

Abstract
Objective:
IL-33 has been shown to play a role in endothelial dysfunction but its role in atherosclerosis is controversial. Therefore, the purpose of this study is to examine its role in vascular wall remodeling following injury.
Approach and Results:
Thrombin induced IL-33 expression in a time dependent manner in human aortic smooth muscle cells (HASMCs) and inhibition of its activity by its neutralizing antibody suppressed thrombin-induced HASMC migration but not DNA synthesis. In exploring the mechanisms, we found that Par1, Gαq/11, PLCβ3, NFATc1, E2F1 and LMCD1 are involved in thrombin-induced IL-33 expression and migration. Furthermore, we identified a NFAT binding site at −100 nt that mediates thrombin-induced IL-33 promoter activity. Interestingly, we observed that NFATc1, E2F1 and LMCD1 bind to NFAT site in response to thrombin and found that LMCD1, while alone has no significant effect, enhanced either NFATc1 or E2F1-dependent IL-33 promoter activity. In addition, we found that guide wire injury induces IL-33 expression in SMC and its neutralizing antibodies substantially reduce SMC migration and neointimal growth in vivo. Increased expression of IL-33 was also observed in human atherosclerotic lesions as compared to arteries without any lesions.
Conclusions:
The above findings reveal for the first time that thrombin-induced HASMC migration and injury-induced neointimal growth require IL-33 expression. In addition, thrombin-induced IL-33 expression requires LMCD1 enhanced combinatorial activation of NFTAc1 and E2F1.
Keywords: Atherosclerosis, E2F1, IL-33, LMCD1, NFATc1, smooth muscle cells
Graphical Abstract
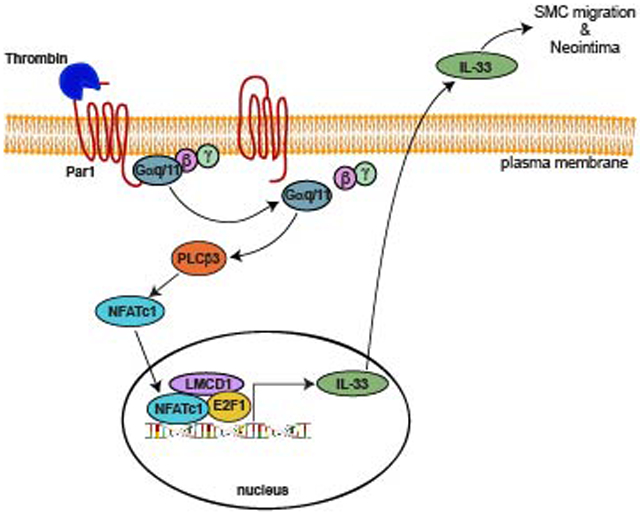
INTRODUCTION
Vascular smooth muscle cell (VSMC) migration and proliferation are important factors in neointima formation and during early stages of atherogenesis [1]. As VSMCs exhibit both phenotypic and functional plasticity, they are able to switch from the quiescent ‘contractile’ state to the ‘proinflammatory’ synthetic state during vascular injury [2, 3]. Although several theories have been proposed to explain the abnormal migration and proliferation of VSMCs [4], the exact cause still remains elusive. Vascular injury elicits production of a wide variety of molecules, which appear to be involved in the modulation of VSMC migration and proliferation [3, 4]. IL-33 is the most recently discovered member of the IL-1 family of cytokines and is secreted from various types of cells during necrotic cell death due to tissue damage, infection, or trauma, but remains intra cellular during apoptosis [5]. Thus, IL-33 functions as ‘alarmin’ to alert immune system after infection or injury [6]. IL-33 can also act as an intracellular transcriptional regulator and extracellular traditional cytokine [6]. Extracellular IL-33 exerts its functions by binding to its transmembrane receptor ST2 and activating ST2-dependent signaling molecules including nuclear factor-κB (NF-κB) [7]. Earlier studies have reported that IL-33 promotes both Th1 and Th2-mediated immunity and thus has a role in a wide variety of inflammatory diseases [6, 8].
Both IL-33 and ST2 are highly expressed in human atherosclerotic plaques [9]. Accumulating evidence suggests that IL-33 also promotes angiogenesis [10] and stimulates vascular endothelial cells towards an inflammatory phenotype by upregulation of inflammatory cytokines like IL-6 and IL-8 [11, 12], adhesion molecules like intercellular adhesion molecule (ICAM) 1 and vascular cell adhesion molecule (VCAM) 1, endothelial (E)-selectin [9], proteolytic factors like urokinase-type plasminogen activator (uPA) and plasminogen activator inhibitor type-1 (PAI-1) [13], chemokines like monocyte chemoattractant protein-1 (MCP-1) [13, 14], growth response genes like growth-regulated oncogene-α (GRO-α) [14], and coagulation factors like tissue factor [15]. Although, IL-33 effects on vascular endothelial cells were well studied, it is not clear whether IL-33 affects VSMCs during vascular wall remodeling. Previously, we have shown that thrombin induces IL-33 in HASMC [16]. We have also shown that thrombin plays a role in neointima formation [17, 18]. Based on these findings, we asked the question whether IL-33 has any role in thrombin-induced neointima formation. In the present study we show that IL-33 was induced by thrombin mediating HASMC migration and involved in injury-induced neointimal growth.
MATERIALS AND METHODS
Reagents:
Anti-β-Actin (SC-47778), anti-E2F1 (SC-251) and anti-PLCβ3 (SC-403) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Gαq (ab210004), anti-Gα11 (ab153951), anti-LMCD1 (ab179454) and anti-rabbit SMCα-actin (ab5694) antibodies were bought from Abcam (Cambridge, MA). Monoclonal anti-SMCα-actin antibodies (A2547) obtained from Sigma-Aldrich (St. Louis, MO). SCH79797 was obtained from TOCRIS Biosciences (Bristol, UK). pGL3 basic vector and Luciferase assay system (E4530) and PCR Master Mix (M750B) were purchased from Promega (Madison, WI). Human Gαq siRNA (ON-TARGETplus SMARTpool J-008562), human Gα11 siRNA (ON-TARGETplus SMARTpool J-010860), and control nontargeting siRNA (D-001810–10) were purchased from Dharmacon RNAi Technologies (Chicago, IL). Anti-IL33 (MBLPM033), anti-NFATc1 (MA3–024), goat anti-rabbit HRP (31460) and goat anti-mouse HRP (31437) antibodies, human E2F1 siRNA (s4405), human LMCD1 siRNA (s26868), human NFATc1 siRNA (s9470), and mouse NFATc1 siRNA (MSS275980), Quickchange Lightning Site-Directed Mutagenesis kit (210518–5), Biotin 3′-end DNA labeling kit (89818) and light-shift chemiluminescent EMSA kit (20148), Ibidi USA CULTURE-INSERTS (50305762), One Shot™ TOP10 Chemically Competent E. coli (C404003), Lipofectamine 3000 transfection reagent (L3000–015), medium 231 (M231–500), smooth muscle growth supplements (S-007–25), and gentamicin/amphotericin solution (R-015–10) were obtained from Thermo Fisher Scientific (Waltham, MA). ECL Western blotting detection reagents (RPN2106) were purchased from GE Healthcare (Pittsburg, PA). Quick Ligation Kit (M2200S) was bought from New England Biolabs (Ipswich, MA). Neutralizing human IL-33 antibody (AF3625) and neutralizing mouse IL-33 antibody (AF3626) were obtained from R&D Systems (Minneapolis, MN). All the primers used for RT-PCR, cloning, site-directed mutagenesis and EMSA were synthesized by IDT (Coralville, IA) and listed in Table 1.
Table 1.
