS/MPN as they confer a worse prognosis and a higher rate of progression to more severe MDS and AML (54).
A recent study analyzed samples from 95 individuals obtained on average 6.3 years before AML diagnosis and found CHIP mutations to be more common in people who developed AML versus healthy controls (73.4% vs. 36.7%). The development of AML was associated with increased clonal size and the number of mutations per individual (58). The authors also found a significant correlation between elevated red cell distribution width and an increased risk of progression to AML, which was previously described in patients with CHIP (58, 59).
4. Role of NGS in risk stratification of MDS patients
A landmark study by Bejar et al analyzed 18 different genes in more than 400 patients with MDS looking for mutations using Sanger sequencing, and assessed their prognostic significance relative to IPSS-based predictions (the study was conducted prior to the introduction of the revised IPSS) (18). This study found that mutations in 5 genes, TP53, ETV6, ASXL1, EZH2, and RUNX1 inferred a worse prognosis independently of an IPSS risk category, and upshifted the risk to the next higher IPSS category (18). A larger follow up study conducted by the International Working Group for MDS Molecular Prognosis (MDS-IWG-PM) sequenced 17 genes from 1996 patients with MDS. This study identified 4 high risk genes, TP53, RUNX1, EZH2, and NRAS, considered to be markers of worse prognosis irrespective of the IPSS-R risk category (60). These findings have been confirmed by other studies (25, 36). By contrast, SF3B1 was present in MDS patients with ring sideroblasts and was associated with both an indolent disease course and more favorable IPSS-R independent outcome (38, 61). Notably, mutations like U2AF1, SRSF2, SF3B1, and ASXL1, which have been associated with an adverse prognosis lost their independent prognostic value in MDS patients with >5% bone marrow blasts. This observation suggests the need for an even more individualized approach to genetic testing in MDS patients (60, 62)
Several models were proposed that incorporated mutational data into existing risk stratification models (39). Xu et al proposed two novel mutation-based risk stratification models, IPSS-M and IPSS-RM, that integrate common MDS mutations into the IPSS and IPSS-R scoring systems, respectively (25, 39, 63). The model’s categories were subdivided into low (no mutations), intermediate-1 (at least one mutation in any gene except genes associated with worse outcomes,) intermediate-2 (2–3 mutations in any genes other than those associated with worse outcomes or 1 poor mutation in genes associated with worse outcomes with 0–2 mutations in any other MDS-related genes), and high (presence of at least 2 mutations in genes associated with worse outcomes). Genes considered to be associated with worse outcomes were TP53, STAG2, DNMT3A, EZH2, RUNX1, ROBO1/2, SRSF2 and WT1.
The addition of the mutation-based scoring system resulted in statistically significant differences in survival curves and AML transformation rate for patients within the same IPSS or IPSS-R categories (39). Nazha et al also proposed a predictive model by incorporating 62 mutated genes into IPSS-R which provides a better prognostic prediction in primary and secondary disease, irrespective of the initial or subsequent MDS therapies (63). The model’s prognostic factors included age, IPSS-R score and somatic mutations in EZH2, SF3B1, and TP53. Furthermore, the model retained its prognostic strength at each point during the disease course, thus allowing one to asses treated MDS patients dynamically, which could provide an advantage in the setting of monitoring for clonal evolution of the disease (63). Haferlach et al also developed a prognostic model that included mutations in 14 genes, age, gender, and IPSS-R parameters, and resulted in four risk groups: low, intermediate, high risk and very high risk with 3-year survival rates of 95.2%, 69.3%,32.8% and 5.3%, respectively (25). The authors also developed a gene only model which was inferior to the combined genetic and clinical model suggesting that somatic mutations are more likely to complement current risk stratification tools rather than replace them (25) In summary, such mutation-based scoring tools might provide helpful information to risk stratify MDS patients and select appropriate treatment options but need to be combined with existing clinical and pathological scoring systems (6, 64–66).
5. The role of NGS in treatment selection and predicting response to therapy
One of the most promising potentials of NGS is determining the best therapy for patients with MDS based on individual genetic profiles (67, 68). Except for IDH1/2 and FLT3 mutations for which specific inhibitors have been approved for AML treatment and which are occasionally used off-label for MDS, the role of genetic testing in treatment selection for MDS remains to be determined (26, 69, 70).
Several studies have established associations between specific genetic mutations and response to therapy. For example, TET2 mutations have been associated with a greater likelihood to respond to the hypomethylating agents (HMA) decitabine and azacitidine (20, 71), whereas mutations in ASXL1 have been shown to predict a lower response to HMA (19). However, the data for ASXL1 mutations is controversial as other studies showed a better outcome in ASXL1-mutated patients treated with HMAs (72). The presence of 4 or more driver mutations in MDS is associated with poor response to HMA and worse overall survival regardless of the IPSS-R risk category (73). MDS patients with mutated TP53 responded equally well to HMA but had significantly shorter duration of response than those without such mutations in one study (74). However, in another study decitabine has been shown to be especially effective in patients with TP53 mutations with response rates being higher than in patients with wild-type TP53 (64% vs. 34%, p<0.001) and a median OS than was comparable to patients with an otherwise more favorable cytogenetic risk profile (12.7 months for patients with TP53 mutations vs. 15.4 months among patients with wild-type TP53, P=0.79) (75).
While lenalidomide has shown impressive results in the treatment of low-risk MDS patients with deletion of the long arm of chromosome 5 (5q-), patients with TP53 mutations treated with lenalidomide are more likely to progress to high risk disease and even to AML than patients with wild-type TP53 (12, 76–78). The mechanism of action of lenalidomide is complex but one of the ways it exerts its effect is via inhibition of CDC25C phosphatase which decreases levels of CK1α by selective degradation and induces cell cycle arrest between the G2 and M phase (12). As 5q- deletion causes a haplodeficiency of CK1α lenalidomide in these patients can induce a functional homozygous loss of CK1α, which leads to activation of TP53 and apoptosis (12, 79, 80). When patients with mutated TP53 are treated with lenalidomide, the mutant clones expand and lead to therapy resistance (12, 81)
Immunosuppressive therapy (IST) is another therapeutic option used in low risk MDS. Komrokji et al investigated the role of somatic gene mutations on response to IST in 66 patients with lower-risk MDS using NGS (82). SF3B1 mutation, which is morphologically associated with ring sideroblasts, was the most common somatic mutation and correlated with IST nonresponse.
