

Abstract
Parkinson’s disease is characterized by the loss of nigrostriatal dopaminergic signaling and the presence of alpha-synuclein aggregates (also called Lewy bodies and neurites) throughout the brain. In 2003, Braak and colleagues created a staging system for Parkinson’s disease describing the connection between the alpha-synuclein pathology and disease severity. Later, they suggested that the pathology might initially be triggered by exogenous insults targeting the gut and olfactory system. In 2008, we and other groups documented Lewy pathology in grafted neurons in people with Parkinson’s disease who had been transplanted over a decade prior to autopsy. We proposed that the Lewy pathology in the grafted neurons was the result of permissive templating or prion-like spread of alpha-synuclein pathology from neurons in the host to those in the grafts. During the following ten years, several studies described the transmission of alpha-synuclein pathology between neurons, both in cell culture and in experimental animals. Recent research has also begun to identify underlying molecular mechanisms. Collectively, these experimental studies tentatively support the idea that the progression from one Braak stage to the next is the consequence of prion-like propagation of Lewy pathology. However, definitive proof that intercellular propagation of alpha-synuclein pathology occurs in Parkinson’s disease cases has proven difficult to secure. In this review, we highlight several open questions that currently prevent us from concluding with certainty that prion-like transfer of alpha-synuclein contributes to the progression of Parkinson’s disease.
Keywords: α-synuclein, aggregation, transmission, Parkinson’s disease
Introduction
People with Parkinson’s disease (PD) progress over time with increasing motor and non-motor signs and symptoms due to the loss of striatal dopaminergic signaling and to the presence of alpha-synuclein-containing Lewy bodies and neurites. The importance of alpha-synuclein (α-syn) to PD pathophysiology is evident in both genetic and pathological realms. After initial discoveries of a mutation in the gene encoding α-syn, SNCA, that confers increased risk of developing PD (Polymeropoulos et al. 1997) and of the presence of α-syn-immunoreactivity in Lewy pathology (Spillantini et al. 1997), Braak and colleagues described staging of pathology in people with PD at autopsy (Braak et al. 2003a). In what was suggested to be the earliest phases of the disease (Braak stage 1 and 2, which actually precede the onset of the classical motor symptoms), α-syn-immunopositive pathology was found in two distinct brain areas, the dorsal motor nucleus of the vagal nerve and the anterior olfactory nucleus. Therefore, the authors proposed that these locations might be initiation sites that direct the spread of the pathology stereotypically throughout the brain (reviewed in (Rey et al. 2016c)). In the same timeframe, Hardy published a permissive templating theory regarding neurodegenerative diseases including Parkinson’s (Hardy 2005).
Braak and colleagues developed a subsequent theory, in which nasal and gastric routes of entry might be used by a pathogen to gain access to neuronal populations. In this hypothesis, a neurotropic pathogen accesses olfactory and gastric environments, and enters the olfactory and gut epithelia. The authors first emphasized a potential neurotropic virus but also suggested briefly, in a passage of their paper that did not get as much attention, that the pathogen might be composed of fragments of misfolded α-syn (Braak et al. 2003b). From that point, infiltration in the gut to submucous plexus could lead to trans-synaptic travel of the pathogen along preganglionic parasympathetic fibers to the dorsal motor nucleus of the vagus nerve. Entry at the olfactory epithelium could induce transit of the pathogen to the olfactory bulb. Thus, in this hypothesis, an external pathogen might induce pathology in the proposed initiation sites identified by Braak and colleagues (reviewed in (Hawkes et al. 2007, 2009)).
These results and theory, in which prion-like mechanisms are proposed to underlie neurodegenerative disorders, fueled speculation about the interpretation of two long-term therapeutic transplantation studies published in 2008 (Kordower et al. 2008; Li et al. 2008). These authors discovered Lewy pathology at autopsy not only in the host neurons of the people with PD but also in young grafted neurons less than two decades of age. At the time, we hypothesized that prion-like transfer of α-syn might underlie the unexpected pathology (Li et al. 2008; Brundin et al. 2008). In fact, earlier work by El Agnaf and colleagues had demonstrated the presence of extracellular α-syn in human plasma and cerebrospinal fluid, thus α-syn could conceivably enter cells from extracellular space (El-Agnaf et al. 2003). These results and many subsequent in vivo studies gave support to the prion-like hypothesis of α-syn transfer from cell to cell (Table 1) (Desplats et al. 2009; Kordower et al. 2011; Luk et al. 2012b; Mougenot et al. 2012; Luk et al. 2012a; Ulusoy et al. 2013; Recasens et al. 2014; Holmqvist et al. 2014; Peelaerts et al. 2015; Paumier et al. 2015; Helwig et al. 2016; Koller et al. 2017; Ulusoy et al. 2017; Abdelmotilib et al. 2017).
Table 1:
Rodent and non-human primate models demonstrating α-syn transmission and neurologic dysfunction
| Animal Model | Pathological α-syn* inoculation site | Timeline post-inoculation | α-syn pathology, neurodegeneration or neurologic dysfunction | Reference(s) |
|---|---|---|---|---|
| WT mice | Intranigral (rAAV of hA53T) | 1, 3, 6 months | Yes | (St Martin et al. 2007) |
| WT rats | Intranigral (rAAV of hA53T) | Varying times up to ~6.75 months | Yes | (Kirik et al. 2002; Yamada et al. 2004) |
| WT mice | Intranigral, intraamygdala and intrastriatal (rAAV of A30P) | Varying times up to 12 months | Yes | (Lauwers et al. 2003) |
| WT rats | Intranigral (rAAV of hA53T, A30P) | 0.75, 1.5, 5 months | Yes | (Lo Bianco et al. 2002) |
| WT rats | Intranigral and intrastriatal (rAAV of α-syn) | Varying times up to 18 months | Yes | (Peelaerts et al. 2015) |
| WT rats | Intranigral and intracerebral (rAAV of α-syn) | ~1.25 months | Yes | (Kordower et al. 2011) |
| WT rats | Intranigral (rAAV of α-syn) | Varying times up to ~1 months | Yes | (Angot et al. 2012) |
| WT Marmosets | Intranigral (rAAV of hA53T) | ~ 4 months | Yes | (Kirik et al. 2003) |
| Mice overexpressing hα-syn | Intrahippocampal (rAAV of α-syn) | Varying times up to 4 months | Yes | (Spencer et al. 2017) |
| WT rats | Vagus nerve (rAAV of α-syn) | Varying times up to ~4.5 months | Yes | (Ulusoy et al. 2013, 2015, 2017) |
| SNCA−/− mice | Vagus nerve (rAAV of α-syn) | Varying times up to 3 months | Yes | (Helwig et al. 2016) |
| WT Marmosets | Intracerebral (rAAV of hA53T) | Varying times up to 12 months | Yes | (Eslamboli et al. 2007) |
| WT mice and macaque monkeys | Intranigral and intrastriatal | Varying times up to ~17 months | Yes | (Recasens et al. 2014) |
| WT mice | Intranigral | 15 months | Yes | (Masuda-Suzukake et al. 2013, 2014) |
| WT mice | Intrastriatal | 1, 6 months | Yes | (Luk et al. 2012a) |
| WT rats | Intrastriatal | 1, 2, 6 months | Yes | (Paumier et al. 2015) |
| WT rats and mice | Intrastriatal | ~ 6 months | Yes | (Abdelmotilib et al. 2017) |
| #Tg(SNCA) Snca0/0 mice | Intrastriatal | 3, 6, 9 months | Yes | (Bernis et al. 2015) |
| WT mice | Intra-OB | Varying times up to 12 months | Yes (olfactory deficits) | (Rey et al. 2013, 2016b) |
| WT rats and mice | Intracerebral | Varying times up to ~1 months | Yes | (Reyes et al. 2014) |
| TgM83 (overexpressing A53T) mice | Intracerebral | 1, 3 months | Yes | (Luk et al. 2012b) |
| TgM83+/+ mice | Intracerebral | Varying times up to ~11 months | Yes | (Mougenot et al. 2012) |
| TgM83+/+ ; TgM83+/− mice | Intracerebral | ~4 months | Yes | (Prusiner et al. 2015) |
| TgM20 mice overexpressing hα-syn | Intracerebral | 1, 2, 4, 8 months | Yes | (Sacino et al. 2013) |
| hα-syn overexpressing mice | Intracerebral | Varying times up to ~13 months | Yes | (Hansen et al. 2011; Osterberg et al. 2015) |
| TgM83+/+ ; TgM83+/− mice | Intracerebral | Varying times up to ~12 months | Yes | (Watts et al. 2013) |
| TgM83 (A53T mutant); TgM47 (E46K mutant) mice | Intracerebral and intrahippocampal | 2, 4 months | Yes | (Sacino et al. 2014a) |
| hα-syn-overexpressing mice | Intrahippocampal | Varying times up to ~1 months | Yes | (Desplats et al. 2009) |
| WT mice | Intrahippocampal | 2, 4 months | Yes | (Koller et al. 2017) |
| TgM83+/+ ; TgM83+/− ; TgM20+/− mice | Intramuscular | Varying times up to 12 months | Yes | (Sacino et al. 2014b) |
| TgM83+/+ ; TgM83+/− mice | Intraperitoneal and intracerebral | Varying times up to 14 months | Yes | (Sargent et al. 2017) |
| Tg(M83+/− ; Gfap-luc+/−) mice | Intraperitoneal and intraglossal | Varying times up to 14 months | Yes | (Breid et al. 2016) |
| WT rats | Intestinal wall | 12, 17, 48 hours | Yes | (Holmqvist et al. 2014) |
WT, Wild type; hα-syn, human α-syn, rAAV, recombinant adeno-associated virus; hA53T, Human A53T; intra-OB, Intra-olfactory bulb.
