

Abstract
Higher order compaction of the eukaryotic genome is key to the regulation of all DNA-templated processes, including transcription. This tightly controlled process involves the formation of mononucleosomes, the fundamental unit of chromatin, packaged into higher-order architectures in an H1 linker histone-dependent process. While much work has been done to delineate the precise mechanism of this event in vitro and in vivo, major gaps still exist, primarily due to a lack of molecular tools. Specifically, there has never been a successful purification and biochemical characterization of all human H1 variants. Here we present a robust method to purify H1 and illustrate its utility in the purification of all somatic variants and one germline variant. In addition, we performed a first ever side-by-side biochemical comparison, which revealed a gradient of nucleosome binding affinities and compaction capabilities. These data provide new insight into H1 redundancy and lay the groundwork for the mechanistic investigation of disease-driving mutations.
Graphical Abstract
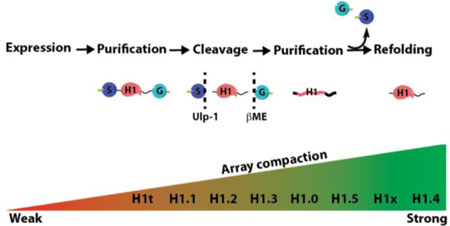
The H1 linker histone family is composed of 11 variants; seven are somatic and include H1.1-H1.5, which are replication-dependent and H1.0 and H1x, which are replication-independent. The four germline variants are replication independent, and include H1t, H1T2 (H1fnt), HILS1 and H1oo (Table 1)1–3. H1 variants differ in their time and patterns of expression2,4. All linker histones share a core globular domain (GD), roughly 80 amino acids in length, containing a ‘winged-helix’ fold found in the FOX/Forkhead-family of DNA-binding factors5. The remaining approximately 130 amino acids are unstructured and make up the N-terminal (25–30 amino acids) and C-terminal (~100 amino acids) tails (see Figure S1 for sequence alignment). Previous work has determined that the GD is responsible for H1 docking to the nucleosome dyad, although whether all variants bind in the same manner is disputed6,7. Recent work suggests that the intrinsically disordered and highly basic C-terminal domain (CTD) adopts a secondary structure upon binding to the linker DNA between two nucleosomes to facilitate compaction8. Moreover, proteomics experiments have identified numerous and diverse post-translational modifications (PTMs) on H1 variants, some of which have been shown to affect its binding to chromatin9–12. However, despite their key functional role in the formation of compacted DNA structures, deletion of individual H1 variants does not induce global changes in transcription, nor is it lethal in most cell types10,13. It has been speculated that the downregulation of a specific H1 variant induces a compensatory response whereby other variants are upregulated on both the transcriptional and translational levels11,12, although other reports suggest this is not always the case10. H1 variants being able to partially compensate for each other is supported by the finding that inactivation of three abundant variants simultaneously (H1.2, H1.3 and H1.4) is fatal in mice14. In addition, single amino acid mutations in individual variants were shown to directly correlate with pathologies such as cancer, suggesting they may drive these disorders15,16. While specific roles for individual H1 variants have been proposed, the extent of their in vivo functional and in vitro biochemical redundancy remain unclear. Existing literature primarily characterizes the properties of one variant at a time using diverse experimental setups, thus making direct comparisons difficult. Moreover, many of the tested H1s are truncated or originate from different species6,7,11,17. Nonetheless, these analyses suggest important and potentially different functions of the H1 variants, which raises the need for a systematic and standardized methodology for evaluating their function.
Table 1: Summary of the human H1 variants.
The table details the molecular and genetic properties of the seven somaticand four germline-specific human linker H1 histone variants. RI, replication-independent; RD, replication-dependent; S, somatic; G, germline; TS, testes; OO, oocytes; pI, isoelectric point; aa, amino acids.