List of primers used in the study
| Gene | Forward primers (5′ → 3′) | Reverse primers (5′ → 3′) |
|---|---|---|
| Primers used for RT-PCR | ||
| Human IL-33 | CAAAGAAGTTTGCCCCATGT | AAGGCAAAGCACTCCACAGT |
| Human β-actin | AGCCATGTACGTTGCTAT | GATGTCCACGTCACACTTCA |
| Primers used for IL-33 promoter cloning | ||
| hIL33p(1.8 kb) | ACGAACGCGTACTAAGAAACTAAGATATGGG | ATTTAGATCTGCTGTGTTCTTCCACTTTGCTGTG |
| hIL33p(1.5 kb) | ACGAACGCGTTTCATTTGGTCCAGAAAGGTG | ATTTAGATCTGCTGTGTTCTTCCACTTTGCTGTG |
| hIL33p(1.3 kb) | ACGAACGCGTGTCAATTCTAATACAGTGG | ATTTAGATCTGCTGTGTTCTTCCACTTTGCTGTG |
| hIL33p(1.0 kb) | ACGAACGCGTGTCGTTACTGACCACGAC | ATTTAGATCTGCTGTGTTCTTCCACTTTGCTGTG |
| hIL33p(0.5 kb) | ACGAACGCGTTGCATGATTGTATGTGCTATACT | ATTTAGATCTGCTGTGTTCTTCCACTTTGCTGTG |
| hIL33p(0.3 kb) | ACGAACGCGTCATTATGCAGTGCAGAGTACCATATC | ATTTAGATCTGCTGTGTTCTTCCACTTTGCTGTG |
| hIL33p(0.2 kb) | ACGAACGCGTCTATATGGTTATGTTACCAATTTG | ATTTAGATCTGCTGTGTTCTTCCACTTTGCTGTG |
| Primers used for site-directed mutagenesis | ||
| hIL33p-NFAT-Mut | CAAACTTTACTGAAATAATTATTTAATCTATATGGTTATG | CATAACCATATAGATTAAATAATTATTTCAGTAAAGTTTG |
| Primers used for EMSA | ||
| hIL33p-NFAT-WT | CAAACTTTACTGAAATAATTATTTCCTCTATATGGTTATG | CATAACCATATAGAGGAAATAATTATTTCAGTAAAGTTTG |
| hIL33p-NFAT-Mut | CAAACTTTACTGAAATAATTATGTAATCTATATGGTTATG | CATAACCATATAGATTACATAATTATTTCAGTAAAGTTTG |
Cell culture:
HASMCs were obtained from Invitrogen and subcultured in medium 231 containing smooth muscle cell growth supplements and gentamicin/amphotericin. Cultures were maintained in a humidified 95% air and 5% CO2 atmosphere at 37ºC. HASMCs between 4–10 passages were growth-arrested overnight in medium 231 without any growth supplements and used for the experiments unless otherwise indicated.
Animals:
C57BL/6 mice were obtained from Charles River Laboratories (Wilmington, MA) and maintained at the University of Tennessee Health Science Center’s animal facilities. The UTHSC LACU facility maintains a 12/12 hours light/dark cycle and the animals have ad libitum access to food and water. Furthermore, the animals were maintained on Teklad Irradiated LM-485 mouse/rat diet (Envigo, Cat # 7912) and Shepherd’s Cob + Plus bedding. Both male and female mice were used. All the experiments involving animals were performed according to protocols approved by the Institutional Animal Care and Use Committee of the University of Tennessee Health Science Center (Memphis, TN).
Cell migration:
Cell migration was measured by wound closure assay as described previously [19]. Briefly, 80 μl of medium 231 containing 2 × 105 cells was added in each chamber of ibidi culture inserts and grown to confluence. After overnight growth-arrest in medium 231, cells were treated with and without thrombin (0.5 U/ml) in the presence of 5 mM hydroxyurea for 24 hrs. In the case of transfections, cells were transfected with non-targeted or on-targeted siRNA, and then plated in ibidi chambers before growing to confluence and growth-arrest. Cells were observed under Nikon Eclipse TS100 microscope before and after the experimental period with 10X/0.25 magnification and images were captured with CCD color camera (KP-D20AU) by using Apple iMovie 7.1.4 software. HASMC migration was expressed as percentage of wound closure (total wound area at 0 hr – wound area at 24 hrs/total area at 0 hr x 100).
DNA synthesis:
HASMC DNA synthesis was measured by [3H]-thymidine incorporation as described previously and expressed as counts/min/dish [16].
RT-PCR:
Total cellular RNA was isolated from HASMCs using TRIzol reagent as per the manufacturer’s instructions. Complementary DNA (cDNA) was prepared using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). IL-33 and β-actin mRNAs were amplified with their specific primers (Table 1) using cDNA as a template. The amplified PCR products were separated on 1.5% agarose gels, stained with ethidium bromide, and the pictures were captured using a Kodak In-Vivo Imaging System (New Haven, CT).
Western blotting:
Cell or tissue extracts containing equal amounts of protein from control and treatments were resolved by electrophoresis on 0.1% (w/v) SDS and 12% (w/v) polyacrylamide gels. The proteins were transferred electrophoretically onto a nitrocellulose membrane. After blocking in 5% (w/v) non-fat dry milk, the membrane was incubated with the appropriate primary antibodies, followed by incubation with horseradish peroxidase-conjugated secondary antibodies. The antigen-antibody complexes were detected with the enhanced chemiluminescence detection reagent kit and the band intensities were quantified by densitometry using NIH ImageJ software.
Coimmunoprecipitation:
After rinsing with cold PBS, cells were lysed in 400 μl of lysis buffer (PBS, 1% Nonidet P40, 0.5% sodium deoxycholate, 0.1% SDS, 100 μg/ml PMSF, 100 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 mM sodium orthovanadate) for 20 min on ice. The cell extracts were cleared by centrifugation at 12,000 rpm for 20 min at 4ºC. The cleared cell extracts containing an equal amount of protein from control and the indicated treatments were incubated with the indicated antibodies overnight at 4ºC, followed by incubation with protein A/G-Sepharose CL4B beads for 3 hrs with gentle rocking. The beads were collected by centrifugation at 1000 rpm for 2 min at 4ºC and washed four times with lysis buffer and once with PBS. The immunocomplexes were released by heating the beads in 40 μl of Laemmli sample buffer and analyzed by Western blotting for the indicated molecules using their specific antibodies.
PLCβ activity assay:
PLCglow (WH-15) fluorescent substrate (KXTbio Inc.) was used to measure PLCβ1 activities as described in the manufacturer’s instructions. Briefly, cell extracts containing an equal amount of protein from control and various treatments were immunoprecipitated with anti-PLCβ3 antibody. The immunocomplexes were suspended in 50 μl of reaction buffer (50 mM HEPES pH 7.2, 250 mM KCl, 1 mM CaCl2, 2 mM DTT, 50 μg/ml BSA and 0.5% sodium cholate) containing 10 μM WH-15 and incubated for 90 min at room temperature with gentle rocking. The reaction was quenched with the addition of 5 volumes of 10 mM EGTA. After centrifugation at 2000 rpm for 5 min, the supernatant was transferred to 96-well plate and the fluorescence intensity was measured using SpectraMax Gemini XPS spectrofluorometer (Molecular Devices) at 344 nm excitation and 530 nm emission.
Transfections:
HASMCs were transfected with scrambled or specific siRNA at a final concentration of 100 nM using Lipofectamine 3000 transfection reagent as per the manufacturer’s instructions. Wherever plasmid vectors were used, cells were transfected with plasmid DNAs at a final concentration of 2.5 μg/60 mm dish or 5 μg/100 mm dish. After 6 hrs of transfection, cells were recovered in complete medium for 24 hrs and then growth-arrested overnight in serum-free medium and used as required.
Cloning and site-directed mutagenesis:
The human IL-33 promoter encompassing −1771 to +77 nt relative to the transcription start site was amplified from human genomic DNA using forward primer hIL33p(1.8 kb) F incorporating a Mlu1 restriction site at the 5′ end and a reverse primer hIL33p(1.8 kb) R incorporating a BglII restriction site at the 5′ end. The PCR product was digested with Mlu1 and BglII and the released fragment was cloned into Mlu1 and BglII sites of the pGL3 basic vector (Promega) to yield pGL3-hIL33p(1.8 kb)-Luc construct. Various truncations of human IL-33 promoter were cloned into pGL3 basic vector using the primers listed in Table 1. Mutations were introduced using QuikChange Lightning Site-Directed Mutagenesis kit. Plasmid DNAs were purified using the EndoFree plasmid maxi kit (Qiagen, Catalog # 12362) and used in the transfection experiments.