Pathological α-syn includes recombinant oligomers and PFFs, as well as brain extracts from sick animals or human patients.
These mice express human wild-type α-syn on a mouse α-syn null background.
In this review, we recognize that the spread of α-syn pathology from one cell to another and even one nervous system structure to another in vivo have already been extensively and convincingly summarized in prior articles and therefore will not be discussed in detail here again (Walker 2016; Dehay et al. 2016; Peelaerts and Baekelandt 2016; Goedert et al. 2017; Valdinocci et al. 2017; George and Brundin 2017; Hasegawa et al. 2017; Stopschinski and Diamond 2017). However, whether this transmission of pathology is the explanation for Braak’s pathological observations is yet unclear. Therefore, the aim of our article is to highlight and discuss some of the outstanding questions that can help define in the future whether prion-like transfer plays an important role in the progression of PD. Specifically we focus on three issues: do non-neuronal (i.e. glial) cells contribute to propagation of pathology; are certain brain areas selectively vulnerable to the prion-like propagation of α-syn aggregates and does the presence of α-syn aggregates explain the worsening and broadening of symptoms as PD progresses.
Do non-neuronal cells play a role in neuron to neuron transmission?
The major risk factor for PD is aging, but genetic predisposition and environmental insults also play key roles (Figure 1) (reviewed in (Hernandez et al. 2016; George and Brundin 2017; Stopschinski and Diamond 2017; Collier et al. 2017)). The brain environment during aging is characterized by inflammation which involves non-neuronal, proliferating, and circulating cells such as microglia, astrocytes, and oligodendrocytes (reviewed in (Chinta et al. 2013)). During aging, these non-neuronal cells release pro-inflammatory agents, which potentially compromises both the function and survival of the neurons that they support (Franceschi et al. 2007; Chung et al. 2009). An age-related elevated inflammatory status might be very important in the context of intercellular protein transfer, since microglia, oligodendrocytes and astroglia (which are all impacted by inflammatory stimuli) are suggested to modulate cell-to-cell transfer of α-syn (reviewed in (Lee et al. 2014)). In this regard, oligodendrocytes, astrocytes, and microglia can take up α-syn from the extracellular space in rodent and organotypic slice models (Reyes et al. 2014; Thakur et al. 2017; Loria et al. 2017). In addition, astrocytes appear to sequester and degrade α-syn assemblies (Loria et al. 2017). It is conceivable that inflammation-induced changes in glia impair both their efficacy to take up extracellular α-syn and their ability to degrade it. Indeed, a recent study has demonstrated that although astrocytes take up a significant amount of aggregated α-syn (i.e. α-syn oligomers) for subsequent degradation, their degradative capacity can become overwhelmed, resulting in limited clearance of α-syn and its associated toxic cellular effects (Lindström et al. 2017). Mechanistically, another study has suggested that overburdened astrocytes can transfer excess aggregated α-syn to other nearby astrocytes either through direct contact or through tunneling nanotubes (Rostami et al. 2017). Moreover, similar to neurons, astrocytes and oligodendrocytes utilize micro-vesicles or exosomes as another secretory mechanism for the bidirectional transfer of cellular organelles such as mitochondria, as well as toxic materials (Frühbeis et al. 2013; Hayakawa et al. 2016). It is possible that exosomes may have a role in the transfer of pathogenic α-syn between neuronal and non-neuronal cells. Indeed, Danzer and colleagues demonstrated that α-syn oligomers can be secreted to the extracellular space in exosomes of neurons and that extracellular exosome-containing α-syn oligomers can be taken up rapidly and induce significant toxicity (Danzer et al. 2012). Both neuron-derived and glia-derived exosomes containing α-syn that are secreted into conditioned medium can be taken up by glial cells and may seed the aggregation of intracellular proteins (Surgucheva et al. 2012; Chang et al. 2013; Chistiakov and Chistiakov 2017). Overall, the spread of excess aggregated α-syn between neuronal and glial cells can contribute to increased propagation and cellular effects of the aggregated protein.
Figure 1: Mechanisms of α-syn aggregation and propagation.
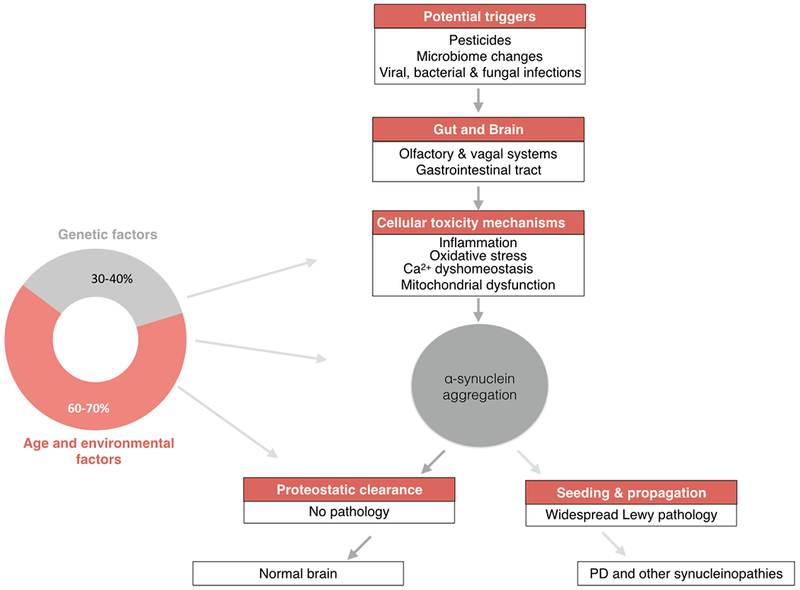
(Left) Schematic showing presumed contributions of different factors to the development of Parkinson’s disease and demonstrating how they influence various pieces of the pathway to Lewy pathology (Keller et al. 2012). (Right) Schematic of development of Lewy pathology by step-wise progression from potential triggers, proposed entry sites and mechanisms that may promote α-syn aggregation to the resultant effects of such aggregation. In a more likely scenario, the system can use proteostatic clearance successfully to remove aggregates. However, when proteostatic clearance is impaired and α-syn aggregation and accumulation proceeds unchecked, the initial aggregates may be transferred in a ‘prion-like’ manner and may seed further aggregation, resulting in widespread Lewy pathology that contributes to the development of PD and other synucleinopathies.