| Variant | Gene | Replication dependency |
Somatic /Germline |
Net Charge |
Pi | CTD (aa) |
Mass (kDa) |
|---|---|---|---|---|---|---|---|
| H1.0 | H1F0 | RI | S | +52 | 10.84 | 97 | 20.7 |
| Hl.l | HIST1H1A | RD | S | +53 | 10.99 | 103 | 21.7 |
| HI.2 | HIST1H1C | RD | S | +54 | 10.94 | 104 | 21.2 |
| HI.3 | HIST1H1D | RD | S | +57 | 11.02 | 111 | 22.2 |
| HI.4 | HIST1H1E | RD | s | +58 | 11.03 | 110 | 21.7 |
| HI.5 | HIST1H1B | RD | s | +59 | 10.91 | 114 | 22.4 |
| HI.6 (Hit) | HIST1H1T | RI | G (TS) | +45 | 11.71 | 94 | 21.8 |
| HI.7 (Hlfnt) | H1FNT | RI | G (TS) | +42 | 11.77 | 60 | 28 |
| HI.8 (Hloo) | H1FOO | RI | G (OO) | +58 | 11.27 | 217 | 35.6 |
| HI.9 (H1 LSI) | HILS1 | RI | G (TS) | +35 | 10.95 | 54 | 25.5 |
| HI.10 (Hlx) | H1FX | RI | S | +40 | 10.76 | 95 | 22.3 |
Technical barriers have hindered the in-depth in vitro analyses of H1 variants. In particular, their highly unstructured, long, lysine-rich, and degradation-prone CTDs yield insoluble and/or truncated proteins upon recombinant expression. While other methods have been developed for H1 purification18,19, these are typically optimized on a single variant, while failing to demonstrate amenability to the purification of all H1 variants. As the complete functional profile of the N/CTDs in linker histones has yet to be elucidated, it is unknown if any attempt to tag them using conventional small epitopes will disrupt native H1 dynamics. To avoid this issue, we utilized two orthogonal “protecting groups” in our development of a novel strategy for a reliable and traceless purification of highly pure, well-folded recombinant linker histones. Specifically, we designed an expression cassette that contains a His-SUMO (Small Ubiquitin-like MOdifier) tag fused to the N-terminal end of H1, to be removed by its specific protease Ulp-1 (ubiquitin-like protease 1)20, and a His-tagged cis intein GyrA (Mycobacterium xenopi gyrA) fused to the C-terminus of H1 (Figure 1a). The SUMO and GyrA domains serve multiple functions: 1) affinity tags, to aid in the initial purification steps, 2) solubility tags, to promote protein expression and stability, thus eliminating the need to purify from inclusion bodies, and 3) traceless removal.
Figure 1: Robust method to generate highly pure and well-folded linker H1 histones.
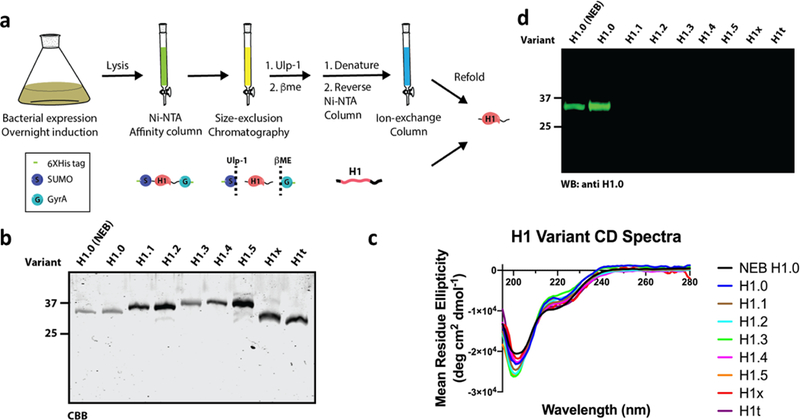
(a) A general scheme for the expression and purification of the H1 variants. Each variant was cloned into a cassette containing an N-terminal 6XHis-SUMO domain (S) and a C-terminal GyrA-6XHis (G). After initial purification by affinity and size exclusion columns, the SUMO and GyrA domains were tracelessly removed by Ulp-1 and β-ME, respectively. The cleaved tags were separated from the H1 by affinity purification under denaturing conditions and, after a final ion exchange, the denaturant was dialyzed out to yield refolded protein. All somatic and a germline H1 were purified and analyzed side-by-side on an SDS-PAGE followed by either coomassie stain for total protein (b) or western blot with anti-H1.0 (d). (c) Circular dichroism spectra of all the H1 variants, including the commercial H1.0.