Luciferase assay:
HASMCs or HEK293T cells were transfected with pGL3 basic vector or the indicated IL-33 promoter-luciferase constructs using Lipofectamine 3000 transfection reagent for 6 hrs. Cells after 24 hrs of recovery in complete medium were growth-arrested in serum-free medium overnight. Cells were then treated with and without thrombin (0.5 U/ml) for 6 hrs, washed with cold PBS, and lysed in 200 μl of lysis buffer. The cell extracts were cleared by centrifugation at 12,000 rpm for 2 min at 4ºC. The supernatants were assayed for luciferase activity using luciferase assay system (Promega) and a single tube luminometer (TD20/20; Turner Designs, Sunnyvale, CA). The values are expressed as relative luciferase units.
EMSA:
HASMC nuclear extracts were prepared using NE-PER nuclear and cytoplasmic extraction kit (Catalog # 78833, Thermo Fisher Scientific) according to the manufacturer’s protocol. The protein content of the nuclear extracts was determined using a micro-BCA method (Pierce Biotechnology). Double stranded oligonucleotides encompassing intact and mutated NFAT binding element located at −100 nt of human IL-33 promoter were used as biotin-labeled probes (Table 1) to measure protein-DNA binding activity. Briefly, 5 μg of nuclear extract was incubated in a binding buffer (10 mM Tris-HCl, pH 7.5, 50 mM KCl, 1 mM DTT, 2.5% glycerol) with 5 nM of biotin-labeled probe and 1 μg of poly(dI-dC) for 30 min at room temperature in a total volume of 20 μl on ice, and the protein-DNA complexes were resolved by electrophoresis on a 6% polyacrylamide gel using Tris borate-EDTA buffer (44.5 mM Tris-HCl, 44.5 mM borate, and 20 mM EDTA, pH 8.0). After separation, the protein-DNA complexes were transferred to nylon membrane using Tris borate-EDTA buffer, UV cross-linked, and visualized by chemiluminescence. To perform a supershift EMSA, the complete reaction mix was incubated with 2 μg of the indicated antibody for 1 hr on ice before separating it by electrophoresis. Normal serum was used as a negative control.
Guide wire injury (GI):
Mice were anesthetized using ketamine/xylazine and subjected to GI as described previously [20]. The left femoral artery was exposed by blunted dissection. Both the vein and artery were looped together proximally and distally with 4–0 silk sutures for blood flow control during the procedure. A small muscular branch of the femoral artery was isolated and a small incision was made on this exposed muscular branch and a straight spring wire (0.38 mm in diameter, No. C-SF-15–15, COOK, Bloomington, IN) was inserted into the lumen of the femoral artery. The wire was moved back and forth two times to denude the artery. After removal of the spring wire, the muscular branch of the artery was ligated. The blood flow in the injured femoral artery was restored by releasing the sutures that were placed in the proximal and distal femoral portions. Whenever siRNA was used, the siRNA was premixed with Invivofectamine 3.0. After injury the indicated siRNA-Invivofectamine 3.0 mix was suspended in 30% Pluronic gel (10 μg in 100 μl) and was applied perivascularly around the injured artery and the skin incision was closed using sutures. The Pluronic gel solidifies upon contact with tissue and forms a gel, which ensures sustained release of siRNA. At the indicated time periods of post GI, the animals were sacrificed with over dose of ketamine/xylazine, and femoral arteries were carefully harvested and either tissue extracts were prepared or fixed overnight in 4% PFA in PBS at 4ºC. After fixing in PFA, the arteries were incubated overnight in 30% sucrose at 4ºC, then embedded in OCT compound and 5–7 μm thick sections were cut in the middle of the artery. The sections were stained with H&E to quantify intimal hyperplasia or co-immuostained for SMCα-actin and IL-33. The intimal (I) and medial (M) areas were measured using NIH ImageJ software and the I/M ratios were calculated.
Injection of antibodies into mice:
One day prior to GI, mice were injected with normal IgG or neutralizing mouse IL-33 antibodies (2 μg/mouse by IP) and thereafter every 3 days for 3 weeks. For in vivo SMC migration assay, mice received a total of 2 injections of neutralizing mouse IL-33 antibodies.
In vivo SMC migration assay:
In vivo SMC migration was measured as described by Bendeck et al with minor modifications [21]. Briefly, 5 days after GI, the femoral arteries were dissected out and fixed in 4% PFA overnight at 4ºC. The middle of the injured femoral arteries was cut and fixed again in cold acetone for 10 min. The artery was then opened longitudinally and pinned down onto an agar plate with the luminal surface facing up. The arteries were rinsed with PBS and treated with 3% H2O2 for 15 min to block the endogenous peroxidase activity. After blocking in 5% goat serum in PBS for 30 min, the arteries were incubated with anti-rabbit SMCα-actin antibody (1:300) overnight at 4ºC, followed by incubation with biotinylated goat anti-rabbit IgG for 30 min. After rinsing with PBS for 5 min, peroxidase labelling was carried out using an ABC kit, and coveslips were placed. The luminal surface of the artery was examined under Nikon Eclipse 50i microscope with 40X/0.25 magnification and images were captured with Nikon Digital Sight DS-L3 color camera and the SMCα-actin-positive cells were counted.
Immunofluorescence:
The human normal and atherosclerotic artery sections were deparaffinized with xylene and then treated with antigen-unmasking solution for 15 min at 95ºC [16]. The sections were permeabilized with 0.5% Triton X-100 for 15 min, and after blocking in normal goat serum, the sections were probed with mouse anti-mouse SMCα-actin with rabbit anti-human IL-33 combination (1:100 dilution), followed by incubation with Alexa Fluor 488-conjugated goat anti-mouse or Alexa Fluor 568-conjugated goat anti-rabbit secondary antibodies (1:300 dilution). In case of mouse femoral artery cryosections, after incubation with 5% normal goat serum for 1 hr the sections were incubated with mouse anti-mouse SMCα-actin and goat anti-mouse IL-33 antibodies (1:100 dilution) overnight followed by incubation with Alexa Fluor 488-conjugated goat anti-mouse or Alexa Fluor 568-conjugated donky anti-goat secondary antibodies (1:300 dilution). The sections were observed under a Zeiss Inverted Microscope (Zeiss AxioObserver Z1; Magnification at 10X/0.25 NA or 40X/0.6 NA), and the fluorescence images were captured with a Zeiss AxioCam MRm camera using the microscope operating software and Image Analysis Software AxioVision 4.7.2 (Carl Zeiss Imaging Solutions).
Statistics:
All the experiments were repeated three times, and the data are presented as Mean ± S.D. The data set was initially analyzed for normality and variance using Minitab 18 software. The normally distributed data with similar variance were then analyzed by one-way ANOVA followed by Tukey’s post hoc test and the p values < 0.05 were considered statistically significant. In the case of EMSA, Western blotting and immunofluorescence, one set of the representative data is presented.
RESULTS
Thrombin induces IL-33 expression in mediating HASMCs migration:
Thrombin generated at the site of vascular injury can induce the expression of several genes and act as a mitogen and chemotactic factor to a variety of cell types, including peripheral blood monocytes and VSMCs [22, 23]. Because increased expression of IL-33 was observed in human atherosclerotic plaques [9, 14, 15] and thrombin-Par1 signaling has been shown to be involved both in neointimal development and atherosclerosis [17, 18, 24], we asked the question whether IL-33 has any role in thrombin-induced vascular wall remodeling. To address this, first we studied a time course effect of thrombin on IL-33 expression in HASMCs. Thrombin (0.5 units/ml) induced IL-33 expression in a time dependent manner both at mRNA and protein levels (Figure 1A & B). Based on these observations, we next examined the functional significance of IL-33 in thrombin-induced HASMC DNA synthesis and migration. Inhibition of IL-33 activity by its neutralizing antibodies without having any effect on DNA synthesis abolished thrombin-induced HASMC migration (Figure 1C & D).
Figure 1. IL-33 mediates thrombin-induced HASMC migration.