However, increased activation of transforming growth factor (TGF)-β pathway signaling has been associated with ineffective erythropoiesis in low-risk MDS and is more common in patients with SF3B1 mutations and >15% ring sideroblasts (83). In a phase II trial of the TGF-ß inhibitor luspatercept patients with a SF3B1 mutation or more than 15% ring sideroblasts, showed a higher response rate than MDS patients without SF3B1 mutation which led to the placebo-controlled MEDALIST trial (84, 85). However, in a larger multi-center study this correlation was not statistically significant (47). Non-SF3B1 mutations were associated with adverse effect on response duration and a higher risk of leukemic progression (82).
In summary, the use of NGS to guide treatment decisions in MDS is not well-supported by the current evidence mainly due to the lack of established predictive biomarkers. For example, ASXL1 mutations have been shown in one study to be a marker suggestive of a lower response rate to HMAs, while the presence of TET2 mutations was associated with a higher response rate (71, 72). Recent studies also suggested that the number of mutations is associated with outcome and response to HMAs with a higher mutational burden pertaining to poorer outcomes and response rates, respectively (73, 86). However, in our opinion patients should be treated with HMAs or IST based on well-established clinical criteria rather than NGS-derived biomarkers and additional studies are warranted to assess the predictive value of these biomarkers if assessed independently or in combination (87).
6. Implications of NGS on referrals for stem cell transplantation and clinical trials
Allogeneic hematopoietic stem cell transplantation (HSCT) is the only curative treatment for MDS and the decision to proceed with HSCT is usually guided by the risk assessment to predict whether survival benefit outweighs the numerous risks associated with HSCT. Appropriate timing of HSCT is also paramount as delays can preclude the candidacy (88–91).
Several factors need to be considered to determine patient eligibility for HSCT such as prognostic scoring systems (IPSS, IPSS-R), comorbidities, extent of cytopenias and the resulting transfusion burden, as well as the presence of somatic mutations predictive of a poor prognosis (89). Molecular characteristics are one major factor to consider as they influence the prognosis of MDS patients (25, 36). Especially mutations in TP53, ETV6, EZH2, ASXL1, and RUNX1 have been associated with an adverse prognosis independent of IPSS risk score and may therefore be considered for an earlier allogeneic HSCT (92). However, genetic mutations can also predict survival following HSCT. Several recent studies have shown that patients with TP53, TET2, DNMT3A, RAS pathway genes or JAK2 mutations have inferior outcomes after HSCT compared to MDS patients who lack such mutations independent of the conditioning regimen used (37, 93, 94). Interestingly, TP53 mutation was a stronger predictor of a poorer outcome than both a monosomal or very complex karyotype (37). In the study by Bejar et al., the median OS following HSCT for patients with TP53 mutations was only 4.6 months which raises the question whether these patients should be referred for HSCT at all given the relevant rate of transplant-related mortality (37). In a larger analysis of 1514 MDS patients who underwent HSCT, the negative impact of TP53 mutations persisted despite the age of patient or intensity of conditioning regimen (93). However, there were patients who had long-term survival despite harboring this mutation suggesting that such patients should not be routinely denied access to the only potentially curative therapy i.e. HSCT. Ideally, patients with high-risk mutations, including TP53, undergoing HSCT should be treated within clinical trials to optimize the conditioning regimen and to study posttransplant treatment strategies that may decrease the risk of relapse (89)
While the influence of NGS on treatment choice in MDS is still limited, it is an important tool to assess clinical trial eligibility and may eventually lead to a more individualized treatment based on a patient’s cytogenetics. For example, a small proportion of MDS patients have been shown to carry IDH1 or IDH2 mutations which can be therapeutically targeted by specific inhibitors that have been FDA-approved for AML and are currently being tested in clinical trials in MDS as well (NCT03383575, NCT02074839). There are several additional trials active that specifically include patients with MDS and certain driver mutations. For example, preclinical studies have shown that vitamin C can restore TET2 function leading to suppression of leukemic stem cell proliferation and leukemia progression (95). This concept is currently tested in a phase Ib/II study evaluating the safety and efficacy of high-dose vitamin C in the treatment of TET2-mutated, intermediate- to high-risk MDS (NCT03433781). Another clinical trial (NCT03072043) is testing APR-246, a compound that restores the function of mutated p53, in combination with azacitidine in patients with hematologic neoplasms including MDS. Monotherapy with APR-246 has previously been shown to be safe and to have biologic effects on gene expression profile which translated into only a limited therapeutic effect in patients with relapsed-refractory AML (96–98). Lastly, early clinical trial data of luspatercept in anemic patients with lower risk MDS suggest enrichment of responders among patients with ring sideroblasts and SF3B1 mutation (84). Indeed, the randomized phase 3 MEDALIST trial (NCT02631070) evaluating luspatercept, has limited enrollment to patients with >15% ring sideroblasts or SF3B1 mutations and preliminary data available showed that 37.9% of patients in the luspatercept group achieved sustained red blood cell transfusion independence compared to 13.2% in the placebo group (p<0.0001) (85). Should this agent be approved for treatment of MDS, assessment of SF3B1 mutation status could be used as a biomarker for patient selection.
7. The role of NGS in measureable residual disease
Monitoring of measurable residual disease (MRD), the detection of disease-specific genetic abnormalities, is being increasingly used for management in both a pre- and post-HSCT setting in AML and MDS and monitoring of MRD by flow cytometry and quantitative PCR is recommended by the European Leukemia Net (99, 100). Compared to AML, measuring MRD in MDS is even more challenging given the genetic heterogeneity of this disease with changes in gene profile during the disease course and the smaller size of the underlying disease clone (101, 102). NGS is therefore a promising technique as it allows for the monitoring of several genetic markers simultaneously and is more sensitive than other techniques like PCR (100).