In addition, both astrocytes and microglia are also clearly capable of migrating over long distances in the adult brain and in the periphery. Animal experiments have suggested that microglia which carry engulfed aggregated tau (another dysfunctional protein similar to α-syn) can contribute to spread of tauopathy by migrating to other brain regions and releasing the aggregates there (Asai et al. 2015). Conceivably, both microglia and astrocytes could play similar roles in the long-distance spread of α-syn pathology. Long-distance transit is controversial, given emerging evidence that the spread of α-syn aggregates follows anatomical pathways in PD (see next section). However, as we elaborate upon in the next section, the notion that the spread of protein aggregates strictly follows neural tracts has also been recently questioned. Indeed, preformed aggregates of both α-syn and tau can spread beyond synaptic connections or anatomical pathways (de Calignon et al. 2012; Sacino et al. 2014a; Peeraer et al. 2015; Asai et al. 2015), suggesting that non-synaptic propagation mechanisms may also exist. Circulating glial cells, particularly microglia, may be key players in such long-distance spread of pathology. For instance, aggregated tau, like α-syn, can be carried by microglia and transduced into cells in other locations in vivo, promoting aggregated protein propagation (Asai et al. 2015). This form of aggregated protein propagation may be inhibited following the depletion of microglia (Asai et al. 2015). In addition, in some models of synucleinopathies, aggregated α-syn injected into the intestinal walls and peritoneal cavity propagates and seeds further aggregation in several parts of the brain (see Table 1). While the mechanism by which this aggregated α-syn is transmitted from the gut into the brain remains to be established, the vagal nerve has long been suggested as an entry site and a possible route by which α-syn may gain access into the brain from the periphery (Braak et al. 2003b). Although it remains to be supported, a role for microglia and other immune cells in such periphery-to-CNS propagation of aggregated α-syn cannot be discounted. Indeed, the transmission of α-syn from the periphery into the brain is often accompanied by increased microglial activity and neuroinflammation (Breid et al. 2016). To sum up, circulating glial cells may play various roles in the spread of excess aggregated α-syn, either over short or long distances.
With respect to the connectome, is there a difference in propagation from different brain areas and cell populations?
The connectome is a comprehensive brain map of the intricate synaptic connections formed between neurons. Functional connectivity changes (i.e. fMRI or specific tracer signal alterations) might track the overall and specific pathological changes in PD, including the spread of α-syn and Lewy pathology or cell death in the substantia nigra pars compacta and other relevant brain areas (Watabe-Uchida et al. 2012; Ogawa et al. 2014; Bellucci et al. 2016).
However, several gaps currently exist in the rationale that functional connectivity changes track specifically with most pathological changes that occur in PD (reviewed in (Surmeier et al. 2017)). Surmeier and coauthors posited that neurons in the substantia nigra pars compacta and other vulnerable neurons possess common anatomical and physiological properties, which might explain not only the pattern of cell death in PD but also the arrangement of Lewy bodies and neurites (Surmeier et al. 2017). Indeed, an elevated vulnerability of substantia nigra pars compacta neurons in PD has long been considered, but other factors that might help connect pathology with functionality are still not verified (Surmeier et al. 2017). While work to fill in the gaps of knowledge continues in animal models of PD and in people with PD, we eagerly await a valid biomarker to track progression of pathology in human PD patients. Essentially, in the absence of a high-resolution in vivo imaging marker for aggregated α-syn (Eberling et al. 2013), it is not possible to define precisely how the Lewy pathology progresses in one individual, and instead currently we have to rely on cross sectional studies describing different patients of varying clinical stages and disease durations. Animal models of PD, with all their possible shortcomings, allow us to examine identically treated and genetically identical animals at different time points after triggering an experimental synucleinopathy, and therefore are the best option for increasing our understanding of how Lewy pathology spreads. Below, we review the animal model-based evidence of propagation of fibrillar α-syn from one brain area to another, and attempt to relate it to connectivity.
We begin with the most caudal site interrogated at this time – the intraperitoneal route of administration. Breid and colleagues injected fibrillar α-syn assemblies (PFFs) via intraperitoneal and intraglossal routes into A53T α-syn-expressing bigenic transgenic (M83+/−:Gfap-luc+/−) mice, and found that intraperitoneal injection of PFFs led to paralysis and the appearance of phosphorylated α-syn pathology in the central nervous system (Breid et al. 2016). Sargent and colleagues also used M83 transgenic mice in combination with intracerebral or systemic (intraperitoneal) injection of brain homogenates from sick mice to demonstrate that the type of inoculum and the genotype (hemizygous vs. homozygous) of the mouse determine the pathology load (phosphorylated α-syn) in the brain (Sargent et al. 2017). Finally, Ulusoy and colleagues performed injections of adeno-associated virus (AAV) 2/6 expressing human α-syn into rat midbrain, which led to the presence of human α-syn in vagal motor neurons and in gastric nerve endings of visceromotor vagal projections (Ulusoy et al. 2017). Taken together, all these results suggest that the dorsal motor nucleus of the vagal nerve could be critical in inducing development of pathology in rostral locations of the brain.
Additionally, the Di Monte group also performed more rostral injections of AAV2/6-human α-syn in the upper vagal nerve and the brain stem of rodents (Ulusoy et al. 2013, 2015; Helwig et al. 2016), and demonstrated that, in each model, phosphorylated- and thioflavin S-labeled α-syn lesions were present throughout the brainstem and forebrain of recipient mice, respectively. In animals where there was inadvertent toxicity due to the virus, which killed brainstem neurons, there was no progression of pathology, indicating that the presence of intact neural connections is a prerequisite for propagation of pathology (Ulusoy et al. 2015). Holmqvist and colleagues similarly demonstrated rostral presence of aggregated α-syn pathology in the dorsal motor nucleus of the vagal nerve after injection of a PD brain lysate into the intestinal wall (Holmqvist et al. 2014). In general, these results lend support to the idea that the dorsal motor nucleus of the vagal nerve is central to α-syn pathology transfer to and from the CNS. The evidence for involvement of dorsal motor nucleus of the vagal nerve in the pathological “network” in PD is worth reviewing. It appears to be both a site of α-syn pathology in PD (Braak et al. 2003a) and a nucleus that contains possible vulnerable neurons with long and branched axons (Surmeier et al. 2017). More research is needed to verify that neurons within dorsal motor nucleus of the vagal nerve are indeed vulnerable to either cell death, or development of α-syn pathology, or both.
Next, we will examine the evidence that α-syn pathology transfers from or appears in the mid- and forebrain after introduction from exogenous sources. One of the first examples of host-to-graft transfer of α-syn in an animal model occurred after transplantation of neural stem cells into the hippocampus of a mouse expressing α-syn (Desplats et al. 2009). Soon after, two groups set up rodent models of protein uptake and host-to-graft transfer, and found that naïve transplanted rodent neurons import human α-syn in a variety of models (Hansen et al. 2011; Kordower et al. 2011; Angot et al. 2012). Other groups have injected patient material or oligomeric/fibrillar α-syn into hippocampus and substantia nigra and found aggregated α-syn lesions (Recasens et al. 2014; Peelaerts et al. 2015; Koller et al. 2017; Abdelmotilib et al. 2017). Even more severe α-syn pathology was found when α-syn was overexpressed and PFFs were injected into the nigra (Thakur et al. 2017). Similar aggregated α-syn results have been observed after injection of PFFs, patient material, and α-syn assemblies into the striatum (Luk et al. 2012a; Recasens et al. 2014; Peelaerts et al. 2015) and cortex (Osterberg et al. 2015). Finally, our group has also shown that PFFs injected into the olfactory bulb of mice led to olfactory deficits and widespread α-syn inclusions throughout the forebrain and midbrain (Rey et al. 2016b).
Regarding the “connectomic” spread of α-syn pathology to distinct brain regions from the injection site, we shall focus our discussion on findings emerging from animal models that received α-syn injections at four commonly investigated sites including: a) vagus nerve, b) substantia nigra, c) striatum, and d) cortex/olfactory bulb (see Table 1). In general, aggregated α-syn has been reported to spread from neurons in the vagus nerve (or medulla oblongata) along stereotypical neural tracts to more rostral brain areas such as pons, locus coeruleus, dorsal raphe, hypothalamus and amygdala twelve months after α-syn injection (Ulusoy et al. 2013, 2015; Helwig et al. 2016). From the substantia nigra, the aggregated protein similarly spreads along neural tracts to multiple brain areas such as amygdala, striatum, hippocampus, dentate gyrus, hypothalamus and the visual, motor, entorhinal, and cingulate cortex (Masuda-Suzukake et al. 2013, 2014; Peelaerts et al. 2015). Similar to those injections in the nigra, injections of aggregated α-syn into the striatum resulted in inclusion formation that spread to several brain areas that project afferent innervations to the striatum, such as the prefrontal, insular, cingulate and motor cortical areas, as well as the substantia nigra, amygdala and olfactory bulb (Luk et al. 2012a; Paumier et al. 2015; Bernis et al. 2015; Abdelmotilib et al. 2017). Moreover, intracerebral injections of α-syn into the cortical areas results in pathological spreading from the cortex to the striatum, thalamus, hypothalamus, locus coeruleus, raphe nucleus, reticular formation, cerebellum, and the spinal cord (Luk et al. 2012b; Mougenot et al. 2012; Watts et al. 2013). Being one of the main predicted entry sites for aggregated α-syn, the olfactory bulb may be quite critical for α-syn propagation (Rey et al. 2016c). Indeed, olfactory bulb injections trigger the spread of aggregated α-syn in an anatomical pattern across several brain areas including the frontal, entorhinal, perirhinal, and parietal cortex, as well as the striatum, amygdala, substantia nigra, and hippocampus (Rey et al. 2016b). Overall, the findings from these studies clearly support the notion that pathological α-syn can spread from neuron to neuron and region to region. It is however, less clear from these in vivo studies, whether there is selective vulnerability and spread of α-syn pathology in some neuronal cell groups over others. What these in vivo studies appear to suggest is that aggregated α-syn, and for that matter Lewy pathology, might spread along defined neural tracts or interconnected brain networks. However, this has so far not been corroborated by data from clinical PD cases, and as outlined above, this will be difficult to do unless a sensitive and specific clinical imaging ligand that identifies α-syn aggregates with very high resolution is developed (Surmeier et al. 2017).