A & B. RT-PCR and Western blot analysis of IL-33 and β-actin mRNA and protein levels, respectively, in control and various time periods of thrombin (0.5 U/ml)-treated HASMCs. C & D. The effects of normal IgG and neutralizing IL-33 antibody (100 ng/ml) on thrombin (0.5 U/ml)-induced HASMC migration and DNA synthesis. The bar graphs represent Mean ± S.D. values of three independent experiments. *, p < 0.05 versus control; **, p < 0.05 versus thrombin.
Thrombin-induced IL-33 expression requires NFAT-binding element:
To understand the molecular mechanisms involved in thrombin-induced IL-33 expression, we have analyzed human IL-33 promoter sequence for transcription factor binding elements using TRANSFAC [25]. We identified four binding sites for nuclear factor of activated T cells (NFAT) (at −49 nt; −100 nt; −1039 nt and −1245 nt); two binding sites for E2F (at −853 nt and −1672 nt) and one binding site for GATA (–253 nt) in IL-33 promoter (Figure 2A). We next cloned 1.8 kb human IL-33 promoter into pGL3 basic vector (pGL3-hIL33p(1.8 kb)-Luc) and studied its activity. We observed a 2-fold increase in IL-33 promoter activity in response to thrombin as compared with vehicle control (Figure 2B). This finding suggests a role for transcriptional mechanisms in the regulation of IL-33 expression by thrombin. To identify the minimal IL-33 promoter region required for thrombin-induced IL-33 promoter activity, we performed serial promoter deletion analysis by subcloning the truncated regions −1384 to +77 nt (1.5 kb), −1194 to +77 nt (1.3 kb), −991 to +77 nt (1.0 kb), −384 to +77 nt (0.5 kb), −183 to +77 nt (0.3 kb), and −98 to +77 nt (0.2 kb) of IL-33 promoter into pGL3 basic vector. The constructs were named as pGL3-hIL33p(1.5 kb)-Luc, pGL3-hIL33p(1.3 kb)-Luc, pGL3-hIL33p(1.0 kb)-Luc, pGL3-hIL33p(0.5 kb)-Luc, pGL3-hIL33p(0.3 kb)-Luc, and pGL3-hIL33p(0.2 kb)-Luc, respectively. HASMCs were transfected with these constructs and their responsiveness to thrombin was measured. Our findings show that thrombin induces IL-33 promoter activity with all the constructs except with pGL3-hIL33p-(0.2 kb)-Luc construct suggesting the presence of thrombin-responsive element(s) between −183 nt to −98 nt of IL-33 promoter from the transcriptional start site (Figure 2B). As the IL-33 promoter region from −183 nt to −98 nt contains only one potential NFAT-binding site at −100 nt, we mutated this site by site-directed mutagenesis and tested its responsiveness to thrombin. Our results reveled that disruption of NFAT-binding element at −100 nt significantly attenuates thrombin-induced IL-33 promoter activity (Figure 2C).
Figure 2. LMCD1 acts as co-activator of E2F1 in thrombin-induced IL-33 promoter activity.

A. TRANSFAC analysis of IL-33 promoter sequence encompassing from −1771 to +77 nt (1.8 kb) for the identification of potential transcriptional factors binding sites. B. IL-33 promoter activity with serial 5′-deletions in response to thrombin in HASMCs. C. Mutation of NFAT-binding site at −100 nt in pGL3-hIL33p(0.3 kb) promoter construct blunts thrombin (0.5 U/ml)-induced luciferase activity. D. Time course effect of thrombin on protein and IL-33 promoter DNA binding activity by EMSA using intact and mutant NFAT binding element at −100 nt as a biotin-labeled probe. E. Supershift EMSA analysis of nuclear extracts of control and thrombin-treated HASMCs for NFATc1, E2F1 and LMCD1 using NFAT binding element at −100 nt as a biotin-labeled probe. F. HEK293T cells (Upper panel) or HASMCs (Lower panel) were transfected with pGL3-basic or pGL3-hIL33p (1.8 kb) vector in combination with the indicated expression vectors and 36 hrs later the luciferase activity was measured. G. HEK293T cells (Upper panel) or HASMCs (Lower panel) were transfected with pGL3-basic or pGL3-hIL33p (1.8 kb) vector along with the indicated siRNA in combination with and without the presence of the indicated expression vector and 36 hrs later luciferase activity was measured. The bar graphs represent Mean ± S.D. values of three independent experiments. *, p < 0.05 versus control or siControl or pCMV; #, p < 0.05 versus pGL3-hIL33p + thrombin; **, p < 0.05 versus pCMV-NFATc1 or pCMV-E2F1 or pCMV-LMCD1 . RLU, relative luciferase units. FP, free probe.
NFATc1, E2F1 and LMCD1 bind to IL-33 promoter and regulate its promoter activity:
To gain mechanistic insights into the role of the NFAT-binding site at −100 nt in thrombin-induced IL-33 promoter activity, we next studied a time course effect of thrombin on NFAT binding to IL-33 promoter by electrophoretic mobility shift assay (EMSA) using the NFAT-binding element at −100 nt as a biotin-labeled double-stranded oligonucleotide probe. Thrombin induced NFAT-DNA binding activity in a time dependent manner with maximum effect at 4 hrs (Figure 2D). To confirm thrombin-induced protein-DNA binding activity with NFAT-binding element as a probe, we mutated this site and used it as a probe for EMSA. Our results showed no protein-DNA binding activity in response to thrombin (Figure 2D). Next to identify which NFAT binds to this site, we performed supershift EMSA using NFATc1 antibodies. Our results show the presence of NFATc1 in thrombin-induced protein-DNA complexes (Figure 2E). As LIM and cysteine-rich domains 1 (LMCD1), a novel member of the LIM protein family, has been shown to play a critical role in the development of cardiac hypertrophy via activation of calcineurin/NFAT signaling pathway [26] and thrombin induced LMCD1 expression in HASMCs [16], we asked the question whether LMCD1 has any role in thrombin-induced NFATc1-DNA binding activity. To test this possibility, we performed supershift EMSA using anti-LMCD1 antibodies and found the presence of LMCD1 in thrombin-induced protein-DNA-complexes (Figure 2E). Since our recent study revealed that LMCD1 acts as a transcriptional co-activator for E2F1 in the modulation of thrombin-induced CDC6 expression in HASMCs [16], we explored whether E2F1 also interacts with LMCD1 in thrombin-induced IL-33 promoter activity. Supershift EMSA using anti-E2F1 antibodies showed the presence of E2F1 in thrombin-induced protein-DNA complexes as well (Figure 2E). Based on these observations, it may be suggested that NFATc1 may interact with both E2F1 and LMCD1 in thrombin-induced IL-33 promoter activity in HASMCs.
LMCD1 acts as co-activator for NFATc1 and E2F1 in thrombin-induced IL-33 promoter activity:
To understand the relative importance of NFATc1, E2F1 and LMCD1 in thrombin-induced IL-33 promoter activity, we co-expressed NFATc1, E2F1 and LMCD1 along with IL-33 promoter in various combinations in HEK293T cells or HASMCs and examined IL-33 promoter activity. While overexpression of NFATc1, E2F1 or LMCD1 alone induced IL-33 promoter activity by 2- to 3-fold (Figure 2F). In addition, coexpression of NFATc1, E2F1 and LMCD1 in various combinations or all together further enhanced the IL-33 promoter activity (Figure 2F). To find whether LMCD1 acts as a co-activator of NFATc1 or E2F1 in thrombin-induced IL-33 promoter activity, we depleted the cellular levels of these factors one at a time along with coexpression of other factors and studied their subsequent effects on IL-33 promoter activity. Our results showed that when cellular NFATc1 or E2F1 levels were depleted, overexpression of LMCD1 had no effect on IL-33 promoter activity, but when cellular LMCD1 levels were depleted, overexpression of NFATc1 but not E2F1 resulted in an increase in IL-33 promoter activity (Figure 2G). These results collectively infer that LMCD1 acts as a co-activator for E2F1 in the regulation of NFTAc1-mediated thrombin-induced IL-33 promoter activity.