Only a minority of MDS patients considered for transplantation achieve complete remission before HSCT, and most patients have evidence of clonal hematopoiesis when evaluated with sensitive NGS methods (37, 88). While the detection of mutations in genes such as TP53, IDH2 and RAS pre-HSCT has been shown to correlate with an adverse outcome posttransplant, it has to be kept in mind that these studies did not explicitly study NGS to detect MRD and that additional studies are needed (93, 103).
Using MRD in the posttransplant setting to monitor for disease relapse and to assess for the need of maintenance therapy is even more challenging as the intensity of the conditioning regimen used for HSCT may influence the time to clearance of MRD (99). Additionally, it can be challenging to distinguish whether the emergence of a new mutations following HSCT is originating from the donor or related to disease relapse (104). The persistence of specific mutations in the post-HSCT setting has been linked to a higher risk of disease progression and poorer OS compared with patients in whom somatic mutations were undetectable following transplant (105–107). Monitoring MRD by NGS could therefore be a valuable tool to monitor AML/MDS patients posttransplant and may enable individualization of follow-up and posttransplant management.
8. Limitations of NGS
Taking full advantage of NGS in clinical management of MDS requires a complex technological infrastructure with a significant ability for data processing and storage. There is also a need for the personnel expertise to comprehensively analyze and interpret volumes of data efficiently, safely and cost effectively. NGS panels tend to vary in their sensitivity, depth and scale of coverage, the number of genes tested and reporting thresholds among institutions (32). While the use of gene mutation profiling is increasing in clinical practice, practice patterns, interpretation and beliefs among providers seems to vary substantially (108). To overcome this, avoid ambiguity, and enable effective sharing of genomic information a uniform nomenclature and standardized criteria will be required, and laboratories would need to remain up-to-date with dynamic changes in the nomenclature maintained by the Human Genome Variation Society (HGVS). The interpretation of sequence variants requires standardized criteria and guidelines and the American Society of Hematology has formed a task force addressing these issues (26). While all variants should be reported, gene variants without a validated phenotype association may have uncertain clinical significance, which can pose a challenge given a high likelihood of misinterpretation by health care providers who may be unfamiliar with genomic sequencing. Variants in healthy or asymptomatic individuals and incidental findings unrelated to the indication for testing should be interpreted cautiously to avoid over-diagnosis and unnecessary interventions.
Given the multitude of known MDS-associated mutations, additional studies are needed that assess the combinatorial effect of various mutations on outcome and treatment choice. It is also important to distinguish between somatic and germline mutations which should be suspected in cases in which the VAF is around 50% (26). Germline mutations of TP53 and RUNX1 underlie specific syndromes such as Li-Fraumeni syndrome that portend an increased risk for other malignancies and inheritance. Detection of such germline mutations should therefore lead to genetic counseling and appropriate screening for other malignancies in the index patient and affected family members (109, 110).
9. Conclusions
Despite a wealth of genetic and mutational data in MDS, established clinico-pathologic risk stratification tools such as IPSS-R remain the preferred basis for establishing prognosis and guide therapy decisions for patients with MDS. Additional studies to determine the role of NGS in the diagnosis, prognosis and treatment of MDS patients are warranted and several challenges remain such as the genetic heterogeneity of the disease and a limited understanding of the precise effect of a mutation alone or especially in combination with other genetic abnormalities. With further advances in diagnostic techniques as well as a better understanding of the prognostic impact and predictive value for response to therapy, NGS has the potential to be an essential tool to personalize treatment for MDS patients in the future. However, as of now NGS is not quite ready for prime time, and treatment choices should be based on well-established criteria such as risk of transformation to AML and transfusion burden rather than mutational testing.
10. Expert opinion
NGS can be a valuable tool to rule out MDS as it has a high negative predictive value for MDS and other hematologic malignancies. Therefore, NGS may proof helpful to distinguish morphologically similar appearing conditions such as AA and hMDS as well as ICUS and CCUS. However, none of the somatic mutations in MDS-related genes, especially in DNMT3A, ASXL1, and TET2, are specific for MDS and can also be detected in patients with other disorders such as MPN/MDS overlap syndromes, AML, and even in neoplastic precursors states such as CCUS and CHIP, which are common in older, otherwise healthy individuals. Only mutations in the spliceosome gene SF3B1 were specific enough to establish a diagnosis of MDS with ring sideroblasts (MDS-RS) in patients with cytopenias, dysplasia, and ringed sideroblasts that constitute at least 5% of all nucleated erythroid cells. Importantly, we want to re-emphasize that genetic testing alone is not sufficient to establish a diagnosis of MDS but can serve as a co-diagnostic criterion in patients with persistent peripheral blood cytopenias who do not fulfill other criteria such as dysplasia or increased percentage of ringed sideroblasts or myeloblasts. The addition of genetic information to established clinical-pathological risk stratification tools has the potential to better determine the individual prognosis of MDS patients and may even aid to guide treatment decisions. However, it remains to be elucidated how mutational testing affects management decisions except for the presence of mutations in IDH1/2 and FLT3 that can be specifically targeted by selective inhibitors. So far, there is insufficient evidence to support treatment selection based on mutational testing although some studies suggested a relationship between the presence of certain mutations and the response rate to treatment. Therefore, it is questionable if NGS should be offered to every MDS patient given its high costs and its limited impact on management decision. Furthermore, additional studies are warranted to assess the combination effect of various mutations and their significance in conjunction with other levels of molecular diagnostics such as RNA sequencing, epigenetic and proteomic profiling.
NGS can also provide a more sensitive method than PCR or FISH to monitor MRD in MDS following treatment although this remains challenging given the genetic heterogeneity of this disease with changes in gene profile during the disease course and the smaller size of the underlying disease clone.