Arguments can be advanced that if indeed Lewy pathology spreads from neuron to neuron along defined interconnected tracts in clinical PD (as is the case in animal models), then from dopaminergic neurons in the substantia nigra pars compacta the pathology must spread to regions robustly innervating the nigral neurons (such as the substantia nigra pars reticulata, subthalamic nuclei and globus pallidus). Yet these regions often have limited pathology, if any (Surmeier et al. 2017). It would appear that Lewy pathology in clinical PD cases may exist in selective cell populations/brain areas, rather than having a widespread presence in the entire brain. Until clinical confirmation, therefore, we cannot say with certainty that the pathological spread of aggregated α-syn between interconnected brain regions from injection/seeding sites observable in the outlined animal models can adequately explain the selective propagation observed in clinical PD cases. Further studies addressing the question of selective propagation of pathological α-syn in both animal models and clinical PD cases may be required, together with more precise animal models and validated methods (Rey et al. 2016a). Assuming that aggregated α-syn could spread from one neuron to another and one brain area to another in both animal models and clinical PD, are there functional consequences or adequate explanations of PD progression? These questions are discussed in the next section.
Does the aggregation and propagation of α-syn correlate with increased PD pathology and symptomatology?
Neuronal loss is a major feature of clinical PD. Nigrostriatal dopaminergic neuronal cell loss, in particular, results in the motor features of PD (Cheng et al. 2010; Spillantini and Goedert 2017). In human PD patients, approximately 30% of nigral dopaminergic neurons, along with 50-60% of their synaptic terminals are lost at the time of onset of motor symptoms (Cheng et al. 2010; Spillantini and Goedert 2017). Major unresolved questions arise as to whether the progressive accumulation of α-syn aggregates contributes to neuronal cell loss in PD and what cellular mechanisms would underlie such a phenomenon (Lashuel et al. 2013; Wong and Krainc 2017). Although Lewy pathology is found in most cases of clinical PD, it is currently quite difficult to tease apart the contribution of only α-syn aggregates towards disease progression in PD patients. Thus, animal models are quite useful in this context for investigating the effect of α-syn aggregates independent of other factors that are also known to influence PD progression.
Several studies conducted on animal models of PD have linked aggregated recombinant α-syn with cytotoxicity. Such PD models often carry mutations in the α-syn gene that favor its aggregation such as the A53T, E46K and A30P point mutations detected in familial PD cases (Bisaglia et al. 2009). While some studies have examined the effect of α-syn aggregation in these point mutation models, others have further injected some of these models with α-syn oligomers and PFFs (Table 1). In particular, studies on animal models of PD and other synucleinopathies have primarily focused on the propagation of α-syn aggregates and therapeutic targeting of this propagation. Thus, much less attention has been given to the in vivo toxicity of α-syn aggregates. Regardless of this, there appears to be enough studies to provide a good picture of the effect of pathogenic α-syn aggregation in vivo. Importantly, in such in vivo studies, α-syn is often administered at higher levels than it is likely present even in a brain affected by a synucleinopathy and the toxic effects are assessed within that context. Drosophila models represent one of the simplest in vivo models used to show that the expression of human α-syn can indeed induce neuronal toxicity. Using such models for investigation, dopaminergic neuron cell loss has frequently been reported (Chen and Feany 2005; Haywood and Staveley 2006; Park and Lee 2006; Kontopoulos et al. 2006; Periquet et al. 2007), although the effects appeared to be quite modest. The nematode C. elegans has also been used to demonstrate that α-syn aggregates do not only spread from neuron to neuron but are also very toxic to dopaminergic neuronal cells and can cause motor deficits in these models (Petrucelli et al. 2002; Lakso et al. 2003; Kuwahara et al. 2006; Tyson et al. 2017). Using transgenic technology and viral vector delivery approaches, rodent and non-human primate mutant α-syn-induced aggregation models have also been generated. Studies introducing α-syn into these animal models using the traditional transgenics approach has produced conflicting results with some evidence for high α-syn toxicity in various parts of brain and the spinal cord, whereas other studies demonstrated moderate to no cell loss in the substantia nigra (Masliah et al. 2000; van der Putten et al. 2000; Kahle et al. 2000; Matsuoka et al. 2001; Richfield et al. 2002; Giasson et al. 2002; Lee et al. 2002). To provide more clarity, the viral vector delivery and stereotaxic PFF injection approaches were adapted to directly deliver α-syn to specific brain areas that are relevant to clinical PD such as the dorsal motor nucleus of the vagus nerve, substantia nigra, striatum and olfactory bulb (as discussed above). Data from this approach have been rather promising with most studies showing the deposition and spread of α-syn, and others demonstrating significant neuronal cell loss in relevant brain areas in mice and rats (Table 1) (Kirik et al. 2002; Giasson et al. 2002; Lo Bianco et al. 2002; Lauwers et al. 2003; Yamada et al. 2004; St Martin et al. 2007) and primates (Kirik et al. 2003; Eslamboli et al. 2007). For example, one study on adult marmosets and monkeys reported α-syn inclusion formation, 30-60% nigral and striatal dopaminergic cell loss, and severe motor impairments 16 weeks after viral vector mediated delivery of human α-syn into the substantia nigra. In these models, the nigrostriatal synucleinopathy developed slowly over time, reminiscent of clinical PD (Kirik et al. 2003). Compared to human PD however, the cell loss and behavioral deficits observed in most of these animal models may be less pronounced. Moreover, the relative toxicity of α-syn in these models is suggested to be detected much more rapidly than is the case in clinical PD (Surmeier et al. 2017). Despite the apparent limitations in studies on animal models, the important observation is that α-syn does not only propagate from neuron to neuron in these models but that it can also evoke toxicity in neuronal cells in the in vivo context when present at non-physiological levels. It is however less clear if the toxicity of human α-syn in cellular and animal models are directly relevant in clinical PD.