NFATc1, E2F1 and LMCD1 regulate thrombin-induced IL-33 expression and HASMCs migration:
In understanding the role of NFATc1 in thrombin-induced IL-33 expression, we found that depletion of NFATc1 attenuates thrombin-induced IL-33 expression at mRNA and protein levels (Figure 3A & B). In addition, NFATc1 knockdown blocked thrombin-induced HASMC migration (Figure 3C). In line with its role in IL-33 promoter activity, depletion of E2F1 levels by its siRNA attenuated thrombin-induced IL-33 expression at mRNA and protein levels (Figure 3D & E). E2F1 depletion also reduced thrombin-induced HASMC migration (Figure 3F). Similarly, knockdown of LMCD1 also inhibited thrombin-induced IL-33 expression and HASMC migration (Figure 4A-C). These findings collectively infer that NFATc1 along with E2F1 and LMCD1 are essential for thrombin-induced IL-33 expression and HASMC migration.
Figure 3. NFATc1 and E2F1 mediate thrombin-induced IL-33 expression and HASMC migration.

A, B, D & E. HASMCs were transfected with the indicated siRNA (100 nmoles), quiesced, treated with and without thrombin (0.5 U/ml) for 6 hrs and either RNA was isolated or cell extracts were prepared. The RNA and cell extracts were analyzed by RT-PCR and Western blotting, respectively, for IL-33 and β-actin mRNA and protein levels using their specific primers or antibodies, respectively. Cell extracts were also analyzed by Western blotting for NFATc1 and E2F1 levels to show the efficacy of their respective siRNA. C & F. HASMCs were transfected with the indicated siRNA, quiesced and subjected to migration assay. The bar graphs represent the Mean ± S.D. values of three independent experiments. *, p < 0.05 versus siControl; **, p < 0.05 versus siControl + thrombin.
Figure 4. LMCD1 and Par1 mediate thrombin-induced IL-33 expression and HASMC migration.

A & B. HASMCs were transfected with the indicated siRNA (100 nmoles), quiesced, treated with and without thrombin (0.5 U/ml) for 6 hrs and either RNA was isolated or cell extracts were prepared. The RNA and cell extracts were analyzed by RT-PCR and Western blotting for IL-33 and β-actin mRNA and protein levels using their specific primers or antibodies, respectively. Cell extracts were also analyzed by Western blotting for LMCD1 levels to show the efficacy of its siRNA. C. HASMCs were transfected with the indicated siRNA, quiesced and subjected to migration assay. D & E. Quiescent HASMCs were treated with and without thrombin (0.5 U/ml) in the presence and absence of Par1 antagonist, SCH79797 (10 μM), for the indicated times and analyzed for Gαq/11 activation or 30 min and assayed for PLCβ3 activity. F-H. The effect of SCH79797 on thrombin-induced IL-33 expression at mRNA and protein levels and HASMC migration. The bar graphs represent Mean ± S.D. values of three independent experiments. *, p < 0.05 versus Control or siControl; **, p < 0.05 versus thrombin or siControl + thrombin.
Par1-Gαq/11-PLCβ3 signaling is required for thrombin-induced IL-33 expression and HASMCs migration:
Previously we have reported that thrombin-induced HASMC proliferation requires Par1-Gαq/11-PLCβ3-mediated CDC6 expression [16]. Therefore, to characterize the upstream signaling mechanisms involved in thrombin-induced NFATc1-E2F1-LMCD1-mediated IL-33 expression, we studied the role of Par1 and its downstream effectors Gαq/11 and PLCβ3. Our results showed that blocking of Par1 with its antagonist SCH79797 substantially diminished thrombin-induced Gαq/11 and PLCβ3 activation, IL-33 expression and HASMC migration (Figure 4D-H). Similarly, siRNA-mediated depletion of Gαq/11 levels diminished thrombin-induced PLCβ3 activation, IL-33 expression and HASMC migration (Figure 5A-G). In line with these observations, downregulation of PLCβ3 levels by its siRNA also reduced thrombin-induced IL-33 expression and HASMC migration (Figure 6A-C). In addition, blockade of Par1 or depletion of Gαq/11 or PLCβ3 levels attenuated thrombin-induced translocation of NFATc1 from cytoplasm to nucleus (Figure 6D). Besides, thrombin induced complex formation of NFATc1 with E2F1 and LMCD1 (Figure 6E). Together, these observations imply that Par1-Gαq/11-PLCβ3 act upstream to NFATc1-E2F1-LMCD1 in thrombin-induced IL-33 expression and HASMC migration. Previously we have demonstrated that Pyk2 via p115 RhoGEF, Rac1, RhoA and Pak1 mediates thrombin-induced neointima formation [27]. Therefore, we asked whether Pyk2 is linked to thrombin-induced IL-33 expression. Depletion of Pyk2 levels had no effect on thrombin-induced IL-33 expression (Figure 6F). These observations infer that thrombin triggers activation of both these signaling events most likely in parallel in the modulation of vascular wall remodeling.
Figure 5. Gαq/11 mediate thrombin-induced IL-33 expression and HASMC migration.

A. HASMCs were transfected with the indicated siRNA (100 nmoles), quiesced, treated with and without thrombin (0.5 U/ml) for 30 min and cell extracts were prepared and analyzed for PLCβ3 activity. B, C, E & F. All the conditions were the same as in panel A except that cells were treated with and without thrombin (0.5 U/ml) for 6 hrs and either RNA was isolated or cell extracts were prepared. The RNA and cell extracts were analyzed by RT-PCR and Western blotting, respectively, for IL-33 and β-actin mRNA and protein levels using their specific primers or antibodies, respectively. Cell extracts were also analyzed by Western blotting for Gαq and Gα11 levels to show the efficacy of their respective siRNA. D & G. HASMCs were transfected with the indicated siRNA, quiesced and subjected to migration assay. The bar graphs represent the Mean ± S.D. values of three independent experiments. *, p < 0.05 versus siControl; **, p < 0.05 versus siControl + thrombin.
Figure 6. PLCβ3 mediate thrombin-induced IL-33 expression and HASMC migration.

A & B. HASMCs were transfected with the indicated siRNA (100 nmoles), quiesced, treated with and without thrombin (0.5 U/ml) for 6 hrs and either RNA was isolated or cell extracts were prepared. The RNA and cell extracts were analyzed by RT-PCR and Western blotting, respectively, for IL-33 and β-actin mRNA and protein levels using their specific primers or antibodies, respectively. Cell extracts were also analyzed by Western blotting for PLCβ3 levels to show the efficacy of its siRNA. C. HASMCs were transfected with the indicated siRNA, quiesced and subjected to migration assay. D. Blockade of Par1 by its antagonist SCH79797 or depletion of Gαq/11 or PLCβ3 levels attenuate thrombin-induced NFATc1 translocation from cytoplasm to nucleus. E. Time course effect of thrombin on complex formation of NFATc1 with E2F1 and LMCD1. F. All the conditions were the same as in panel B except that siControl and siPyk2 were tested on thrombin-induced IL-33 levels and the blot was reprobed for Pyk2 and β-actin levels. The bar graphs represent Mean ± S.D. values of three independent experiments. *, p < 0.05 versus siControl; **, p < 0.05 versus siControl + thrombin.
IL-33 mediates neointimal development:
To understand the role of IL-33 in neointimal growth, we used mouse femoral artery guide wire injury (GI) model. Western blot analysis showed increased expression of IL-33 in injured femoral artery as compared to uninjured artery (Figure 7A). In addition, siRNA-mediated knockdown of NFATc1 or E2F1 levels inhibited GI-induced IL-33 expression (Figure 7B). Double immunofluorescence staining of femoral artery cross sections exhibited a robust IL-33 expression in SMC of injured arteries as compared to uninjured arteries (Figure 7C). H&E staining of the femoral artery further revealed a significant increase in neointimal growth after 3 wks of GI (Figure 7D). In addition, nIL-33 Ab significantly reduced neointimal growth (Figure 7D). Migration of SMC from media to intima is a pathological manifestation of restenosis after angioplasty. As IL-33 has no influence on HASMC proliferation, we tested its role in SMC migration in vivo. Femoral arteries from mice with and without nIL-33 Ab treatment were collected at day 5 after GI, opened longitudinally and stained for SMCα-actin. We noted a significantly reduced SMC on the luminal surface of injured femoral arteries that received nIL-33 Ab as compared with injured arteries that received control IgG (Figure 7E). These observations clearly demonstrate a role for IL-33 in SMC migration during neointimal development. Since SMC migration is a common phenomenon in early development of atherosclerosis, we examined human normal and atherosclerotic coronary artery sections for IL-33. We observed increased colocalization of IL-33 with SMC in human atherosclerotic lesions as compared arteries without lesions (Figure 7F).