Systematized nomenclature and standardized criteria are essential to get NGS integrated into clinical settings. Currently, NGS panels test for specific mutations in a selected group of driver genes. As we learn more about the pathogenesis of MDS and discover novel mutations, NGS panels will have to expand and be updated frequently and perhaps at some point transition to whole exome or even whole genome testing. This will add significant complexity to data interpretation to isolate actionable variants and requires technological infrastructure, rigorous training of laboratory personnel and clinicians and correlation with clinical outcomes.
Despite these limitations, NGS and advanced molecular assays have the potential to lead to a deeper understanding of the molecular underpinnings of MDS and potentially lead to a new era of effective targeted therapy but additional studies to link its use to improved clinical outcome are warranted.
Figure 1: Pros and cons of next-generation sequencing in MDS.
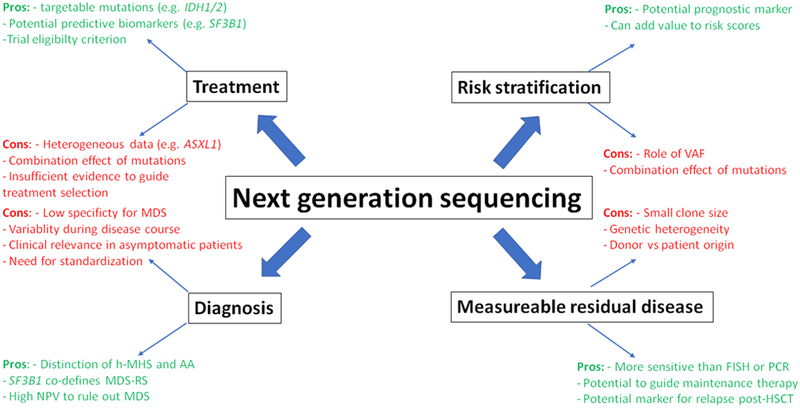
Next-generation sequencing (NGS) has several advantages and promising features in the diagnosis, treatment, and prognosis of MDS patients. However, several challenges remain which currently limit its use in routine clinical practice.
Table 2.
The most common gene mutations in MDS and the prognostic values
| Epigenetic Regulators | Prognostic Impact |
Comments |
|---|---|---|
| TET2 | worse | associated with higher response to HMA; inferior post HCT outcomes |
| EZH2 | worse | |
| ASXL1 | worse | Associated with lower response to HMA |
| DNMT3A | worse | Inferior outcomes after HSCT |
| IDH1/IDH2 | unknown | Substrate for potential targeted therapy with enasidenib |
| Spliceosomal genes | ||
| SF3B1 | better | Associated with ring sideroblasts and IST non-response |
| U2AF1 | worse | |
| SRSF2 | worse | |
| ZRSR2 | unknown | |
| Cytoplasmic tyrosine kinases | ||
| JAK2 | worse | Inferior outcomes after HSCT |
| Signaling molecules | ||
| SETBP1 | worse | |
| NRAS | worse | Inferior outcomes after HSCT |
| Transcriptional factors | ||
| ETV6 | worse | |
| RUNX1 | worse | |
| Tumor suppressors | ||
| TP53 | worse | Higher risk for leukemia transformation with lenalidomide; worse post HSCT outcomes |
| ROBO1/ROBO2 | worse | |
| Chromatid cohesion | ||
| STAG2 | worse | |
HMA: hypomethylating agents; HSCT: hemopoietic stem cell transplantation
Article Highlights.
Next generation sequencing (NGS) is a highly sensitive method to detect specific mutations in the blood and bone marrow of patients with hematologic disorders and can be helpful to distinguish morphologically similar appearing conditions.
NGS may have additional diagnostic, therapeutic, and prognostic value when combined with established risk stratification tools
As of now, there is insufficient evidence to base treatment choices on NGS-based mutational testing, although there is increasing data that show that the presence or absence of specific genetic mutations may confer a higher or lower likelihood of response to a certain treatment.
Major limitations to the routine use of NGS in MDS include the genetic heterogeneity, the small clonal size, a lack of understanding of the impact of a certain mutation and the combination of various mutations on prognosis and treatment response, and the high cost in the absence of a clear benefit for patients.
Acknowledgments
Funding
AM Zeidan is a Leukemia and Lymphoma Society Scholar in Clinical Research and is also supported by a NCI’s Cancer Clinical Investigator Team Leadership Award (CCITLA). Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number P30 CA016359. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Declaration of interests
AM Zeidan has received research funding (institutional) from Celgene, Acceleron, Abbvie, Otsuka, Pfizer, Medimmune/AstraZeneca, Boehringer-Ingelheim, Trovagene, Incyte, Takeda, and ADC Therapeutics. AM Zeidan had a consultancy with and received honoraria from AbbVie, Otsuka, Pfizer, Celgene, Ariad, Incyte, Agios, Boehringer-Ingelheim, Novartis, Acceleron, Astellas, Daiichi Sankyo, Cardinal Health, Seattle Genetics, BeyondSpring, and Takeda. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or conflict with the subject matter or materials discussed in this manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (*) or of considerable interest (**) to readers.