To verify α-syn toxicity in clinical PD and to determine whether Lewy pathology precedes cellular dysfunction and neurodegeneration, a few studies have been conducted on PD patients and on postmortem tissues (these studies are rather challenging to perform in clinical cases due to current technological limitations). The findings from these studies have been inconclusive. In this regard, some of these studies have examined toxicity in brain areas having significant Lewy pathology and reported nigral dopamine cell loss (Halliday et al. 1996; Damier et al. 1999; Milber et al. 2012; Dijkstra et al. 2014), as well as a modest loss of glutamatergic neurons in the basolateral amygdala and thalamus (Henderson et al. 2000; Harding et al. 2002) and cholinergic neurons in the peduncolopontine nucleus (Halliday et al. 1990) in early clinical PD cases. In late clinical PD cases, there appears to be extensive neuronal cell loss in α-syn inclusion-rich areas including a significant loss of neurons in parts of the hypothalamus (Halliday et al. 1990; Kremer and Bots 1993; Thannickal et al. 2007; Fronczek et al. 2008). In contrast, some studies have failed to observe significant neuronal loss in brain areas that had α-syn inclusions and Lewy pathology including the motor cortex and neocortical areas (Halliday et al. 1990; Ansorge et al. 1997; MacDonald and Halliday 2002; Pedersen et al. 2005). In addition, one study reported no significant glutamatergic or GABAergic cell loss in the pedunculopontine nucleus (Halliday et al. 1990). There are also indications that some PD patients with widespread α-syn inclusions and Lewy pathology display no observable neuropsychiatric and motor symptoms (Jellinger 2009). In addition, there has been a report that significant neuronal cell loss and cellular dysfunction precede α-syn and Lewy pathology in some PD cases who are in Braak PD stages 1 and 2 (Milber et al. 2012). Taken together, the correlation between Lewy pathology and neuronal cell loss in clinical PD is not yet clear. Development of robust technology and biochemical approaches (e.g. biomarkers that can reliably track pathology development and disease progression in humans) may be useful in clarifying this. However, it must be mentioned that recent studies have identified polymorphs (i.e. different strains) of aggregated α-syn with the oligomeric and fibrillar strains being particularly toxic (Stöckl et al. 2013; Peelaerts et al. 2015; Walker 2016). Thus, not all α-syn inclusions may lead to pathology. It is currently uncertain if these polymorphs do exist in clinical PD, if it does it could at least explain in part the possible disconnect between Lewy pathology and neuronal cell loss reported in some clinical PD cases. In addition, α-syn levels that are too low to cause disease may also fail to result in observable pathological changes. So, while there could be detectable misfolded α-syn to seed aggregation in patients, there may not necessarily be enough to cause severe detectable neuronal cell loss and associated neurological dysfunction. Moreover, in humans, unlike in animal models, it is possible that it takes much longer for enough α-syn to aggregate to significant levels that could cause appreciable pathology (Tamgüney and Korczyn 2017).
Given that various lines of evidence from rodent models to human PD cases position aggregated α-syn as a potentially toxic protein in synucleinopathies, the logical question that emerges is how, and in which context would the aggregated protein be toxic to neuronal cells (for review, see (Wong and Krainc 2017)). The toxicity of α-syn may be induced through several mechanisms: a) binding and inhibition of lysosomal function (Cuervo et al. 2004; Flavin et al. 2017), b) inhibition of proteasome activity (Tanaka et al. 2001; Stefanis et al. 2001; Petrucelli et al. 2002; Lindersson et al. 2004; Smith et al. 2005), c) induction of oxidative stress, Ca2+ dyshomeostasis and mitochondrial dysfunction (Cappai et al. 2005; Smith et al. 2005; Surmeier et al. 2017), and d) pathogenic redistribution of membrane proteins (Shrivastava et al. 2015; Mao et al. 2016; Shrivastava et al. 2017). In particular, α-syn oligomer and A53T overexpression in cultured cells significantly increase oxidative stress by enhancing the intracellular levels of reactive oxygen species (ROS) (Cappai et al. 2005; Smith et al. 2005; Reeve et al. 2015). The generation of ROS promotes mitochondrial and neuronal damage. In nigral neurons, a major source of ROS is the metabolism of dopamine. Interestingly, exposure of α-syn mutant cells to dopamine results in increased cell loss (Cappai et al. 2005). Perhaps, this could be related to the increased vulnerability of dopaminergic cells to α-syn induced toxicity. In addition, α-syn in A53T mutant cells may, in some conditions localize to the mitochondria, where it causes the release of cytochrome c from the mitochondria (Smith et al. 2005; Parihar et al. 2008). The accumulation of cytochrome c in the cytosol of cultured cells has been associated with increased activities of caspase 3 and 9 (Smith et al. 2005). Moreover, in line with its effect on mitochondrial homeostasis, the expression of α-syn in A53T mutants have been strongly associated with higher concentrations of mitochondrial Ca2+ levels and increased DNA damage (Martin et al. 2006; Chen et al. 2015). Abnormally high levels of mitochondrial Ca2+ triggers apoptosis. Therefore, α-syn may induce apoptotic cell degradation in a mitochondrial-linked pathway and caspase activation (Smith et al. 2005; Parihar et al. 2008), making mitochondria cellular targets of α-syn accumulation and neurotoxicity (Di Maio et al. 2016; Wong and Krainc 2017; Tapias et al. 2017). A mitochondrial-independent pathway has also been suggested, whereby α-syn oligomers are thought to interfere with the normal functions of cell membranes and form pore-like structures in lipid bilayers that leads to abnormal Ca2+ influx and subsequent neuronal cell damage (Tsigelny et al. 2012). In addition, strong interactions between fibrillar α-syn assemblies and the plasma membrane might disrupt the Na+ gradient (Shrivastava et al. 2015). Overall, it appears that multiple systems can be affected by α-syn aggregation and accumulation, which can trigger cellular pathways that results in neuronal cell loss. This view links primary protein aggregation to cellular targets of the protein. Therefore, determining which events are actually primary (i.e. α-syn aggregation vs. mitochondrial, proteasomal and lysosomal dysfunction) and which ones are secondary would be crucial for deciding the most important aspects of the cell death process that should be targeted to hinder disease progression (Wong and Krainc 2017; Brundin and Melki 2017; Surmeier et al. 2017).
Conclusions
Given that this field offers many reviews on the prion-like spread of α-syn pathology, we focused on unaddressed and under published questions. Our goal was to direct prion-like attention to the role of non-neuronal cells, the “connectomic” propagation, and whether α-syn pathology always begets dysfunction. Although we find that there is still a lack of tools to help connect pathology to dysfunction and disease state in clinical PD cases, we express optimism for the discovery of biomarkers that can reliably track pathology development and disease progression in humans.
Acknowledgement
We acknowledge the Van Andel Research Institute and the many individuals and corporations that financially support research into neurodegenerative diseases at Van Andel Research Institute. P.B. is supported by grants from the National Institutes of Health (1R01DC016519-01; 1R21NS105436-01; 5R21NS093993-02) and the Department of Defense (W81XWH-17-1-0535). P.B. also reports additional grants from The Michael J Fox Foundation, National Institutes of Health, and Cure Parkinson’s Trust UK.
Footnotes
Conflicts of Interest
P.B. has received commercial support as a consultant from Renovo Neural, Inc., Roche, Cellular Dynamics International Inc, Teva Inc, Lundbeck A/S, AbbVie, ClearView Healthcare, FCB Health, IOS Press Partners, and Capital Technologies, Inc. Additionally, he has received commercial support for grants/research from Renovo and Teva/Lundbeck. Furthermore, P.B. has ownership interest in Acousort AB. J.A.S. and E.Q. report no conflicts of interest.