As a proof-of-concept, we began with the purification of H1.0 (UniProtKB P07305), a commonly used and commercially available H1 variant. For best expression, we used Rosetta DE3 E. coli cells, which were inoculated and grown to OD600 of 0.6. Expression was then induced with IPTG at 16 oC for 12 hours. Cells were harvested and lysed in a mild neutral buffer (20 mM Tris pH 7.5, 200 mM NaCl, 1 mM PMSF). After clearing the lysate, the doubly His-tagged protein was incubated with Ni-NTA agarose beads for 1 hour at 4 oC. The bound protein was washed, eluted and loaded on a size-exclusion column (preparative scale S200 10/300) to remove any degradation and early termination products. For optimal cleavage of the N-terminal SUMO tag, the full-length H1-enriched fractions were pooled and treated with Ulp-1 for 1 hour at room temperature. This was followed by a 6 hour treatment at room temperature with 500 mM β-ME to promote the rapid auto-excision of the GyrA intein, leaving a free acid C-terminus21. To efficiently separate the tags from the full-length H1, the sample was supplemented with denaturant (8 M urea) immediately followed by a Ni-NTA column, this time collecting the column flow through, containing the purified H1. To completely remove any residual tags and concentrate the sample, it was loaded onto a cation exchange column (5 mL HiTrap HP SP) after adjustment of the pH to 9, which was key to ensuring binding. In alignment with their similar net charge, all the different H1 variants eluted at ~650 mM NaCl on a 0–1 M salt gradient. Pure fractions were pooled and refolded in a stepwise buffer exchange (8, 4, 2, 0 M urea) dialysis (Figure 1a, Figure S2).
In order to verify our purification regime produces intact and highly pure protein, following the purification of H1.0, we compared its migration on an SDS-PAGE alongside a commercial H1.0 (New England Biolabs, cat. #M2501S) and observed near-identical purity and migration pattern (Figure 1b). Since our purification scheme includes unfolding and refolding steps, we next aimed to verify that our final purified H1.0 protein contains the expected H1 random coil and alpha helix secondary structures. Turning to circular dichroism (CD) spectroscopy, which utilizes circularly polarized light to probe the secondary structure of proteins, we analyzed the purified H1.0 variant and the commercial H1.0. Similar traces, with regards to alpha helix and random coil signatures, were recorded between the two samples (Figure 1c).
To illustrate the robustness of our optimized methodology we next purified the remaining somatic H1 variants: H1.1, H1.2, H1.3, H1.4, H1.5 and H1x (UniProtKB Q02539, P16403, P16402, P10412, P16401, and Q92522, respectively). Using the described protocol, we were able to obtain the above variants at over 95 % purity, with an average yield of 1 mg at 10 μM (Figure 1b). In addition, we purified the germline-specific variant, H1t (UniProtKB P22492) with similar yield. To verify that our purification produces H1 variants that are recognized by commercially available specific antibodies, we first tested their reactivity with anti H1.0 (Invitrogen cat. # PA530055). The results presented in Figure 1d show that our purified H1.0 is recognized by the commercial anti H1.0 antibody equally well relative to the commercial H1.0 (Figure 1d). Importantly, this analysis demonstrates the high selectivity for this antibody as it does not cross-react with any of the other variants. In contrast, we tested a pan-H1 antibody (Active Motif cat. #39707), which displayed overall lower selectivity but variable affinity between the variants (Figure S3). The CD spectra for all variants shared a signature trace (Figure 1c), indicating they are well-folded.
After obtaining this library of H1 variants we next aimed to characterize their affinity to the fundamental nucleosomal subunit. We turned to the well-established recombinant nucleosome assembly protocol17 to assess H1 variant binding in vitro. The advantage of using the reconstituted nucleosome is that it is a defined substrate containing no histone or DNA modifications that could alter specific variant binding. They can therefore be used to test H1 binding uncoupled from its variant-specific regulation and compaction functions. We assembled octamers using recombinantly purified core histones and used them to reconstitute nucleosome core particles (NCPs) in the presence of the “601” strong nucleosome positioning DNA sequence. To enhance H1 binding, we added a 30 bp linker at the 3’ end of the 601, which was shown to create a docking surface for H117,22,23. Using our purified H1 and the assembled NCPs, we turned to the quantitative Biolayer Interferometry (BLI) assay, which relies on the optical measurement of kon and koff rates to calculate Kd values (Figure 2a). Briefly, we immobilized 5’ biotinylated NCPs to streptavidin sensors and measured H1 binding kinetics using the Octet Red96e system (Figure 2b, Figures S4 and S5 and Table S3). This analysis reveals that while most variants have similar affinities, others bind less tightly despite having nearly identical folds (Figure 2c).