- 1.Shallis RM, Ahmad R, Zeidan AM. The genetic and molecular pathogenesis of myelodysplastic syndromes. Eur J Haematol. 2018;101(3):260–71. [DOI] [PubMed] [Google Scholar]
- 2.Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89(6):2079–88. [PubMed] [Google Scholar]
- 3.Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malcovati L, Germing U, Kuendgen A, Della Porta MG, Pascutto C, Invernizzi R, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol. 2007;25(23):3503–10. [DOI] [PubMed] [Google Scholar]
- 5.Kantarjian H, O’Brien S, Ravandi F, Cortes J, Shan J, Bennett JM, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer. 2008;113(6):1351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeidan AM, Sekeres MA, Wang XF, Al Ali N, Garcia-Manero G, Steensma DP, et al. Comparing the prognostic value of risk stratifying models for patients with lower-risk myelodysplastic syndromes: Is one model better? Am J Hematol. 2015;90(11):1036–40. [DOI] [PubMed] [Google Scholar]
- 7.Lee EJ, Podoltsev N, Gore SD, Zeidan AM. The evolving field of prognostication and risk stratification in MDS: Recent developments and future directions. Blood Rev. 2016;30(1):1–10. [DOI] [PubMed] [Google Scholar]
- 8.Santos FP, Kantarjian H, Garcia-Manero G, Ravandi F. The search for better prognostic models in myelodysplastic syndromes. Curr Hematol Malig Rep. 2011;6(1):13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeidan AM, Komrokji RS. There’s risk, and then there’s risk: The latest clinical prognostic risk stratification models in myelodysplastic syndromes. Curr Hematol Malig Rep. 2013;8(4):351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swerdlow SH CE, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues WHO Classification of Tumours, Revised 4th Edition. 2017;Volume 2. [Google Scholar]
- 11.Zeidan AM, Shallis RM, Wang R, Davidoff A, Ma X. Epidemiology of myelodysplastic syndromes: Why characterizing the beast is a prerequisite to taming it. Blood Rev. 2018. [DOI] [PubMed] [Google Scholar]
- 12.Stahl M, Zeidan AM. Lenalidomide use in myelodysplastic syndromes: Insights into the biologic mechanisms and clinical applications. Cancer. 2017;123(10):1703–13. [DOI] [PubMed] [Google Scholar]
- 13.Zeidan AM, Al Ali NH, Padron E, Lancet J, List A, Komrokji RS. Lenalidomide Treatment for Lower Risk Nondeletion 5q Myelodysplastic Syndromes Patients Yields Higher Response Rates When Used Before Azacitidine. Clin Lymphoma Myeloma Leuk. 2015;15(11):705–10. [DOI] [PubMed] [Google Scholar]
- 14.Abou Zahr A, Saad Aldin E, Komrokji RS, Zeidan AM. Clinical utility of lenalidomide in the treatment of myelodysplastic syndromes. J Blood Med. 2015;6:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405. [DOI] [PubMed] [Google Scholar]
- 16.Della Porta MG, Travaglino E, Boveri E, Ponzoni M, Malcovati L, Papaemmanuil E, et al. Minimal morphological criteria for defining bone marrow dysplasia: a basis for clinical implementation of WHO classification of myelodysplastic syndromes. Leukemia. 2015;29(1):66–75. [DOI] [PubMed] [Google Scholar]
- 17.Mangan JK, Speck NA. RUNX1 mutations in clonal myeloid disorders: from conventional cytogenetics to next generation sequencing, a story 40 years in the making. Critical reviews in oncogenesis. 2011;16(1–2):77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496–506.**This is the landmark study that first investigated the prognostic relevance of certain somatic mutations in MDS and paved the way for further studies and increasingly personalized treatment approaches to MDS patients.
- 19.Traina F, Visconte V, Elson P, Tabarroki A, Jankowska AM, Hasrouni E, et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia. 2014;28(1):78–87. [DOI] [PubMed] [Google Scholar]
- 20.Bejar R, Lord A, Stevenson K, Bar-Natan M, Perez-Ladaga A, Zaneveld J, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. 2014;124(17):2705–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abou Zahr A, Saad Aldin E, Barbarotta L, Podoltsev N, Zeidan AM. The clinical use of DNA methyltransferase inhibitors in myelodysplastic syndromes. Expert Rev Anticancer Ther. 2015;15(9):1019–36. [DOI] [PubMed] [Google Scholar]
- 22.DiNardo CD, Patel KP, Garcia-Manero G, Luthra R, Pierce S, Borthakur G, et al. Lack of association of IDH1, IDH2 and DNMT3A mutations with outcome in older patients with acute myeloid leukemia treated with hypomethylating agents. Leukemia & lymphoma. 2014;55(8):1925–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coombs CC, Sallman DA, Devlin SM, Dixit S, Mohanty A, Knapp K, et al. Mutational correlates of response to hypomethylating agent therapy in acute myeloid leukemia. Haematologica. 2016;101(11):e457–e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bejar R Myelodysplastic Syndromes Diagnosis: What Is the Role of Molecular Testing? Curr Hematol Malig Rep. 2015;10(3):282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steensma DP. How I use molecular genetic tests to evaluate patients who have or may have myelodysplastic syndromes. Blood. 2018;132(16):1657–63.**This is an excellent contemporary review on molecular testing in MDS patients illustrated by various clinical cases.