References
- Abdelmotilib H, Maltbie T, Delic V, Liu Z, Hu X, Fraser KB, Moehle MS, Stoyka L, Anabtawi N, Krendelchtchikova V, Volpicelli-Daley LA, West A (2017) α-Synuclein fibril-induced inclusion spread in rats and mice correlates with dopaminergic Neurodegeneration. Neurobiol Dis 105:84–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angot E, Steiner JA, Tomé CML, Ekström P, Mattsson B, Björklund A, Brundin P (2012) Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. Plos One 7:e39465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansorge O, Daniel SE, Pearce RK (1997) Neuronal loss and plasticity in the supraoptic nucleus in Parkinson’s disease. Neurology 49:610–613 [DOI] [PubMed] [Google Scholar]
- Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kügler S, Ikezu T (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18:1584–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellucci A, Mercuri NB, Venneri A, Faustini G, Longhena F, Pizzi M, Missale C, Spano P (2016) Review: Parkinson’s disease: from synaptic loss to connectome dysfunction. Neuropathol Appl Neurobiol 42:77–94 [DOI] [PubMed] [Google Scholar]
- Bernis ME, Babila JT, Breid S, Wüsten KA, Wüllner U, Tamgüney G (2015) Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol Commun 3:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisaglia M, Mammi S, Bubacco L (2009) Structural insights on physiological functions and pathological effects of alpha-synuclein. FASEB J 23:329–340 [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E (2003a) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211 [DOI] [PubMed] [Google Scholar]
- Braak H, Rüb U, Gai WP, Del Tredici K (2003b) Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm 110:517–536 [DOI] [PubMed] [Google Scholar]
- Breid S, Bernis ME, Babila JT, Garza MC, Wille H, Tamgüney G (2016) Neuroinvasion of α-synuclein prionoids after intraperitoneal and intraglossal inoculation. J Virol 90:9182–9193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundin P, Li J-Y, Holton JL, Lindvall O, Revesz T (2008) Research in motion: the enigma of Parkinson’s disease pathology spread. Nat Rev Neurosci 9:741–745 [DOI] [PubMed] [Google Scholar]
- Brundin P, Melki R (2017) Prying into the prion hypothesis for Parkinson’s disease. J Neurosci 37:9808–9818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappai R, Leck S-L, Tew DJ, Williamson NA, Smith DP, Galatis D, Sharples RA, Curtain CC, Ali FE, Cherny RA, Culvenor JG, Bottomley SP, Masters CL, Barnham KJ, Hill AF (2005) Dopamine promotes alpha-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J 19:1377–1379 [DOI] [PubMed] [Google Scholar]
- Chang C, Lang H, Geng N, Wang J, Li N, Wang X (2013) Exosomes of BV-2 cells induced by alpha-synuclein: important mediator of neurodegeneration in PD. Neurosci Lett 548:190–195 [DOI] [PubMed] [Google Scholar]
- Chen L, Feany MB (2005) Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci 8:657–663 [DOI] [PubMed] [Google Scholar]
- Chen L, Xie Z, Turkson S, Zhuang X (2015) A53T human α-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J Neurosci 35:890–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H-C, Ulane CM, Burke RE (2010) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67:715–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Lieu CA, Demaria M, Laberge R-M, Campisi J, Andersen JK (2013) Environmental stress, ageing and glial cell senescence: a novel mechanistic link to Parkinson’s disease? J Intern Med 273:429–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakov DA, Chistiakov AA (2017) α-Synuclein-carrying extracellular vesicles in Parkinson’s disease: deadly transmitters. Acta Neurol Belg 117:43–51 [DOI] [PubMed] [Google Scholar]
- Chung HY, Cesari M, Anton S, Marzetti E, Giovannini S, Seo AY, Carter C, Yu BP, Leeuwenburgh C (2009) Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res Rev 8:18–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier TJ, Kanaan NM, Kordower JH (2017) Aging and Parkinson’s disease: Different sides of the same coin? Mov Disord 32:983–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305:1292–1295 [DOI] [PubMed] [Google Scholar]
- Damier P, Hirsch EC, Agid Y, Graybiel AM (1999) The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 122:1437–1448 [DOI] [PubMed] [Google Scholar]
- Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ (2012) Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener 7:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT (2012) Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73:685–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, Vila M, Bezard E, Brundin P, Kordower JH (2016) Alpha-synuclein propagation: new insights from animal models. Mov Disord 31:161–168 [DOI] [PubMed] [Google Scholar]
- Desplats P, Lee H-J, Bae E-J, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee S-J (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc Natl Acad Sci USA 106:13010–13015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, Hu X, McCoy J, Chu CT, Burton EA, Hastings TG, Greenamyre JT (2016) α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci Transl Med 8:342ra78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra AA, Voorn P, Berendse HW, Groenewegen HJ, Netherlands Brain Bank, Rozemuller AJM, van de Berg WDJ (2014) Stage-dependent nigral neuronal loss in incidental Lewy body and Parkinson’s disease. Mov Disord 29:1244–1251 [DOI] [PubMed] [Google Scholar]
- Eberling JL, Dave KD, Frasier MA (2013) α-synuclein imaging: a critical need for Parkinson’s disease research. J Park Dis 3:565–567 [DOI] [PubMed] [Google Scholar]
- El-Agnaf OMA, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ, Curran MD, Court JA, Mann DMA, Ikeda S, Cookson MR, Hardy J, Allsop D (2003) Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J 17:1945–1947 [DOI] [PubMed] [Google Scholar]
- Eslamboli A, Romero-Ramos M, Burger C, Bjorklund T, Muzyczka N, Mandel RJ, Baker H, Ridley RM, Kirik D (2007) Long-term consequences of human alpha-synuclein overexpression in the primate ventral midbrain. Brain 130:799–815 [DOI] [PubMed] [Google Scholar]
- Flavin WP, Bousset L, Green ZC, Chu Y, Skarpathiotis S, Chaney MJ, Kordower JH, Melki R, Campbell EM (2017) Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol 134:629–653 [DOI] [PubMed] [Google Scholar]
- Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S (2007) Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev 128:92–105 [DOI] [PubMed] [Google Scholar]
- Fronczek R, Overeem S, Lee SYY, Hegeman IM, van Pelt J, van Duinen SG, Lammers GJ, Swaab DF (2008) Hypocretin (orexin) loss and sleep disturbances in Parkinson’s Disease. Brain 131:e88. [DOI] [PubMed] [Google Scholar]
- Frühbeis C, Fröhlich D, Kuo WP, Krämer-Albers E-M (2013) Extracellular vesicles as mediators of neuron-glia communication. Front Cell Neurosci 7:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S, Brundin P (2017) Solving the conundrum of insoluble protein aggregates. Lancet Neurol 16:258–259 [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM-Y (2002) Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34:521–533 [DOI] [PubMed] [Google Scholar]
- Goedert M, Masuda-Suzukake M, Falcon B (2017) Like prions: the propagation of aggregated tau and α-synuclein in neurodegeneration. Brain 140:266–278 [DOI] [PubMed] [Google Scholar]
- Halliday GM, Li YW, Blumbergs PC, Joh TH, Cotton RG, Howe PR, Blessing WW, Geffen LB (1990) Neuropathology of immunohistochemically identified brainstem neurons in Parkinson’s disease. Ann Neurol 27:373–385 [DOI] [PubMed] [Google Scholar]
- Halliday GM, McRitchie DA, Cartwright H, Pamphlett R, Hely MA, Morris JG (1996) Midbrain neuropathology in idiopathic Parkinson’s disease and diffuse Lewy body disease. J Clin Neurosci 3:52–60 [DOI] [PubMed] [Google Scholar]
- Hansen C, Angot E, Bergström A-L, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li J-Y, Brundin P (2011) α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest 121:715–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding AJ, Stimson E, Henderson JM, Halliday GM (2002) Clinical correlates of selective pathology in the amygdala of patients with Parkinson’s disease. Brain 125:2431–2445 [DOI] [PubMed] [Google Scholar]
- Hardy J (2005) Expression of normal sequence pathogenic proteins for neurodegenerative disease contributes to disease risk: “permissive templating” as a general mechanism underlying neurodegeneration. Biochem Soc Trans 33:578–581 [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Nonaka T, Masuda-Suzukake M (2017) Prion-like mechanisms and potential therapeutic targets in neurodegenerative disorders. Pharmacol Ther 172:22–33 [DOI] [PubMed] [Google Scholar]
- Hawkes CH, Del Tredici K, Braak H (2007) Parkinson’s disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol 33:599–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes CH, Del Tredici K, Braak H (2009) Parkinson’s disease: the dual hit theory revisited. Ann N Y Acad Sci 1170:615–622 [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH (2016) Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535:551–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haywood AFM, Staveley BE (2006) Mutant alpha-synuclein-induced degeneration is reduced by parkin in a fly model of Parkinson’s disease. Genome 49:505–510 [DOI] [PubMed] [Google Scholar]
- Helwig M, Klinkenberg M, Rusconi R, Musgrove RE, Majbour NK, El-Agnaf OMA, Ulusoy A, Di Monte DA (2016) Brain propagation of transduced α-synuclein involves non-fibrillar protein species and is enhanced in α-synuclein null mice. Brain 139:856–870 [DOI] [PubMed] [Google Scholar]