Figure 2: The different H1 variants exhibit a range of affinities to mononucleosomes.
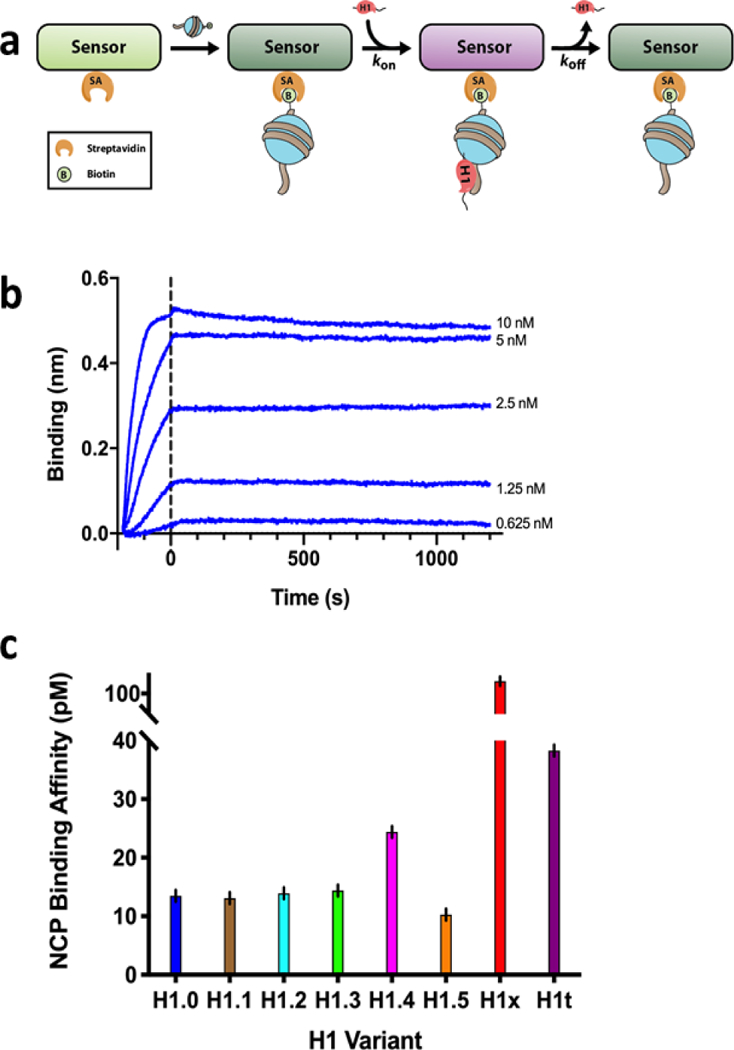
(a) Schematic model of biolayer interferometry experiments. Streptavidin biosensors were incubated with biotinylated NCPs, followed by H1 exposure to measure association kinetics, and then removal from H1 to measure dissociation. (b) Representative plot of BLI data for H1.0 dilution series. (c) Bar graph showing the calculated binding affinity constants for each H1 variant measured by BLI. Error bars represent standard error from a global fit of five replicates at differing concentrations.
While these experiments determined the affinity of the H1 variants to mononucleosomes containing linker DNA, we next aimed to measure the compaction abilities of each histone in the context of a chromatin fiber. In order to systematically compare the H1 variants compaction capacities, we used assembled nucleosome arrays as substrates. This 12-mer array mimics a chromatin fiber segment capable of adopting a higher order fold7. The arrays were assembled using the same recombinant core histone octamers as described previously, but with a DNA fragment containing 12 repeats of the 601-sequence spaced by the same 30 bp linker DNA present in the BLI experiments23. Assemblies also contained a weak nucleosome positioning buffer DNA (mouse mammary tumor virus, or MMTV) to quench any excess octamers used, thus ensuring stoichiometric occupancy. The MMTV mononucleosomes and buffer DNA were subsequently removed after assembly. To test our library of H1 variants, we assembled the arrays in the presence of a slight excess of each of the H1 variants to ensure full array saturation, as stoichiometric assemblies have been shown to require chaperones in vitro 24. In order to characterize the compaction state of the arrays, we turned to two complementary in vitro array architecture assays: magnesium precipitation and micrococcal nuclease (MNase) digestion25. Divalent cations are known to interact with the linker DNA between NCPs and induce higher-order folding, thus leading to the oligomerization of 12-mer arrays and triggering precipitation out of solution26 (Figure 3a). The concentration of Mg2+ required for sedimentation is therefore in direct correlation to the solvent accessibility of the linker DNA in the arrays. Given that H1 binds linker DNA in chromatin and induces a more compact state, we anticipated that arrays occupied with H1 would require less Mg2+ to induce precipitation. Indeed, H1-occupied arrays precipitated at a lower Mg2+ concentration relative to arrays without H1 (Figure 3b and 3c). In addition, our side-by-side comparison of the entire H1 variant library allowed us to subdivide the variants into two categories: strong (Figure 3b) and weak (Figure 3c) compactors. H1.1 and H1t are weak compactors, while the remaining variants displayed stronger compaction ability, in alignment with previous studies8,27–30.