- 27.Abou Zahr A, Bernabe Ramirez C, Wozney J, Prebet T, Zeidan AM. New Insights into the Pathogenesis of MDS and the rational therapeutic opportunities. Expert Rev Hematol. 2016;9(4):377–88. [DOI] [PubMed] [Google Scholar]
- 28.Moyo TK, Savona MR. Molecular Testing in Patients with Suspected Myelodysplastic Syndromes. Curr Hematol Malig Rep. 2016;11(6):441–8. [DOI] [PubMed] [Google Scholar]
- 29.Valent P, Orazi A, Steensma DP, Ebert BL, Haase D, Malcovati L, et al. Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions. Oncotarget. 2017;8(43):73483–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeidan AM, Stahl M, Sekeres MA, Steensma DP, Komrokji RS, Gore SD. A call for action: Increasing enrollment of untreated patients with higher-risk myelodysplastic syndromes in first-line clinical trials. Cancer. 2017;123(19):3662–72. [DOI] [PubMed] [Google Scholar]
- 31.Yohe S, Thyagarajan B. Review of Clinical Next-Generation Sequencing. Archives of Pathology & Laboratory Medicine. 2017;141(11):1544–57. [DOI] [PubMed] [Google Scholar]
- 32.Hardwick SA, Deveson IW, Mercer TR. Reference standards for next-generation sequencing. Nat Rev Genet. 2017;18(8):473–84. [DOI] [PubMed] [Google Scholar]
- 33.Kohlmann A, Grossmann V, Nadarajah N, Haferlach T. Next-generation sequencing - feasibility and practicality in haematology. Br J Haematol. 2013;160(6):736–53. [DOI] [PubMed] [Google Scholar]
- 34.Behjati S, Tarpey PS. What is next generation sequencing? Arch Dis Child Educ Pract Ed. 2013;98(6):236–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steensma DP. The evolving role of genomic testing in assessing prognosis of patients with myelodysplastic syndromes. Best Pract Res Clin Haematol. 2017;30(4):295–300. [DOI] [PubMed] [Google Scholar]
- 36.Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616–27; quiz 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bejar R, Stevenson KE, Caughey B, Lindsley RC, Mar BG, Stojanov P, et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J Clin Oncol. 2014;32(25):2691–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mossner M, Jann JC, Wittig J, Nolte F, Fey S, Nowak V, et al. Mutational hierarchies in myelodysplastic syndromes dynamically adapt and evolve upon therapy response and failure. Blood. 2016;128(9):1246–59. [DOI] [PubMed] [Google Scholar]
- 39.Xu F, Wu LY, He Q, Wu D, Zhang Z, Song LX, et al. Exploration of the role of gene mutations in myelodysplastic syndromes through a sequencing design involving a small number of target genes. Sci Rep. 2017;7:43113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu SJ, Kuo YY, Hou HA, Li LY, Tseng MH, Huang CF, et al. The clinical implication of SRSF2 mutation in patients with myelodysplastic syndrome and its stability during disease evolution. Blood. 2012;120(15):3106–11. [DOI] [PubMed] [Google Scholar]
- 41.Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7:12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bartels S, Schipper E, Hasemeier B, Kreipe H, Lehmann U. Routine clinical mutation profiling using next generation sequencing and a customized gene panel improves diagnostic precision in myeloid neoplasms. Oncotarget. 2016;7(21):30084–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lew JL, Fenderson JL, Carmichael MG. Next-Generation Gene Sequencing Differentiates Hypoplastic Myelodysplastic Syndrome from Aplastic Anemia. Hawaii J Med Public Health. 2017;76(11 Suppl 2):10–2. [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang X, List AF, Lancet JE, Jinming S, Moscinski LC, Sokol L, et al. Clinicopathological Features and Mutational Analysis of Patients with Aplastic Anemia and Hypoplastic Myelodysplastic Syndrome. Blood. 2016;128(22):1993.27737848 [Google Scholar]
- 45.Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yoshida K, Townsley D, et al. Somatic Mutations and Clonal Hematopoiesis in Aplastic Anemia. N Engl J Med. 2015;373(1):35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shallis RM, Chokr N, Stahl M, Pine AB, Zeidan AM. Immunosuppressive therapy in myelodysplastic syndromes: a borrowed therapy in search of the right place. Expert Rev Hematol. 2018;11(9):715–26. [DOI] [PubMed] [Google Scholar]
- 47.Stahl M, DeVeaux M, de Witte T, Neukirchen J, Sekeres MA, Brunner AM, et al. The use of immunosuppressive therapy in MDS: clinical outcomes and their predictors in a large international patient cohort. Blood Adv. 2018;2(14):1765–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kulasekararaj AG, Jiang J, Smith AE, Mohamedali AM, Mian S, Gandhi S, et al. Somatic mutations identify a sub-group of aplastic anemia patients that progress to myelodysplastic syndrome. Blood. 2014:blood-2014–05-574889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malcovati L, Cazzola M. The shadowlands of MDS: idiopathic cytopenias of undetermined significance (ICUS) and clonal hematopoiesis of indeterminate potential (CHIP). Hematology Am Soc Hematol Educ Program. 2015;2015:299–307. [DOI] [PubMed] [Google Scholar]
- 50.Kwok B, Hall JM, Witte JS, Xu Y, Reddy P, Lin K, et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood. 2015;126(21):2355–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cargo CA, Rowbotham N, Evans PA, Barrans SL, Bowen DT, Crouch S, et al. Targeted sequencing identifies patients with preclinical MDS at high risk of disease progression. Blood. 2015;126(21):2362–5. [DOI] [PubMed] [Google Scholar]
- 52.Malcovati L, Galli A, Travaglino E, Ambaglio I, Rizzo E, Molteni E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. 2017;129(25):3371–8.* This paper showed that the presence of somatic mutations in patients with cytopenias is associated with a higher risk of progression to MDS
- 53.Heuser M, Thol F, Ganser A. Clonal Hematopoiesis of Indeterminate Potential. Dtsch Arztebl Int. 2016;113(18):317–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sellar RS, Jaiswal S, Ebert BL. Predicting progression to AML. Nature Medicine. 2018;24(7):904–6. [DOI] [PubMed] [Google Scholar]
- 56.Hansen JW, Westman MK, Sjo LD, Saft L, Kristensen LS, Orskov AD, et al. Mutations in idiopathic cytopenia of undetermined significance assist diagnostics and correlate to dysplastic changes. Am J Hematol. 2016;91(12):1234–8. [DOI] [PubMed] [Google Scholar]