Figure 7. IL-33 mediates injury-induced neointima formation.

A. Tissue extracts prepared from uninjured and 3 days of post guide wire-injured mouse femoral arteries were analyzed by Western blotting for IL-33 levels using its specific antibodies and normalized to α-tubulin levels. B. An equal amount of protein from 3 days of post guide wire-injured arteries that received either siControl, siNFATc1 or siE2F1 (10 μg/artery) or uninjured arteries were analyzed by Western blotting for IL-33 using its specific antibody and the blot was reprobed for NFATc1, E2F1 or α-tubulin to show the efficacy of the siRNA on its target and off target molecule levels. C. Uninjured and 3 wks of post guide wire-injured mouse femoral artery cross sections were co-immunostained for IL-33 (red) and SMC α-actin (green). The sections of injured artery were also stained for control mouse and goat IgG. D. Mice were administered with normal IgG or neutralizing IL-33 antibodies and subjected to GI. Femoral arteries from uninjured and 3 wks of post-GI mice were dissected out, fixed, cross sections were made and stained with H&E. The I/M ratios were calculated. Representative pictures of GI femoral artery cross-sections that were stained with H&E are shown and bar graph shows Mean ± S.D. values of the I/M ratios of the femoral arteries from injured and injured + IL-33 nAb administered mice (n = 6). E. Left panel: Femoral arteries from 5 days of post-GI mice that were administered with IL-33 nAb or normal IgG (2 μg/mouse by IP) were isolated, opened longitudinally and stained for SMCα-actin. Right panel: the bar graph shows Mean ± S.D. values of the SMCα-actin-positive cells from injured and injured + IL-33 nAb administered mice (n = 6). F. Paraffin-embedded human normal and atherosclerotic artery sections were co-immunostained for IL-33 (red) and SMCα-actin (green). The normal human artery sections were also stained for control mouse and rabbit IgG. Scale bars are 50 μm (panels C, D & F) and 20 μm (panel E). *, p < 0.05 versus injured + nIL-33 Ab.
DISCUSSION
Some of the earlier studies have reported an atheroprotective role of IL-33 [28, 29]. Administration of murine IL-33 to ApoE−/− mice fed with high-fat diet reduced plaque size in the aortic sinus via enhancing polarization of the immune response towards the Th2 phenotype, decreased macrophage foam cell formation and increased auto-antibodies targeting oxidized low density lipoproteins [28, 29]. In contrast, Martin et al have reported lack of effect of IL-33/ST2 signaling on the development of atherosclerosis in ApoE−/− mice [30]. However, increased IL-33 expression has been reported in human atherosclerotic plaques [9, 14, 15]. Increased IL-33 expression has also been reported in the atherosclerotic aorta of ApoE−/− mice fed with high-fat diet as compared to ApoE−/− mice fed chow diet or WT mice [28]. Various clinical studies have further shown correlation between circulating IL-33 levels and atherosclerosis [31-33]. Consistent with these observations, our findings clearly show an increased IL-33 expression in SMC during GI-induced neointimal development and human atherosclerotic plaques. More interestingly, neutralizing IL-33 antibodies blunted SMC migration and reduced neointimal development. Since neutralizing IL-33 antibodies were attenuating SMC migration and neointima formation, it is likely that these antibodies were blocking extracellular and/or circulating IL-33. Based on these previous and present findings, it may be suggested that IL-33 by promoting endothelial dysfunction and VSMC migration might be playing a role in the pathophysiology of vascular diseases, such as restenosis and atherosclerosis.
In regard to regulation of IL-33 expression, some reports showed that IFNγ induces IL-33 in cardiac myocytes and fibroblasts [34]. On the other hand, other reports have shown that IFNγ appears to attenuate IL-33 expression in macrophages and microglial cells [35, 36]. Furthermore, it was demonstrated that IFN regulatory factor-3 (IRF-3) and CREB mediate IL-33 expression by TLR and non-TLR agonists [37]. Recently, Yamazumi et al have reported MEX3B-mediated post-transcriptional regulation of IL-33 during allergic airway inflammation [38]. Besides, negative regulation of IL-33 by Rac1 and STAT1 during airway epithelial cell inflammation and viral infection, respectively, has also been reported [39, 40]. In addition, it was shown that Ras-mediated NIH-3T3 cell transformation requires IL-33 expression [41]. Towards exploring the mechanisms, we found that interactions between NFATc1, E2F1 and LMCD1 are required for thrombin-induced IL-33 expression in HASMCs. Specifically, our promoter reporter gene analysis reveals the presence of thrombin-responsive regulatory element(s) between −183 nt to −98 nt of IL-33 promoter. As this region contains only one potential NFAT-binding site at −100 nt and site-directed mutagenesis of this site attenuates its responsiveness to thrombin, it can be speculated that IL-33 expression in HASMCs depends on this regulatory element. NFATs (NFATc1-c4) are a family of transcription factors that belong to Rel group, mediate cytokine gene expression in T cells [42]. In addition, NFATc1 has been shown to play a role in cardiovascular development [43]. NFATc1 has also been shown to be involved in linking CD137 to the progression of atherosclerotic plaques [44]. Selective inhibition of NFAT suppressed proliferative SMC phenotype and accelerated atherosclerosis in diabetic mice [45, 46]. Previous studies from our laboratory have revealed the importance of NFTAc1 in vascular wall remodeling and explored the underlying mechanisms [19, 20, 47-49]. The present study identifies IL-33 as another target of NFATc1 in mediating vascular wall remodeling.
The supershift EMSA results showed the presence of E2F1 and LMCD1 in thrombin-induced protein and IL-33 promoter DNA complexes, it is likely that E2F1 and LMCD1 interact with NFATc1 in the transcriptional regulation of IL-33 expression. Furthermore, while over-expression of NFATc1 or E2F1 alone induced IL-33 promoter activity 4-fold, LMCD1 enhanced its activity only by 2-fold. In addition, when cellular NFATc1 or E2F1 levels were depleted, overexpression of LMCD1 had no effect on IL-33 promoter activity. However, when cellular LMCD1 levels were depleted, overexpression of NFATc1 but not E2F1 resulted in an increase in IL-33 promoter activity. These results collectively infer that LMCD1 acts as a co-activator for E2F1 in the regulation of NFTAc1-mediated and thrombin-induced IL-33 expression. These observations are consistent with our recent findings that LMCD1 acts as a transcriptional co-activator for E2F1 in CDC6 expression by thrombin [16]. The proteins containing LIM domains play a key role in diverse cellular functions such as signal transduction, gene expression, cytoskeleton remodeling and cell adhesion [50]. Although several LIM proteins have been functionally well studied, little is known about LMCD1. It has been reported that LMCD1 plays a role in the development of cardiac hypertrophy via activation of calcineurin/NFAT signaling pathway [26]. LMCD1 was also shown to act as a repressor for GATA6, a zinc-finger transcription factor, in lung and cardiac tissues [51]. While our recent findings show that LMCD1 acts as a transcriptional co-activator for E2F1 in thrombin-induced CDC6 expression mediating HASMC proliferation [16], the present study explores a novel role of LMCD1 in thrombin-induced IL-33 expression in modulating HASMC migration. In regard to mechanisms of IL-33 induction by thrombin, our results show that Par1, Gαq/11 and PLCβ3 are involved in this GPCR agonist-induced NFATc1-E2F1-LMCD1-mediated IL-33 expression and HASMC migration. The role of Par1, Gαq/11 and PLCβ3 in the regulation of HASMC growth has been reported previously [16, 52, 53]. Based on our previous and present findings, it may be suggested that thrombin via inducing CDC6 and IL-33 expression in parallel downstream to Par1, Gαq/11, PLCβ3, NFATc1, E2F1 and LMCD1 signaling modulates HASMC proliferation and migration, respectively, in enhancing vascular wall remodeling.
Supplementary Material
HIGHLIGHTS.