Figure 3: The different H1 variants exhibit a range of chromatin compaction capabilities.
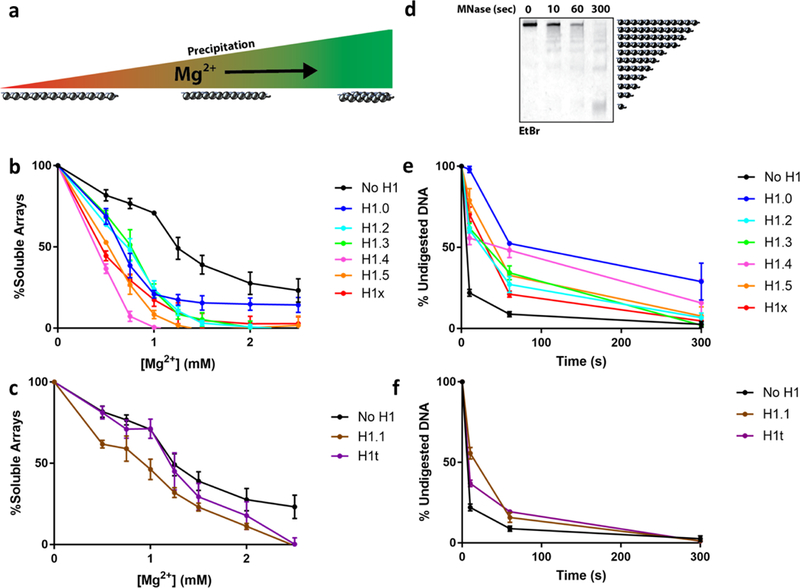
(a) Schematic model for the Mg2+ precipitation assay. 12-mer nucleosome arrays precipitate in the presence of divalent salts such as Mg2+ as a function of their compaction state. The arrays were assembled in the presence of a slight stoichiometric excess of H1, followed by step-wise precipitation by Mg2+. The H1 variants were subcategorized into strong (b) and weak (c) array compactors. (d) Representative 5 % acrylamide TBE native gel of MNase digestion of arrays assembled with H1t. MNase digestions were performed on arrays assembled with all the purified H1 variants, quantified by densitometry and plotted as percentage of undigested array band. The data was plotted based on the Mg2+ subdivision of strong (e) and weak (f) compactors. Error bars indicate the standard deviation of three separate experiments.
In parallel, we performed micrococcal nuclease (MNase) digestion of the arrays to measure the degree of DNA accessibility. Since MNase is unable to digest histone-bound DNA, the rate of array digestion is indicative of linker-DNA accessibility31. We hypothesized that H1 binding would hinder linker DNA from MNase cleavage in two ways. First, it does so indirectly by inducing chromatin fiber compaction, thus shielding a larger portion of the linker DNA from digestion. Secondl, it has been suggested that the H1 CTD wraps around the linker DNA following NCP docking, which would directly protect it from MNase cleavage6. Indeed, performing the MNase assay on arrays assembled with the library of H1 variants revealed a clustering of compaction abilities in agreement with the results from the Mg2+ precipitation experiments (Figure 3d–f, Figure S6). Together, these assays indicate that members of the linker histone family display differential abilities to compact chromatin.
Altogether, we present here a robust and traceless purification strategy whose broad applicability we illustrate by producing a library of human H1 variants and systematically comparing them. Both the purification method and the resulting library of H1s are important biochemical tools for the field that will open new lines of research. Moreover, although developed for the purification of H1, this technique could be amenable to many different insoluble and/or intrinsically disordered proteins.