- 57.Snetsinger B, Heath E, Rauh MJ. Suspicious, Non-MDS-Diagnostic Bone Marrows Have a High Incidence of Clonal Hematopoiesis (CHIP), with MDS-like Clone Size but Restricted Mutation Burden. Blood. 2015;126(23):1668. [Google Scholar]
- 58.Abelson S, Collord G, Ng SWK, Weissbrod O, Mendelson Cohen N, Niemeyer E, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. 2018;559(7714):400–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. New England Journal of Medicine. 2014;371(26):2488–98.*This study showed that clonal hematopoiesis is a common phenomenon in elderly patients and although it does not necessarily constitute a pre-leukemic state it is associated with adverse outcomes
- 60.Bejar R, Papaemmanuil E, Haferlach T, Garcia-Manero G, Maciejewski JP, Sekeres MA, et al. Somatic Mutations in MDS Patients Are Associated with Clinical Features and Predict Prognosis Independent of the IPSS-R: Analysis of Combined Datasets from the International Working Group for Prognosis in MDS-Molecular Committee. Blood. 2015;126(23):907. [Google Scholar]
- 61.Malcovati L, Karimi M, Papaemmanuil E, Ambaglio I, Jadersten M, Jansson M, et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood. 2015;126(2):233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bejar R Implications of molecular genetic diversity in myelodysplastic syndromes. Curr Opin Hematol. 2017;24(2):73–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nazha A, Narkhede M, Radivoyevitch T, Seastone DJ, Patel BJ, Gerds AT, et al. Incorporation of molecular data into the Revised International Prognostic Scoring System in treated patients with myelodysplastic syndromes. Leukemia. 2016;30(11):2214–20. [DOI] [PubMed] [Google Scholar]
- 64.Zeidan AM, Gore SD, Padron E, Komrokji RS. Current state of prognostication and risk stratification in myelodysplastic syndromes. Curr Opin Hematol. 2015;22(2):146–54. [DOI] [PubMed] [Google Scholar]
- 65.Zeidan AM, Sekeres MA, Garcia-Manero G, Steensma DP, Zell K, Barnard J, et al. Comparison of risk stratification tools in predicting outcomes of patients with higher-risk myelodysplastic syndromes treated with azanucleosides. Leukemia. 2016;30(3):649–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zeidan AM, Smith BD, Komrokji RS, Gore SD. Prognostication in myelodysplastic syndromes: beyond the International Prognostic Scoring System (IPSS). Am J Med. 2013;126(4):e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nazha A, Sekeres MA, Gore SD, Zeidan AM. Molecular Testing in Myelodysplastic Syndromes for the Practicing Oncologist: Will the Progress Fulfill the Promise? Oncologist. 2015;20(9):1069–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee EJ, Zeidan AM. Genome sequencing in myelodysplastic syndromes: can molecular mutations predict benefit from hypomethylating agent therapy? Expert Rev Hematol. 2015;8(2):155–8. [DOI] [PubMed] [Google Scholar]
- 69.DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N Engl J Med. 2018;378(25):2386–98. [DOI] [PubMed] [Google Scholar]
- 70.Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Itzykson R, Kosmider O, Cluzeau T, Mansat-De Mas V, Dreyfus F, Beyne-Rauzy O, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011;25(7):1147–52. [DOI] [PubMed] [Google Scholar]
- 72.Tobiasson M, McLornan DP, Karimi M, Dimitriou M, Jansson M, Ben Azenkoud A, et al. Mutations in histone modulators are associated with prolonged survival during azacitidine therapy. Oncotarget. 2016;7(16):22103–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takahashi K, Wang F, Sahil S, Zhang J, Gumbs C, Issa GC, et al. Presence of 4 or More Driver Mutations Predicts Poor Response to Hypomethylating Agent (HMA) Therapy and Poor Overall Survival in MDS. Blood. 2015;126(23):1663. [Google Scholar]
- 74.Takahashi K, Kantarjian HM, Patel K, Bueso-Ramos CE, Kadia T, Jabbour E, et al. TP53 Mutated MDS Patients Respond Equally to Hypomethylating Agents but Have Significantly Shorter Response Duration Compared to Patients with Wild Type TP53. Blood. 2015;126(23):1681. [Google Scholar]
- 75.Welch JS, Petti AA, Miller CA, Fronick CC, O’Laughlin M, Fulton RS, et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. New England Journal of Medicine. 2016;375(21):2023–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jadersten M, Saft L, Smith A, Kulasekararaj A, Pomplun S, Gohring G, et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J Clin Oncol. 2011;29(15):1971–9. [DOI] [PubMed] [Google Scholar]
- 77.List A, Dewald G, Bennett J, Giagounidis A, Raza A, Feldman E, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355(14):1456–65. [DOI] [PubMed] [Google Scholar]
- 78.Fenaux P, Giagounidis A, Selleslag D, Beyne-Rauzy O, Mufti G, Mittelman M, et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with Low-/Intermediate-1-risk myelodysplastic syndromes with del5q. Blood. 2011;118(14):3765–76. [DOI] [PubMed] [Google Scholar]
- 79.Wei S, Chen X, McGraw K, Zhang L, Komrokji R, Clark J, et al. Lenalidomide promotes p53 degradation by inhibiting MDM2 auto-ubiquitination in myelodysplastic syndrome with chromosome 5q deletion. Oncogene. 2013;32(9):1110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schneider RK, Schenone M, Ferreira MV, Kramann R, Joyce CE, Hartigan C, et al. Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A9. Nat Med. 2016;22(3):288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lode L, Menard A, Flet L, Richebourg S, Loirat M, Eveillard M, et al. Emergence and evolution of TP53 mutations are key features of disease progression in myelodysplastic patients with lower-risk del(5q) treated with lenalidomide. Haematologica. 2018;103(4):e143–e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Komrokji RS, Haider M, Al Ali NH, Lancet JE, Zhang Q, Epling-Burnette PK, et al. Somatic Gene Mutations Serve As Molecular Biomarkers Predictive for Response to Immunosuppressive Therapy (IST) in Myelodysplastic Syndromes (MDS). Blood. 2015;126(23):1664. [Google Scholar]
- 83.Mies A, Platzbecker U. Increasing the effectiveness of hematopoiesis in myelodysplastic syndromes: erythropoiesis-stimulating agents and transforming growth factor-beta superfamily inhibitors. Semin Hematol. 2017;54(3):141–6. [DOI] [PubMed] [Google Scholar]
- 84.Platzbecker U, Germing U, Gotze KS, Kiewe P, Mayer K, Chromik J, et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017;18(10):1338–47. [DOI] [PubMed] [Google Scholar]