Our findings show that IL-33 expression is required thrombin-induced HASMC migration and vascular wall remodeling following injury.
Interactions between NFATc1, E2F1 and LMCD1 were required for thrombin-induced IL-33 expression.
LMCD1 acts as a co-activator for E2F1 in the regulation of NFTAc1-mediated and thrombin-induced IL-33 expression.
Thrombin-induced IL-33 expression and HASMC migration require Par1-Gαq/11-PLCβ3 signaling.
ACKNOWLEDGMENTS
The present work was supported by a grant HL069908 from NIH to GNR.
Nonstandard Abbreviations and Acronyms
- EMSA
Electrophoretic mobility shift assay
- Gαq/11
Gα protein q/11
- IFNγ
Interferon γ
- IL-33
Interleukin-33
- LMCD1
LIM and cysteine-rich domains protein 1
- NFATc1
Nuclear factor of activated T cells
- Par1
Protease-activated receptor 1
- PLCβ3
Phospholipase Cβ3
- Rac1
Ras-related C3 botulinum toxin substrate 1
- Ras
Rat sarcoma
- STAT1
Signal transducer and activator of transcription 1
- VSMC
Vascular smooth muscle cells
Footnotes
DISCLOSURES
The authors have nothing to disclose.
REFERENCES
- 1.Schwartz SM, de Blois D, O’Brien ER. The intima. Soil for atherosclerosis and restenosis. Circ Res. 1995;77:445–465. [DOI] [PubMed] [Google Scholar]
- 2.Bennett MR, Sinha S, Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res. 2016;118:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berk BC. Vascular smooth muscle growth. Autocrine growth mechanisms. Physiol Rev. 2001;81:999–1030. [DOI] [PubMed] [Google Scholar]
- 4.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. [DOI] [PubMed] [Google Scholar]
- 5.Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity. 2015;42:1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cayrol C, Girard JP. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol Rev. 2018;281:154–168. [DOI] [PubMed] [Google Scholar]
- 7.Funakoshi-Tago M, Tago K, Hayakawa M, Tominaga S, Ohshio T, Sonoda Y, Kasahara T. TRAF6 is a critical signal transducer in IL-33 signaling pathway. Cell Signal. 2008;20:1679–1686. [DOI] [PubMed] [Google Scholar]
- 8.Miller AM. Role of IL-33 in inflammation and disease. J Inflamm (Lond). 2011;8:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Demyanets S, Konya V, Kastl SP, Kaun C, Rauscher S, Niessner A, Pentz R, Pfaffenberger S, Rychli K, Lemberger CE, de Martin R, Heinemann A, Huk I, Gröger M, Maurer G, Huber K, Wojta J. Interleukin-33 induces expression of adhesion molecules and inflammatory activation in human endothelial cells and in human atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2011;31:2080–2089. [DOI] [PubMed] [Google Scholar]
- 10.Choi YS, Choi HJ, Min JK, Pyun BJ, Maeng YS, Park H, Kim J, Kim YM, Kwon YG. Interleukin-33 induces angiogenesis and vascular permeability through ST2/TRAF6-mediated endothelial nitric oxide production. Blood. 2009;114:3117–126. [DOI] [PubMed] [Google Scholar]
- 11.Pollheimer J, Bodin J, Sundnes O, Edelmann RJ, Skånland SS, Sponheim J, Brox MJ, Sundlisaeter E, Loos T, Vatn M, Kasprzycka M, Wang J, Küchler AM, Taskén K, Haraldsen G, Hol J. Interleukin-33 drives a proinflammatory endothelial activation that selectively targets nonquiescent cells. Arterioscler Thromb Vasc Biol. 2013;33:e47–e55. [DOI] [PubMed] [Google Scholar]
- 12.Yagami A, Orihara K, Morita H, Futamura K, Hashimoto N, Matsu- moto K, Saito H, Matsuda A. IL-33 mediates inflammatory responses in human lung tissue cells. J Immunol. 2010;185:5743–5750. [DOI] [PubMed] [Google Scholar]
- 13.Stojkovic S, Kaun C, Heinz M, Krychtiuk KA, Rauscher S, Lem-berger CE, de Martin R, Gröger M, Petzelbauer P, Huk I, Huber K, Wojta J, Demyanets S. Interleukin-33 induces urokinase in human endothelial cells: possible impact on angiogenesis. J Thromb Haemost. 2014;12:948–957. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto M, Umebashi K, Tokito A, Imamura J, Jougasaki M. Interleukin-33 induces growth-regulated oncogene-α expression and secretion in human umbilical vein endothelial cells. Am J Physiol Regul Integr Comp Physiol. 2017;313:R272–R279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stojkovic S, Kaun C, Basilio J, Rauscher S, Hell L, Krychtiuk KA, Bonstingl C, de Martin R, Gröger M, Ay C, Holnthoner W, Eppel W, Neumayer C, Huk I, Huber K, Demyanets S, Wojta J. Tissue factor is induced by interleukin-33 in human endothelial cells: a new link between coagulation and inflammation. Sci Rep. 2016;6:25171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janjanam J, Zhang B, Mani AM, Singh NK, Traylor JG Jr, Orr AW, Rao GN. LIM and cysteine-rich domains 1 is required for thrombin induced smooth muscle cell proliferation and promotes atherogenesis. J Biol Chem. 2018;293:3088–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang D, Paria BC, Zhang Q, Karpurapu M, Li Q, Gerthoffer WT, Nakaoka Y, Rao GN. A role for Gab1/SHP2 in thrombin activation of PAK1: gene transfer of kinase-dead PAK1 inhibits injury-induced restenosis. Circ Res. 2009;104:1066–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao H, Dronadula N, Rizvi F, Li Q, Srivastava K, Gerthoffer WT, Rao GN. Novel role for STAT-5B in the regulation of Hsp27-FGF-2 axis facilitating thrombin-induced vascular smooth muscle cell growth and motility. Circ Res. 2006;98:913–922. [DOI] [PubMed] [Google Scholar]
- 19.Kundumani-Sridharan V, Van Quyen D, Subramani J, Singh NK, Chin YE, Rao GN. Novel interactions between NFATc1 (Nuclear Factor of Activated T cells c1) and STAT-3 (Signal Transducer and Activator of Transcription-3) mediate G protein-coupled receptor agonist, thrombin-induced biphasic expression of cyclin D1, with first phase influencing cell migration and second phase directing cell proliferation. J Biol Chem. 2012;287:22463–22482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kundumani-Sridharan V, Singh NK, Kumar S, Gadepalli R, Rao GN. Nuclear factor of activated T cells c1 mediates p21-activated kinase 1 activation in the modulation of chemokine-induced human aortic smooth muscle cell F-actin stress fiber formation, migration, and proliferation and injury-induced vascular wall remodeling. J Biol Chem. 2013;26;288:22150–22162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bendeck MP, Zempo N, Clowes AW, Galardy RE, Reidy MA. Smooth muscle cell migration and matrix metalloproteinase expression after arterial injury in the rat. Circ Res. 1994;75:539–545. [DOI] [PubMed] [Google Scholar]
- 22.Bar-Shavit R, Kahn A, Fenton JW 2nd, Wilner GD. Chemotactic response of monocytes to thrombin. J Cell Biol. 1983;96:282–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McNamara CA, Sarembock IJ, Gimple LW, Fenton JW 2nd Coughlin SR, Owens GK. Thrombin stimulates proliferation of cultured rat aortic smooth muscle cells by a proteolytically activated receptor. J Clin Invest. 1993;91:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raghavan S, Singh NK, Mani AM, Rao GN. Protease-activated receptor 1 inhibits cholesterol efflux and promotes atherogenesis via cullin 3-mediated degradation of the ABCA1 transporter. J Biol Chem. 2018;293:10574–10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, Ignatieva EV, Ananko EA, Podkolodnaya OA, Kolpakov FA, Podkolodny NL, Kolchanov NA. Databases on transcriptional regulation. TRANSFAC, TRRD, and COMPEL. Nucleic Acids Res. 1998;26:362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bian ZY, Huang H, Jiang H, Shen DF, Yan L, Zhu LH, Wang L, Cao F, Liu C, Tang QZ, Li H. LIM and cysteine-rich domains 1 regulates cardiac hypertrophy by targeting calcineurin/nuclear factor of activated T cells signaling. Hypertension. 2010;55:257–263. [DOI] [PubMed] [Google Scholar]