With this new library of highly pure human H1 variants we performed the first side-by-side analysis of all the somatic variants as well as a germline variant. This characterization revealed that although they all have similar biochemical properties, they display different capabilities in both binding to the nucleosome core particle and compacting synthetic chromatin fragments. In terms of binding to the minimal monoucleosomal substrate, for example, the determined binding affinities range from single-digit to over 100 pM in value. This is particularly interesting because it was shown that binding to nucleosomes is primarily determined by the globular domain, which is very similar between the variants. Although varied, these Kd values are within the range of others reported previously17,32–34. One explanation for our observed variability in affinities could be whether the H1 binds on or off the nucleosome dyad6,7. Our comparison indicates that binding and compaction do not necesasarily correlate. For example, H1.1 and H1.5 exhibit similar nucleosome binding (Figure 2), but while H1.5 is a strong compactor, H1.1 is a weak one (Figure 3). Conversely, H1x is the weakest nucleosome binder, but clusters with the strong compactors based on our analyses. Notably, H1x is the least conserved somatic variant (Figure S1). Its weak nucleosome binding affinity could be ascribed to the fact that it is missing two residues previously described to be critical for the high affinity of H1 to chromatin in vivo35.
While it is presently unclear what is causing the disparity between binding affinity and compaction capabilities, one hypothesis is that the CTDs, although similar in composition, are capable of assuming H1 variant-specific folds upon chromatin binding3,6. Furthermore, it is possible that different H1 variants induce alternative array conformations upon binding, which would not be captured by the assays described herein. It is noteworthy that our observed hierarchy of H1 variants based on compaction strength, largely aligns with previous reports. For example, our measurements determined that H1.2 is a weaker compactor than H1.4, which is in complete agreement with previous work comparing these two variants36.
Despite its critical role in the formation of the higher-order chromatin structures, as mentioned before, the knockout of any single H1 variant is not lethal13,37. This may be due to the compensatory expression of other H1 variants, suggesting the H1 variants have possible functional redundancy. Interestingly, it was shown that the upregulated H1 variant often falls under our “strong compactors” category, such as H1.437. On the other hand, specific mutations that highly correlate with pathologies, specifically cancer, include very strong (H1.4) and mild (H1.2) compactors. This which might suggest additional functions for these variants in transcription and/or signaling15,16.
Finally, applying a powerful chemical biology technique such as intein splicing as part of our methodology facilitates chemical flexibility for the introduction of synthetic moieties into these proteins. This provides a new set of tools to study linker histones in great detail. For example, expressed protein ligation can be used to generate modified and semi-synthetic H1 proteins in order to study H1 PTMs, dynamics and interactors in vitro. Similarly, protein trans-splicing can be used to study these phenomena in cells38. This opens the door to thorough characterization of linker histones in both native and pathological contexts.
Supplementary Material
ACKNOWLEDGMENT
The authors would like to thank Robert Thompson, Alexey Soshnev, C. David Allis, Trudy Ramlall , David Eliezer, Elizabeth Orth, Jia Ma, and the Columbia University Precision Biocharacterization Facility as well as members of the David lab for technical and intellectual support.
Funding Sources
A.O. is supported by the National Science Foundation Graduate Research Fellowship Grant Number 2016217612. N.A.P. is supported by the National Science Foundation Graduate Research Fellowship Grant Number 2017239554. A.O. and N.A.P. are supported by the NIH T32 GM115327-Tan chemistry-biology interface training grant. D.M.R. is supported by a Medical Scientist Training Program grant from the National Institute of General Medical Sciences of the National Institutes of Health under award number: T32GM007739 to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD Program. This work was supported by the Josie Robertson Foundation and the CCSG core grant P30 CA008748.