- 85.Fenaux P, Platzbecker U, Mufti GJ, Garcia-Manero G, Buckstein R, Santini V, et al. The Medalist Trial: Results of a Phase 3, Randomized, Double-Blind, Placebo-Controlled Study of Luspatercept to Treat Anemia in Patients with Very Low-, Low-, or Intermediate-Risk Myelodysplastic Syndromes (MDS) with Ring Sideroblasts (RS) Who Require Red Blood Cell (RBC) Transfusions. Blood. 2018;132(Suppl 1):1.29976776 [Google Scholar]
- 86.Montalban-Bravo G, Takahashi K, Patel K, Wang F, Xingzhi S, Nogueras GM, et al. Impact of the number of mutations in survival and response outcomes to hypomethylating agents in patients with myelodysplastic syndromes or myelodysplastic/myeloproliferative neoplasms. Oncotarget. 2018;9(11):9714–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Greenberg PL, Stone RM, Al-Kali A, Barta SK, Bejar R, Bennett JM, et al. Myelodysplastic Syndromes, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2017;15(1):60–87. [DOI] [PubMed] [Google Scholar]
- 88.Saber W, Horowitz MM. Transplantation for myelodysplastic syndromes: who, when, and which conditioning regimens. Hematology Am Soc Hematol Educ Program. 2016;2016(1):478–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de Witte T, Bowen D, Robin M, Malcovati L, Niederwieser D, Yakoub-Agha I, et al. Allogeneic hematopoietic stem cell transplantation for MDS and CMML: recommendations from an international expert panel. Blood. 2017;129(13):1753–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zeidan AM, Linhares Y, Gore SD. Current therapy of myelodysplastic syndromes. Blood Rev. 2013;27(5):243–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zeidan AM, Gore SD. Should elderly patients with higher-risk myelodysplastic syndromes undergo allogeneic hematopoietic stem cell transplantation? Expert Rev Hematol. 2013;6(5):539–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Malcovati L, Papaemmanuil E, Ambaglio I, Elena C, Galli A, Della Porta MG, et al. Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood. 2014;124(9):1513–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N Engl J Med. 2017;376(6):536–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Della Porta MG, Galli A, Bacigalupo A, Zibellini S, Bernardi M, Rizzo E, et al. Clinical Effects of Driver Somatic Mutations on the Outcomes of Patients With Myelodysplastic Syndromes Treated With Allogeneic Hematopoietic Stem-Cell Transplantation. J Clin Oncol. 2016;34(30):3627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell. 2017;170(6):1079–95 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang Q, Bykov VJN, Wiman KG, Zawacka-Pankau J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018;9(5):439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Deneberg S, Cherif H, Lazarevic V, Andersson PO, von Euler M, Juliusson G, et al. An open-label phase I dose-finding study of APR-246 in hematological malignancies. Blood Cancer J. 2016;6(7):e447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lehmann S, Bykov VJ, Ali D, Andren O, Cherif H, Tidefelt U, et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol. 2012;30(29):3633–9. [DOI] [PubMed] [Google Scholar]
- 99.Shapiro RM, Kim DDH. Next-generation sequencing-based minimal residual disease monitoring in patients receiving allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia or myelodysplastic syndrome. Curr Opin Hematol. 2018;25(6):425–32. [DOI] [PubMed] [Google Scholar]
- 100.Schuurhuis GJ, Heuser M, Freeman S, Bene MC, Buccisano F, Cloos J, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2018;131(12):1275–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Grimwade D, Freeman SD. Defining minimal residual disease in acute myeloid leukemia: which platforms are ready for “prime time”? Blood. 2014;124(23):3345–55. [DOI] [PubMed] [Google Scholar]
- 102.Nassereddine S, Nishihori T, Padron E, Mahfouz R, Bazarbachi A, Komrokji RS, et al. Integrating Genomics in Myelodysplastic Syndrome to Predict Outcomes After Allogeneic Hematopoietic Cell Transplantation. Clin Lymphoma Myeloma Leuk. 2017;17(1):7–13. [DOI] [PubMed] [Google Scholar]
- 103.Kharfan-Dabaja MA, Komrokji RS, Zhang Q, Kumar A, Tsalatsanis A, Perkins J, et al. TP53 and IDH2 Somatic Mutations Are Associated With Inferior Overall Survival After Allogeneic Hematopoietic Cell Transplantation for Myelodysplastic Syndrome. Clin Lymphoma Myeloma Leuk. 2017;17(11):753–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gibson CJ, Kennedy JA, Nikiforow S, Kuo FC, Alyea EP, Ho V, et al. Donor-engrafted CHIP is common among stem cell transplant recipients with unexplained cytopenias. Blood. 2017;130(1):91–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Thol F, Gabdoulline R, Liebich A, Klement P, Schiller J, Kandziora C, et al. Measurable residual disease monitoring by NGS before allogeneic hematopoietic cell transplantation in AML. Blood. 2018;132(16):1703–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Duncavage EJ, Jacoby MA, Chang GS, Miller CA, Edwin N, Shao J, et al. Mutation Clearance after Transplantation for Myelodysplastic Syndrome. N Engl J Med. 2018;379(11):1028–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fu Y, Schroeder T, Zabelina T, Badbaran A, Bacher U, Kobbe G, et al. Postallogeneic monitoring with molecular markers detected by pretransplant next-generation or Sanger sequencing predicts clinical relapse in patients with myelodysplastic/myeloproliferative neoplasms. Eur J Haematol. 2014;92(3):189–94. [DOI] [PubMed] [Google Scholar]
- 108.Pine AB CN, Stahl M, Steensma DP, Sekeres MA, Litzow MR, Luger S, Stone RM, Greenberg PL, Bejar R, Gore SD and Zeidan AM. . Wide Variation in Use and Interpretation of Gene Mutation Profiling Panels Among Health Care Providers of Patients with Myelodysplastic Syndromes (MDS): Results of a Large Web-Based Survey. . Blood. 2018;132:1825. [DOI] [PubMed] [Google Scholar]
- 109.Kanagal-Shamanna R, Loghavi S, DiNardo CD, Medeiros LJ, Garcia-Manero G, Jabbour E, et al. Bone marrow pathologic abnormalities in familial platelet disorder with propensity for myeloid malignancy and germline RUNX1 mutation. Haematologica. 2017;102(10):1661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kratz CP, Achatz MI, Brugieres L, Frebourg T, Garber JE, Greer MC, et al. Cancer Screening Recommendations for Individuals with Li-Fraumeni Syndrome. Clin Cancer Res. 2017;23(11):e38–e45. [DOI] [PubMed] [Google Scholar]