- 27.Gadepalli R, Singh NK, Kundumani-Sridharan V, Heckle MR, Rao GN. Novel role of proline-rich nonreceptor tyrosine kinase 2 in vascular wall remodeling after balloon injury. Arterioscler Thromb Vasc Biol. 2012;32:2652–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller AM, Xu D, Asquith DL, Denby L, Li Y, Sattar N, Baker AH, McInnes IB, Liew FY. IL-33 reduces the development of atherosclerosis. J Exp Med. 2008;205:339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aimo A, Migliorini P, Vergaro G, Franzini M, Passino C, Maisel A, Emdin M. The IL-33/ST2 pathway, inflammation and atherosclerosis: Trigger and target? Int J Cardiol. 2018;267:188–192. [DOI] [PubMed] [Google Scholar]
- 30.Martin P, Palmer G, Rodriguez E, Woldt E, Mean I, James RW, Smith DE, Kwak BR, Gabay C. Atherosclerosis severity is not affected by a deficiency in IL-33/ST2 signaling. Immun Inflamm Dis. 2015;3:239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen J, Shang Q, Wong CK, Li EK, Wang S, Li RJ, Lee KL, Leung YY, Ying KY, Yim CW, Kun EW, Leung MH, Li M, Li TK, Zhu TY, Yu SL, Kuan WP, Yu CM, Tam LS. IL-33 and soluble ST2 levels as novel predictors for remission and progression of carotid plaque in early rheumatoid arthritis: A prospective study. Semin Arthritis Rheum. 2015;45:18–27. [DOI] [PubMed] [Google Scholar]
- 32.Demyanets S, Tentzeris I, Jarai R, Katsaros KM, Farhan S, Wonnerth A, Weiss TW, Wojta J, Speidl WS, Huber K. An increase of interleukin-33 serum levels after coronary stent implantation is associated with coronary in-stent restenosis. Cytokine 2014;67:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dhillon OS, Narayan HK, Khan SQ, Kelly D, Quinn PA, Squire IB, Davies JE, Ng LL. Pre-discharge risk stratification in unselected STEMI: is there a role for ST2 or its natural ligand IL-33 when compared with contemporary risk markers? Int J Cardiol. 2013;167:2182–2188. [DOI] [PubMed] [Google Scholar]
- 34.Demyanets S, Kaun C, Pentz R, Krychtiuk KA, Rauscher S, Pfaffenberger S, Zuckermann A, Aliabadi A, Gröger M, Maurer G, Huber K, Wojta J. Components of the interleukin-33/ST2 system are differentially expressed and regulated in human cardiac cells and in cells of the cardiac vasculature. J Mol Cell Cardiol. 2013;60:16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Erp K, Dach K, Koch I, Heesemann J, Hoffmann R Role of strain differences on host resistance and the transcriptional response of macrophages to infection with Yersinia enterocolitica. Physiol Genomics. 2006;25:75–84. [DOI] [PubMed] [Google Scholar]
- 36.Rock RB, Hu S, Deshpande A, Munir S, May BJ, Baker CA, Peterson PK, Kapur V. Transcriptional response of human microglial cells to interferon-γ. Genes Immun. 2005;6:712–719. [DOI] [PubMed] [Google Scholar]
- 37.Polumuri SK, Jayakar GG, Shirey KA, Roberts ZJ, Perkins DJ, Pitha PM, Vogel SN. Transcriptional regulation of murine IL-33 by TLR and non-TLR agonists. J Immunol. 2012;189;50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamazumi Y, Sasaki O, Imamura M, Oda T, Ohno Y, Shiozaki-Sato Y, Nagai S, Suyama S, Kamoshida Y, Funato K, Yasui T, Kikutani H, Yamamoto K, Dohi M, Koyasu S, Akiyama T. The RNA Binding Protein Mex-3B Is Required for IL-33 Induction in the Development of Allergic Airway Inflammation. Cell Rep. 2016;16:2456–2471. [DOI] [PubMed] [Google Scholar]
- 39.Juncadella IJ, Kadl A, Sharma AK, Shim YM, Hochreiter-Hufford A, Borish L, Ravichandran KS. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature. 2013;493:547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stier MT, Goleniewska K, Cephus JY, Newcomb DC, Sherrill TP, Boyd KL, Bloodworth MH, Moore ML, Chen K, Kolls JK, Peebles RS Jr. STAT1 Represses Cytokine-Producing Group 2 and Group 3 Innate Lymphoid Cells during Viral Infection. J Immunol. 2017;199:510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohta S, Tago K, Funakoshi-Tago M, Matsugi J, Yanagisawa K. Intracellular NF-HEV/IL-33 harbors essential roles in Ras-induced cellular transformation by contributing to cyclin D1 protein synthesis. Cell Signal. 2016;28:1025–1036. [DOI] [PubMed] [Google Scholar]
- 42.Wu H, Peisley A, Graef IA, Crabtree GR. NFAT signaling and the invention of vertebrates. Trends Cell Biol 2007;17:251e260. [DOI] [PubMed] [Google Scholar]
- 43.Schulz RA, Yutzey KE. Calcineurin signaling and NFAT activation in cardiovascular and skeletal muscle development. Dev Biol. 2004;266:1–16. [DOI] [PubMed] [Google Scholar]
- 44.Weng J, Wang C, Zhong W, Li B, Wang Z, Shao C, Chen Y, Yan J. Activation of CD137 signaling promotes angiogenesis in atherosclerosis via modulating endothelial Smad1/5-NFATc1 pathway. J Am Heart Assoc. 2017;6:e004756. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Shiny A, Regin B, Mohan V, Balasubramanyam M. Coordinated augmentation of NFAT and NOD signaling mediates proliferative VSMC phenotype switch under hyperinsulinemia. Atherosclerosis. 2016;246:257–266. [DOI] [PubMed] [Google Scholar]
- 46.Zetterqvist AV, Berglund LM, Blanco F, Garcia-Vaz E, Wigren M, Dunér P, Andersson AM, To F, Spegel P, Nilsson J, Bengtsson E, Gomez MF. Inhibition of nuclear factor of activated T-Cells (NFAT) suppresses accelerated atherosclerosis in diabetic mice. PLoS ONE. 2013;8(6):e65020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh NK, Kundumani-Sridharan V, Kumar S, Verma SK, Kotla S, Mukai H, Heckle MR, Rao GN. Protein kinase N1 is a novel substrate of NFATc1-mediated cyclin D1-CDK6 activity and modulates vascular smooth muscle cell division and migration leading to inward blood vessel wall remodeling. J Biol Chem. 2012;287:36291–36304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karpurapu M, Wang D, Singh NK, Li Q, Rao GN. NFATc1 targets cyclin A in the regulation of vascular smooth muscle cell multiplication during restenosis. J Biol Chem. 2008;283:26577–26590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karpurapu M, Wang D, Van Quyen D, Kim TK, Kundumani-Srid-haran V, Pulusani S, Rao GN. Cyclin D1 is a bona fide target gene of NFATc1 and is sufficient in the mediation of injury-induced vascular wall remodeling. J Biol Chem. 2010;285:3510–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kadrmas JL, Beckerle MC. The LIM domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell Biol. 2004;5:920–931. [DOI] [PubMed] [Google Scholar]
- 51.Rath N, Wang Z, Lu MM, Morrisey EE. LMCD1/Dyxinis a novel transcriptional cofactor that restricts GATA6 function by inhibiting DNA binding. Mol Cell Biol. 2005;25:8864–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanski WJ, Roztocil E, Hemady EA, Williams JA, Davies MG. Role of Galphaq in smooth muscle cell proliferation. J Vasc Surg. 2004;39:639–644. [DOI] [PubMed] [Google Scholar]
- 53.Martorell L, Martínez-González J, Rodríguez C, Gentile M, Calvayrac O, Badimon L. Thrombin and protease-activated receptors (PARs) in atherothrombosis. Thromb Haemost. 2008;99:305–315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.