Footnotes
The authors declare no competing financial interests.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Detailed materials and methods, and representative gels of all quantified assays. (PDF)
REFERENCES
- (1).Izzo A, Kamieniarz-Gdula K, Ramírez F, Noureen N, Kind J, Manke T, van Steensel B, and Schneider R (2013) The Genomic Landscape of the Somatic Linker Histone Subtypes H1.1 to H1.5 in Human Cells. Cell Rep. 3, 2142–2154. [DOI] [PubMed] [Google Scholar]
- (2).Hergeth SP, and Schneider R (2015) The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle. EMBO Rep. 16, 1439–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Fyodorov DV, Zhou B-R, Skoultchi AI, and Bai Y (2018) Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol. 19, 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Pan C, and Fan Y (2016) Role of H1 linker histones in mammalian development and stem cell differentiation. Biochim. Biophys. Acta BBA-Gene Regul. Mech. 1859, 496–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ramakrishnan V, Finch JT, Graziano V, Lee PL, and Sweet RM (1993) Crystal structure of globular domain of histone H5 and its implications for nucleosome binding. Nature 362, 219–223. [DOI] [PubMed] [Google Scholar]
- (6).Bednar J, Garcia-Saez I, Boopathi R, Cutter AR, Papai G, Reymer A, Syed SH, Lone IN, Tonchev O, Crucifix C, Menoni H, Papin C, Skoufias DA, Kurumizaka H, Lavery R, Hamiche A, Hayes JJ, Schultz P, Angelov D, Petosa C, and Dimitrov S (2017) Structure and Dynamics of a 197 bp Nucleosome in Complex with Linker Histone H1. Mol. Cell 66, 384–397.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu R-M, Zhu P, and Li G (2014) Cryo-EM Study of the Chromatin Fiber Reveals a Double Helix Twisted by Tetranucleosomal Units. Science 344, 376–380. [DOI] [PubMed] [Google Scholar]
- (8).Vyas P, and Brown DT (2012) N- and C-terminal Domains Determine Differential Nucleosomal Binding Geometry and Affinity of Linker Histone Isotypes H10 and H1c. J. Biol. Chem. 287, 11778–11787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wiśniewski JR, Zougman A, Krüger S, and Mann M (2007) Mass Spectrometric Mapping of Linker Histone H1 Variants Reveals Multiple Acetylations, Methylations, and Phosphorylation as Well as Differences between Cell Culture and Tissue. Mol. Cell. Proteomics 6, 72–87. [DOI] [PubMed] [Google Scholar]
- (10).Sancho M, Diani E, Beato M, and Jordan A (2008) Depletion of Human Histone H1 Variants Uncovers Specific Roles in Gene Expression and Cell Growth. PLOS Genet. 4, e1000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mishra LN, Shalini V, Gupta N, Ghosh K, Suthar N, Bhaduri U, and Rao MRS (2018) Spermatid-specific linker histone HILS1 is a poor condenser of DNA and chromatin and preferentially associates with LINE-1 elements. Epigenetics Chromatin 11, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Parseghian MH (2015) What is the role of histone H1 heterogeneity? A functional model emerges from a 50 year mystery. Biophys. 2015 Vol 2 Pages 724–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lin Q, Sirotkin A, and Skoultchi AI (2000) Normal Spermatogenesis in Mice Lacking the Testis-Specific Linker Histone H1t. Mol. Cell. Biol. 20, 2122–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Fan Y, Nikitina T, Morin-Kensicki EM, Zhao J, Magnuson TR, Woodcock CL, and Skoultchi AI (2003) H1 Linker Histones Are Essential for Mouse Development and Affect Nucleosome Spacing In Vivo. Mol. Cell. Biol. 23, 4559–4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Li H, Kaminski MS, Li Y, Yildiz M, Ouillette P, Jones S, Fox H, Jacobi K, Saiya-Cork K, Bixby D, Lebovic D, Roulston D, Shedden K, Sabel M, Marentette L, Cimmino V, Chang AE, and Malek SN (2014) Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood 123, 1487–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Scaffidi P (2016) Histone H1 alterations in cancer. Biochim. Biophys. Acta BBA - Gene Regul. Mech. 1859, 533–539. [DOI] [PubMed] [Google Scholar]
- (17).White AE, Hieb AR, and Luger K (2016) A quantitative investigation of linker histone interactions with nucleosomes and chromatin. Sci. Rep. 6, 19122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ivic N, Bilokapic S, and Halic M (2017) Preparative two-step purification of recombinant H1.0 linker histone and its domains. PLOS ONE 12, e0189040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ryan DP, and Tremethick DJ (2016) A dual affinity-tag strategy for the expression and purification of human linker histone H1.4 in Escherichia coli. Protein Expr. Purif. 120, 160–168. [DOI] [PubMed] [Google Scholar]