- Henderson JM, Carpenter K, Cartwright H, Halliday GM (2000) Degeneration of the centré median-parafascicular complex in Parkinson’s disease. Ann Neurol 47:345–352 [PubMed] [Google Scholar]
- Hernandez DG, Reed X, Singleton AB (2016) Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J Neurochem 139 Suppl 1:59–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Björklund T, Wang Z-Y, Roybon L, Melki R, Li J-Y (2014) Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol 128:805–820 [DOI] [PubMed] [Google Scholar]
- Jellinger KA (2009) Formation and development of Lewy pathology: a critical update. J Neurol 3:270–279 [DOI] [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, van Der Putten H, Probst A, Kremmer E, Kretzschmar HA, Haass C (2000) Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J Neurosci 20:6365–6373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller MF, Saad M, Bras J, Bettella F, Nicolaou N, Simón-Sánchez J, Mittag F, Büchel F, Sharma M, Gibbs JR, Schulte C, Moskvina V, Durr A, Holmans P, Kilarski LL, Guerreiro R, Hernandez DG, Brice A, Ylikotila P, Stefánsson H, Majamaa K, Morris HR, Williams N, Gasser T, Heutink P, Wood NW, Hardy J, Martinez M, Singleton AB, Nalls MA; International Parkinson’s Disease Genomics Consortium (IPDGC); Wellcome Trust Case Control Consortium 2 (WTCCC2) (2012) Using genome-wide complex trait analysis to quantify ‘missing heritability’ in Parkinson’s disease. Hum Mol Genet 21:4996–5009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Annett LE, Burger C, Muzyczka N, Mandel RJ, Björklund A (2003) Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: a new primate model of Parkinson’s disease. Proc Natl Acad Sci USA 100:2884–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ, Björklund A (2002) Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci 22:2780–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller EJ, Brooks MMT, Golde TE, Giasson BI, Chakrabarty P (2017) Inflammatory pre-conditioning restricts the seeded induction of α-synuclein pathology in wild type mice. Mol Neurodegener 12:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontopoulos E, Parvin JD, Feany MB (2006) Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet 15:3012–3023 [DOI] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 14:504–506 [DOI] [PubMed] [Google Scholar]
- Kordower JH, Dodiya HB, Kordower AM, Terpstra B, Paumier K, Madhavan L, Sortwell C, Steece-Collier K, Collier TJ (2011) Transfer of host-derived alpha synuclein to grafted dopaminergic neurons in rat. Neurobiol Dis 43:552–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer HP, Bots GT (1993) Lewy bodies in the lateral hypothalamus: do they imply neuronal loss? Mov Disord 8:315–320 [DOI] [PubMed] [Google Scholar]
- Kuwahara T, Koyama A, Gengyo-Ando K, Masuda M, Kowa H, Tsunoda M, Mitani S, Iwatsubo T (2006) Familial Parkinson mutant alpha-synuclein causes dopamine neuron dysfunction in transgenic Caenorhabditis elegans. J Biol Chem 281:334–340 [DOI] [PubMed] [Google Scholar]
- Lakso M, Vartiainen S, Moilanen A-M, Sirviö J, Thomas JH, Nass R, Blakely RD, Wong G (2003) Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J Neurochem 86:165–172 [DOI] [PubMed] [Google Scholar]
- Lashuel HA, Overk CR, Oueslati A, Masliah E (2013) The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 14:38–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauwers E, Debyser Z, Van Dorpe J, De Strooper B, Nuttin B, Baekelandt V (2003) Neuropathology and neurodegeneration in rodent brain induced by lentiviral vector-mediated overexpression of alpha-synuclein. Brain Pathol 13:364–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H-J, Bae E-J, Lee S-J (2014) Extracellular α--synuclein-a novel and crucial factor in Lewy body diseases. Nat Rev Neurol 10:92–98 [DOI] [PubMed] [Google Scholar]
- Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL (2002) Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 --> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci USA 99:8968–8973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J-Y, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, Widner H, Revesz T, Lindvall O, Brundin P (2008) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 14:501–503 [DOI] [PubMed] [Google Scholar]
- Lindersson E, Beedholm R, Højrup P, Moos T, Gai W, Hendil KB, Jensen PH (2004) Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biol Chem 279:12924–12934 [DOI] [PubMed] [Google Scholar]
- Lindström V, Gustafsson G, Sanders LH, Howlett EH, Sigvardson J, Kasrayan A, Ingelsson M, Bergström J, Erlandsson A (2017) Extensive uptake of α-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol Cell Neurosci 82:143–156 [DOI] [PubMed] [Google Scholar]
- Lo Bianco C, Ridet J-L, Schneider BL, Deglon N, Aebischer P (2002) alpha-Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson’s disease. Proc Natl Acad Sci USA 99:10813–10818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria F, Vargas JY, Bousset L, Syan S, Salles A, Melki R, Zurzolo C (2017) α-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol 134:789–808 [DOI] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM-Y (2012a) Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VMY (2012b) Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med 209:975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald V, Halliday GM (2002) Selective loss of pyramidal neurons in the pre-supplementary motor cortex in Parkinson’s disease. Mov Disord 17:1166–1173 [DOI] [PubMed] [Google Scholar]
- Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin JH, Kang HC, Zhang J, Xu J, Chen R, Park H, Andrabi SA, Kang SU, Gonçalves RA, Liang Y, Zhang S, Qi C, Lam S, Keiler JA, Tyson J, Kim D, Panicker N, Yun SP, Workman CJ, Vignali DA, Dawson VL, Ko HS, Dawson TM (2016) Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353(6307): aah3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK (2006) Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci 26:41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L (2000) Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 287:1265–1269 [DOI] [PubMed] [Google Scholar]
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DMA, Hasegawa M (2013) Prion-like spreading of pathological α-synuclein in brain. Brain 136:1128–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Kubo M, Shimozawa A, Akiyama H, Hasegawa M (2014) Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol Commun 2:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y, Vila M, Lincoln S, McCormack A, Picciano M, LaFrancois J, Yu X, Dickson D, Langston WJ, McGowan E, Farrer M, Hardy J, Duff K, Przedborski S, Di Monte DA (2001) Lack of nigral pathology in transgenic mice expressing human alpha-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis 8:535–539 [DOI] [PubMed] [Google Scholar]
- Milber JM, Noorigian JV, Morley JF, Petrovitch H, White L, Ross GW, Duda JE (2012) Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology 79:2307–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mougenot A-L, Nicot S, Bencsik A, Morignat E, Verchère J, Lakhdar L, Legastelois S, Baron T (2012) Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging 33:2225–2228 [DOI] [PubMed] [Google Scholar]
- Ogawa SK, Cohen JY, Hwang D, Uchida N, Watabe-Uchida M (2014) Organization of monosynaptic inputs to the serotonin and dopamine neuromodulatory systems. Cell Rep 8:1105–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterberg VR, Spinelli KJ, Weston LJ, Luk KC, Woltjer RL, Unni VK (2015) Progressive aggregation of alpha-synuclein and selective degeneration of Lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep 10:1252–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P (2008) Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol Life Sci 65:1272–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SS, Lee D (2006) Selective loss of dopaminergic neurons and formation of Lewy body-like aggregations in alpha-synuclein transgenic fly neuronal cultures. Eur J Neurosci 23:2908–2914 [DOI] [PubMed] [Google Scholar]
- Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ, Steece-Collier K, Kemp CJ, Celano S, Schulz E, Sandoval IM, Fleming S, Dirr E, Polinski NK, Trojanowski JQ, Lee VM, Sortwell CE (2015) Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis 82:185–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen KM, Marner L, Pakkenberg H, Pakkenberg B (2005) No global loss of neocortical neurons in Parkinson’s disease: a quantitative stereological study. Mov Disord 20:164–171 [DOI] [PubMed] [Google Scholar]
- Peelaerts W, Baekelandt V (2016) ɑ-Synuclein strains and the variable pathologies of synucleinopathies. J Neurochem 139 Suppl 1:256–274 [DOI] [PubMed] [Google Scholar]
- Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van den Haute C, Melki R, Baekelandt V (2015) α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522:340–344 [DOI] [PubMed] [Google Scholar]
- Peeraer E, Bottelbergs A, Van Kolen K, Stancu IC, Vasconcelos B, Mahieu M, Duytschaever H, Ver Donck L, Torremans A, Sluydts E, Van Acker N, Kemp JA, Mercken M, Brunden KR, Trojanowski JQ, Dewachter I, Lee VM, Moechars D (2015) Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol Dis 73:83–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periquet M, Fulga T, Myllykangas L, Schlossmacher MG, Feany MB (2007) Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J Neurosci 27:3338–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucelli L, O’Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR (2002) Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 36:1007–1019 [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047 [DOI] [PubMed] [Google Scholar]
- Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, Patel S, Oehler A, Lowe JK, Kravitz SN, Geschwind DH, Glidden DV, Halliday GM, Middleton LT, Gentleman SM, Grinberg LT, Giles K (2015) Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci USA 112:E5308–E5317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recasens A, Dehay B, Bové J, Carballo-Carbajal I, Dovero S, Pérez-Villalba A, Fernagut P-O, Blesa J, Parent A, Perier C, Fariñas I, Obeso JA, Bezard E, Vila M (2014) Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 75:351–362 [DOI] [PubMed] [Google Scholar]
- Reeve AK, Abramov AY, Klenerman D, Turnbull DM, Simcox EM, Horrocks MH, Ludtmann MH, Angelova PR, Gandhi S (2015) Aggregated α-synuclein and complex I deficiency: exploration of their relationship in differentiated neurons. Cell Death Dis 6:e1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey NL, George S, Brundin P (2016a) Review: Spreading the word: precise animal models and validated methods are vital when evaluating prion-like behaviour of alpha-synuclein. Neuropathol Appl Neurobiol 42:51–76 [DOI] [PubMed] [Google Scholar]
- Rey NL, Petit GH, Bousset L, Melki R, Brundin P (2013) Transfer of human α-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol 126:555–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ, Lee VM-Y, Brundin P (2016b) Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med 213:1759–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey NL, Wesson DW, Brundin P (2016c) The olfactory bulb as the entry site for prion-like propagation in neurodegenerative diseases. Neurobiol Dis 109: 226–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes JF, Rey NL, Bousset L, Melki R, Brundin P, Angot E (2014) Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 62:387–398 [DOI] [PubMed] [Google Scholar]
- Richfield EK, Thiruchelvam MJ, Cory-Slechta DA, Wuertzer C, Gainetdinov RR, Caron MG, Di Monte DA, Federoff HJ (2002) Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp Neurol 175:35–48 [DOI] [PubMed] [Google Scholar]
- Rostami J, Holmqvist S, Lindström V, et al. (2017) Human astrocytes transfer aggregated alpha-synuclein via tunneling nanotubes. J Neurosci 37(49): 11835–11853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacino AN, Brooks M, McGarvey NH, McKinney AB, Thomas MA, Levites Y, Ran Y, Golde TE, Giasson BI (2013) Induction of CNS α-synuclein pathology by fibrillar and non-amyloidogenic recombinant α-synuclein. Acta Neuropathol Commun 1:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacino AN, Brooks M, Thomas MA, McKinney AB, Lee S, Regenhardt RW, McGarvey NH, Ayers JI, Notterpek L, Borchelt DR, Golde TE, Giasson BI (2014a) Amyloidogenic α-synuclein seeds do not invariably induce rapid, widespread pathology in mice. Acta Neuropathol 127:645–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacino AN, Brooks M, Thomas MA, McKinney AB, McGarvey NH, Rutherford NL, Ceballos-Diaz C, Robertson J, Golde TE, Giasson BI (2014b) Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc Natl Acad Sci USA 111:10732–10737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent D, Verchère J, Lazizzera C, Gaillard D, Lakhdar L, Streichenberger N, Morignat E, Bétemps D, Baron T (2017) “Prion-like” propagation of the synucleinopathy of M83 transgenic mice depends on the mouse genotype and type of inoculum. J Neurochem 143:126–135 [DOI] [PubMed] [Google Scholar]
- Shrivastava AN, Redeker V, Fritz N, Pieri L, Almeida LG, Spolidoro M, Liebmann T, Bousset L, Renner M, Léna C, Aperia A, Melki R, Triller A (2015) α-synuclein assemblies sequester neuronal α3-Na+/K+-ATPase and impair Na+ gradient. EMBO J 34:2408–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava R, Singh V, Asif M, Negi MPS, Bhadauria S (2017) Oncostatin M upregulates HIF-1α in breast tumor associated macrophages independent of intracellular oxygen concentration. Life Sci 194:59–66 [DOI] [PubMed] [Google Scholar]
- Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A, Dawson VL, Dawson TM, Ross CA (2005) Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet 14:3801–3811 [DOI] [PubMed] [Google Scholar]
- Spencer B, Valera E, Rockenstein E, Overk C, Mante M, Adame A, Zago W, Seubert P, Barbour R, Schenk D, Games D, Rissman RA, Masliah E (2017) Anti-α-synuclein immunotherapy reduces α-synuclein propagation in the axon and degeneration in a combined viral vector and transgenic model of synucleinopathy. Acta Neuropathol Commun 5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M (2017) Neurodegeneration and the ordered assembly of α-synuclein. Cell Tissue Res 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840 [DOI] [PubMed] [Google Scholar]
- St Martin JL, Klucken J, Outeiro TF, Nguyen P, Keller-McGandy C, Cantuti-Castelvetri I, Grammatopoulos TN, Standaert DG, Hyman BT, McLean PJ (2007) Dopaminergic neuron loss and up-regulation of chaperone protein mRNA induced by targeted over-expression of alpha-synuclein in mouse substantia nigra. J Neurochem 100:1449–1457 [DOI] [PubMed] [Google Scholar]
- Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA (2001) Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci 21:9549–9560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöckl MT, Zijlstra N, Subramaniam V (2013) α-Synuclein oligomers: an amyloid pore? Mol Neurobiol 47:613–621 [DOI] [PubMed] [Google Scholar]
- Stopschinski BE, Diamond MI (2017) The prion model for progression and diversity of neurodegenerative diseases. Lancet Neurol 16:323–332 [DOI] [PubMed] [Google Scholar]
- Surgucheva I, Sharov VS, Surguchov A (2012) γ-Synuclein: seeding of α-synuclein aggregation and transmission between cells. Biochemistry 51:4743–4754 [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Obeso JA, Halliday GM (2017) Parkinson’s disease is not simply a prion disorder. J Neurosci 37:9799–9807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamgüney G, Korczyn AD (2017) A critical review of the prion hypothesis of human synucleinopathies. Cell Tissue Res 1–8 [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa A, L Dawson V, Dawson TM, Ross CA (2001) Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet 10:919–926 [DOI] [PubMed] [Google Scholar]
- Tapias V, Hu X, Luk KC, Sanders LH, Lee VM, Greenamyre JT (2017) Synthetic alpha-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cell Mol Life Sci 74:2851–2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur P, Breger LS, Lundblad M, Wan OW, Mattsson B, Luk KC, Lee VMY, Trojanowski JQ, Björklund A (2017) Modeling Parkinson’s disease pathology by combination of fibril seeds and α-synuclein overexpression in the rat brain. Proc Natl Acad Sci USA 114:E8284–E8293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal TC, Lai Y-Y, Siegel JM (2007) Hypocretin (orexin) cell loss in Parkinson’s disease. Brain 130:1586–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsigelny IF, Sharikov Y, Wrasidlo W, Gonzalez T, Desplats PA, Crews L, Spencer B, Masliah E (2012) Role of α-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J 279:1000–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson T, Senchuk M, Cooper JF, George S, Van Raamsdonk JM, Brundin P (2017) Novel animal model defines genetic contributions for neuron-to-neuron transfer of α-synuclein. Sci Rep 7:7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulusoy A, Musgrove RE, Rusconi R, Klinkenberg M, Helwig M, Schneider A, Di Monte DA (2015) Neuron-to-neuron α-synuclein propagation in vivo is independent of neuronal injury. Acta Neuropathol Commun 3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulusoy A, Phillips RJ, Helwig M, Klinkenberg M, Powley TL, Di Monte DA (2017) Brain-to-stomach transfer of α-synuclein via vagal preganglionic projections. Acta Neuropathol 133:381–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulusoy A, Rusconi R, Pérez-Revuelta BI, Musgrove RE, Helwig M, Winzen-Reichert B, Monte DAD (2013) Caudo-rostral brain spreading of α-synuclein through vagal connections. EMBO Mol Med 5:1051–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdinocci D, Radford RAW, Siow SM, Chung RS, Pountney DL (2017) Potential Modes of Intercellular α-Synuclein Transmission. Int J Mol Sci 18(2):E469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Putten H, Wiederhold KH, Probst A, Barbieri S, Mistl C, Danner S, Kauffmann S, Hofele K, Spooren WP, Ruegg MA, Lin S, Caroni P, Sommer B, Tolnay M, Bilbe G (2000) Neuropathology in mice expressing human alpha-synuclein. J Neurosci 20:6021–6029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LC (2016) Proteopathic Strains and the Heterogeneity of Neurodegenerative Diseases. Annu Rev Genet 50:329–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N (2012) Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron 74:858–873 [DOI] [PubMed] [Google Scholar]
- Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM, DeArmond SJ, Prusiner SB (2013) Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci USA 110:19555–19560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Krainc D (2017) α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med 23:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Iwatsubo T, Mizuno Y, Mochizuki H (2004) Overexpression of alpha-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of alpha-synuclein and activation of caspase-9: resemblance to pathogenetic changes in Parkinson’s disease. J Neurochem 91:451–461 [DOI] [PubMed] [Google Scholar]