- (20).Mossessova E, and Lima CD (2000) Ulp1-SUMO Crystal Structure and Genetic Analysis Reveal Conserved Interactions and a Regulatory Element Essential for Cell Growth in Yeast. Mol. Cell 5, 865–876. [DOI] [PubMed] [Google Scholar]
- (21).Shah NH, and Muir TW (2013) Inteins: nature’s gift to protein chemists. Chem. Sci. 5, 446–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lowary PT, and Widom J (1998) New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning11Edited by T. Richmond. J. Mol. Biol. 276, 19–42. [DOI] [PubMed] [Google Scholar]
- (23).Fierz B, Chatterjee C, McGinty RK, Bar-Dagan M, Raleigh DP, and Muir TW (2011) Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat. Chem. Biol. 7, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Feng H, Zhou B-R, and Bai Y (2018) Binding Affinity and Function of the Extremely Disordered Protein Complex Containing Human Linker Histone H1.0 and Its Chaperone ProTα. Biochemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Debelouchina GT, Gerecht K, and Muir TW (2017) Ubiquitin utilizes an acidic surface patch to alter chromatin structure. Nat. Chem. Biol. 13, 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Schwarz PM, Felthauser A, Fletcher TM, and Hansen JC (1996) Reversible oligonucleosome self-association: dependence on divalent cations and core histone tail domains. Biochemistry 35, 4009–4015. [DOI] [PubMed] [Google Scholar]
- (27).Khadake JR, and Rao MRS (1995) DNA- and chromatin-condensing properties of rat testes H1a and H1t compared to those of rat liver H1bdec: H1t is a poor condenser of chromatin. Biochemistry 34, 15792–15801. [DOI] [PubMed] [Google Scholar]
- (28).Raghuram N, Carrero G, Stasevich TJ, McNally JG, Th’ng J, and Hendzel MJ (2010) Core Histone Hyperacetylation Impacts Cooperative Behavior and High-Affinity Binding of Histone H1 to Chromatin. Biochemistry 49, 4420–4431. [DOI] [PubMed] [Google Scholar]
- (29).Th’ng JPH, Sung R, Ye M, and Hendzel MJ (2005) H1 Family Histones in the Nucleus control of binding and localization by the c-terminal domain. J. Biol. Chem. 280, 27809–27814. [DOI] [PubMed] [Google Scholar]
- (30).Hannon R, Bateman E, Allan J, Harborne N, and Gould H (1984) Control of RNA polymerase binding to chromatin by variations in linker histone composition. J. Mol. Biol. 180, 131–149. [DOI] [PubMed] [Google Scholar]
- (31).Simpson RT (1978) Structure of the chromatosome, a chromatin particle containing 160 base pairs of DNA and all the histones. Biochemistry 17, 5524–5531. [DOI] [PubMed] [Google Scholar]
- (32).Zhou B-R, Feng H, Kato H, Dai L, Yang Y, Zhou Y, and Bai Y (2013) Structural insights into the histone H1-nucleosome complex. Proc. Natl. Acad. Sci. 110, 19390–19395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hieb AR, D’Arcy S, Kramer MA, White AE, and Luger K (2012) Fluorescence strategies for high-throughput quantification of protein interactions. Nucleic Acids Res. 40, e33–e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Caterino TL, Fang H, and Hayes JJ (2011) Nucleosome Linker DNA Contacts and Induces Specific Folding of the Intrinsically Disordered H1 Carboxyl-Terminal Domain. Mol. Cell. Biol. 31, 2341–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Happel N, Schulze E, and Doenecke D (2005) Characterisation of human histone H1x. Biol. Chem. 386, 541–551. [DOI] [PubMed] [Google Scholar]
- (36).Clausell J, Happel N, Hale TK, Doenecke D, and Beato M (2009) Histone H1 Subtypes Differentially Modulate Chromatin Condensation without Preventing ATP-Dependent Remodeling by SWI/SNF or NURF. PLOS ONE 4, e0007243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sirotkin AM, Edelmann W, Cheng G, Klein-Szanto A, Kucherlapati R, and Skoultchi AI (1995) Mice develop normally without the H1(0) linker histone. Proc. Natl. Acad. Sci. 92, 6434–6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).David Y, Vila-Perelló M, Verma S, and Muir TW (2015) Chemical tagging and customizing of cellular chromatin states using ultrafast trans-splicing inteins. Nat. Chem. 7, 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.